

Abstract
Dual-acting kappa opioid receptor (KOR) agonist and mu opioid receptor (MOR) partial agonist ligands have been put forward as potential treatment agents for cocaine and other psychostimulant abuse. Members of the orvinol series of ligands are known for their high binding affinity to both KOR and MOR, but efficacy at the individual receptors has not been thoroughly evaluated. In this study, it is shown that a predictive model for efficacy at KOR can be derived, with efficacy being controlled by the length of the group attached to C20 and by the introduction of branching into the side chain. In vivo evaluation of two ligands with the desired in vitro profile confirms both display KOR, and to a lesser extent MOR, activity in an analgesic assay suggesting that, in this series, in vitro measures of efficacy using the [35S]GTPγS assay are predictive of the in vivo profile.
Introduction
The continuing illicit use of psychoactive substances, with the resulting health and social consequences, emphasizes the need for improved pharmacotherapies for drug abuse. Treatments for opiate abuse, such as methadone and buprenorphine (1n), are available and have proven successful against a range of addict populations. However, there are no such approved pharmacotherapies for cocaine abuse, though a wide range of possible treatment agents have been evaluated in the laboratory and in clinical trials.1
There has been interest in the use of kappa opioid receptor (KOR) agonists as potential pharmacotherapies for cocaine and other psychostimulant abuse,2−7 particularly as repeated administration of KOR agonists has been shown to prevent or reduce many of cocaine’s behavioral effects.4,8−10 Primarily due to the dysphoric effects produced by KOR-agonists, the development of a KOR-agonist pharmacotherapy for human use is not straightforward. For example, while enadoline (CI-977), a high efficacy, selective KOR-agonist, appeared to be better tolerated in subjects with a history of drug use compared to naive individuals, it still caused some dysphoria.11 Overall, the evidence suggests that higher efficacy KOR-agonists with some additional mu opioid receptor (MOR)-agonist activity, such as ethylketazocine (EKC), are more effective in reducing cocaine self-administration, and display fewer side effects, than their more KOR-selective counterparts such as enadoline and spiradoline.4,12 Presumably, the presence of some MOR-agonist effects helps attenuate any dysphoria induced by the KOR-agonism.
In the treatment of opiate abuse, a long-duration of action is of benefit for successful treatment agents. The main pharmacotherapies methadone and buprenorphine (1n), but also LAAM, are long acting, and their success is at least partly as a result of this property.13 Together with its reduced efficacy, the slow onset of effects displayed by buprenorphine (1n) appears to reduce its abuse potential. EKC and the other KOR-agonists effective in reducing cocaine self-administration all have short duration of action.4 A strategy for the treatment of cocaine abuse could therefore be the utilization of mixed KOR-agonists/MOR-partial agonists having extended duration of action. Of interest in this regard are the orvinols, a series of opioids displaying a range of pharmacological profiles including the long-acting MOR partial agonist 1n.14 The orvinols have not previously been thoroughly evaluated as KOR-agonists, but it has become clear that a number of them do display substantial KOR-efficacy in vivo and that their lack of absolute KOR-selectivity, in particular over MOR, makes them of particular interest to this project. An early example was the isopentyl orvinol M320 (5: Table 1), which was the subject of a detailed pharmacological study by Boura and Fitzgerald.15 Compound 5 shows a KOR/MOR profile similar to that of EKC, but it is more potent and very much longer acting. In this article, we describe our initial work toward analogues of 5 and 1n as potential therapies for cocaine abuse. There is a particular focus on orvinols having a branched chain attached to C20 as these regions (above and away from C6/C7 and below C8) are associated with efficacy at KOR.16,17
Table 1. Binding Affinities of Ligands to Opioid Receptors and Stimulation of [35S]GTPγS Binding to CHO-KOR and C6-MOR Membranesa.
| R | Ki/nM – MOR | Ki/nM – KOR | EC50/nM, % stim MOR | EC50/nM, % stim KOR | |
|---|---|---|---|---|---|
| 1a | iPr | 0.15 ± 0.013 | 0.051 ± 0.024 | 2.1 ± 1.4, 12 ± 4.4 | 0.083 ± 0.048, 30 ± 5.3 |
| 1ab | iPr | 0.60 ± 0.05 | 0.40 ± 0.20 | ||
| 1be | ibutyl | 0.20 ± 0.037 | 0.092 ± 0.025 | 1.2 ± 0.56, 14 ± 1.0 | 0.057 ± 0.029, 84 ± 4.1 |
| 1ce | ipentyl | 0.22 ± 0.018 | 0.060 ± 0.013 | 0.19 ± 0.049, 33 ± 7.6 | 0.071 ± 0.027, 69 ± 5.1 |
| 1d | cpentyl | 0.21 ± 0.042 | 0.074 ± 0.024 | 2.8 ± 1.7, 25 ± 3.7 | 0.068 ± 0.045, 46 ± 4.9 |
| 1e | cpentylmethyl | 0.29 ± 0.063 | 0.069 ± 0.019 | 0.56 ± 0.12, 38 ± 0.35 | 0.011 ± 0.0033, 93 ± 1.2 |
| 1f | cpentylethyl | 0.62 ± 0.17 | 0.12 ± 0.019 | 2.9 ± 0.51, 56 ± 4.7 | 0.027 ± 0.0067, 82 ± 5.3 |
| 1g | chexyl | 0.21 ± 0.069 | 0.10 ± 0.033 | 0.43 ± 0.10, 35 ± 2.8 | 0.031 ± 0.0061, 53 ± 0.22 |
| 1h | chexylmethyl | 0.34 ± 0.12 | 0.051 ± 0.016 | 1.7 ± 0.86, 63 ± 5.7 | 0.017 ± 0.0012, 75 ± 1.6 |
| 1i | chexylethyl | 1.0 ± 0.14 | 0.18 ± 0.022 | 4.8 ± 1.5, 43 ± 4.6 | 0.041 ± 0.035, 89 ± 1.7 |
| 1j | benzyl | 0.15 ± 0.036 | 0.082 ± 0.025 | 1.3 ± 0.98, 75 ± 8.6 | 0.071 ± 0.011, 44 ± 6.6 |
| 1k | phenethyl | 0.32 ± 0.067 | 0.065 ± 0.022 | 4.9 ± 3.0, 47 ± 8.6 | 0.016 ± 0.011, 89 ± 6.9 |
| 1lb | nPropyl | 0.90 ± 0.20 | 1.2 ± 0.10 | N.D. | N.D. |
| 1mb | nPentyl | 2.4 ± 1.0 | 4.7 ± 0.10 | N.D. | N.D. |
| 1n | tButyl | 0.19 ± 0.018 | 0.067 ± 0.021 | 0.27 ± 0.094, 18 ± 0.99 | -, 0 |
| 2a | ipropyl | 0.091 ± 0.011 | 0.11 ± 0.024 | 1.27 ± 0.72, 11 ± 1.1 | 0.46 ± 0.13, 53 ± 2.6 |
| 2b | ibutyl | 0.19 ± 0.036 | 0.11 ± 0.024 | 0.85 ± 0.59, 7.6 ± 1.7 | 0.18 ± 0.078, 89 ± 4.0 |
| 2c | ipentyl | 0.21 ± 0.099 | 0.15 ± 0.044 | 0.43 ± 0.098, 25 ± 1.3 | 0.16 ± 0.084, 96 ± 11 |
| 2d | cpentyl | 0.18 ± 0.048 | 0.15 ± 0.037 | 1.0 ± 0.66, 9.3 ± 2.1 | 0.083 ± 0.016, 95 ± 5.2 |
| 2e | chexylmethyl | 0.37 ± 0.12 | 0.22 ± 0.057 | 0.72 ± 0.015, 46 ± 1.8 | 0.31 ± 0.17, 87 ± 1.5 |
| 2f | chexylethyl | 0.47 ± 0.21 | 0.16 ± 0.012 | 2.50 ± 0.98, 8.5 ± 1.8 | 0.46 ± 0.15, 72 ± 2.4 |
| 2gb | nPropyl | 1.3 ± 0.0 | 2.0 ± 0.50 | N.D. | N.D. |
| 2hb | nPentyl | 2.7 ± 0.85 | 5.5 ± 0.90 | N.D. | N.D. |
| 2ib | tButyl | 0.40 ± 0.05 | 0.50 ± 0.10 | N.D. | N.D. |
| 3a | ipropyl | 0.33 ± 0.16 | 0.067 ± 0.011 | 1.0 ± 0.41, 6.9 ± 3.8 | 0.63 ± 0.22, 49 ± 3.1 |
| 3b | ibutyl | 0.054 ± 0.013 | 0.059 ± 0.014 | -, 0c | 0.17 ± 0.045, 89 ± 3.9 |
| 3c | ipentyl | 0.031 ± 0.0086 | 0.033 ± 0.011 | 1.5 ± 0.99, 39 ± 4.8 | 0.042 ± 0.0027, 89 ± 2.0 |
| 3d | cpentyl | 0.31 ± 0.23 | 0.078 ± 0.015 | 0.60 ± 0.057, 11 ± 1.2 | 0.29 ± 0.12, 71 ± 5.4 |
| 3e | chexyl | 0.15 ± 0.035 | 0.090 ± 0.042 | 0.26 ± 0.063, 48 ± 3.8 | 0.053 ± 0.0088, 78 ± 1.9 |
| 3f | chexylmethyl | 0.043 ± 0.016 | 0.015 ± 0.0043 | 0.56 ± 0.098, 45 ± 4.5 | 0.19 ± 0.037, 81 ± 4.6 |
| 3g | chexylethyl | 0.060 ± 0.021 | 0.019 ± 0.0030 | 2.0 ± 0.45, 23 ± 1.6 | 0.24 ± 0.027, 94 ± 9.3 |
| 3h | benzyl | 0.038 ± 0.018 | 0.020 ± 0.0066 | 0.32 ± 0.11, 41 ± 2.4 | 0.077 ± 0.039, 87 ± 8.7 |
| 3i | phenethyl | 0.029 ± 0.0035 | 0.025 ± 0.0039 | -, 0c | 0.052 ± 0.020, 109 ± 8.2 |
| 4a | ipropyl | 0.88 ± 0.19 | 0.16 ± 0.079 | -, 0 | 0.035 ± 0.018, 59 ± 7.2 |
| 4b | ibutyl | 0.68 ± 0.20 | 0.057 ± 0.025 | 0.48 ± 0.24, 20 ± 2.0 | 0.013 ± 0.0092, 107 ± 7.3 |
| 4c | ipentyl | 0.12 ± 0.0074 | 0.067 ± 0.023 | 0.16 ± 0.059, 65 ± 2.0 | 0.0061 ± 0.002, 113 ± 3.3 |
| 4d | cpentyl | 1.0 ± 0.48 | 0.086 ± 0.048 | 0.29 ± 0.12, 35 ± 35 | 0.020 ± 0.080, 104 ± 12 |
| 4e | cpentylmethyl | 0.36 ± 0.067 | 0.086 ± 0.040 | 0.16 ± 0.060, 80 ± 3.3 | 0.007 ± 0.0007, 103 ± 3.9 |
| 4f | cpentylethyl | 0.49 ± 0.058 | 0.062 ± 0.017 | 0.16 ± 0.026, 83 ± 7.8 | 0.0077 ± 0.0045, 95 ± 8.1 |
| 4g | chexyl | 0.41 ± 0.14 | 0.12 ± 0.0082 | 0.12 ± 0.0070, 73 ± 8.1 | 0.0098 ± 0.003, 102 ± 6.2 |
| 4h | chexylmethyl | 0.27 ± 0.12 | 0.12 ± 0.028 | 0.094 ± 0.029, 91 ± 9.3 | 0.029 ± 0.015, 108 ± 1.9 |
| 4i | chexylethyl | 0.23 ± 0.17 | 0.086 ± 0.016 | 0.20 ± 0.10, 34 ± 2.9 | 0.047 ± 0.003, 83 ± 6.7 |
| 4j | benzyl | 0.030 ± 0.0041 | 0.041 ± 0.0060 | 0.23 ± 0.13, 45 ± 4.0 | 0.076 ± 0.043, 63 ± 7.3 |
| 4k | phenethyl | 0.029 ± 0.0058 | 0.047 ± 0.0054 | 21.0 ± 17.5, 7.5 ± 2.0 | 0.73 ± 0.58, 58 ± 4.9 |
| 5 | iPentyld | 0.18 ± 0.068 | 0.082 ± 0.019 | 1.0 ± 0.42, 34 ± 3.2 | 0.051 ± 0.022, 91 ± 3.7 |
| EKC | 0.39 ± 0.11 | 4.4 ± 2.5, 26 ± 1.0 | 2.2 ± 0.66, 93 ± 1.3 |
Ki (nM) versus [3H]diprenorphine; values are an average ± SEM from three separate experiments. Percent maximal stimulation (% stim) with respect to the standard agonists DAMGO (MOR) and U69,593 (KOR); values are an average ± SEM from three separate experiments; N.D., not determined.
Binding to Hartley guinea pig membranes, Ki (nM) versus [3H]DAMGO (MOR) and [3H]U69,593 (KOR).
3b Ke (versus DAMGO) 0.50 ± 0.08 nM and 3i Ke (versus DAMGO) 0.35 ± 0.11.
C6–C14 etheno bridged analogue of 1c.
Affinities for 1b and 1c at DOR were determined by displacement of [3H]-diprenorphine binding from C6-rat glioma cells expressing recombinant DOR; Ki values were 0.34 ± 0.07 nM and 0.42 ± 0.18 nM, respectively. Efficacy at DOR was 1b, 2.3 ± 1.3%; EC50 was not determined; and 1c, 7.9 ± 1.7%; EC50 values were not determined.
Synthesis
The compounds were prepared using the standard techniques for orvinol synthesis.14,18,19 Only one significant change has been made to the synthetic route and relates to how and when the N-methyl group is replaced with cyclopropylmethyl in series 1. We have found that this process is most reliably performed (simpler and higher yielding) with diisopropyl azodicarboxylate (DIAD) on methyl ketone 6 (Scheme 1). Grignard addition to ketone 8 provided series 1 via 9. Series 2–4 were all synthesized from aldehyde 10, itself prepared from N-cyclopropylnorthebaine,20 the initial step being the addition of the appropriate Grignard reagent, which gave a mixture of diastereomeric secondary alcohols, with the major isomer (11a) as shown (Scheme 2). This major product in each case was isolated in sufficient quantity to allow 3-O-demethylation to series 4. Swern oxidation of 11a alone or the mixture of diastereomeric alcohols gave ketones 12 that on treatment with methylmagnesium bromide and subsequent 3-O-demethylation gave the diastereomeric 3°-alcohol series (2). Reduction of 12 gave the 2° alcohols 11b (major product) and 11a (minor product), with 3-O-demethylation of 11b giving access to series 3.
Scheme 1.

Scheme 2.

Results
Four series of orvinols were studied. These were tertiary alcohols having the same relative stereochemistry as buprenorphine (1a–1n, Scheme 1), a more limited series of diastereomeric tertiary alcohols (2a–2i), secondary alcohols with the same relative stereochemistry as buprenorphine (3a–3i), and finally a series of diastereomeric secondary alcohols (4a–4k) (Scheme 2).
Binding affinities of the new compounds for MOR and KOR were determined by displacement of [3H]-diprenorphine binding from C6-rat glioma cells expressing recombinant rat MOR and CHO cells expressing recombinant human KOR. Details of the assay have been described previously.21 Delta opioid receptor (DOR) activity of the new ligands was only determined for the ligands progressed to in vivo studies as in no case has DOR activity proved significant in the pharmacological profile of orvinol based ligands. As expected, all of the ligands bound with high to very high affinity to both KOR and MOR with little or no selectivity for one receptor over the other (Table 1). If any selectivity was observed, it was for the KOR (e.g., 4b and 4d had 11-fold higher affinity for KOR than for MOR). Affinities for KOR were in the subnanomolar range (0.015 to 0.22 nM) for all of the ligands, with affinities for the MOR in the range 0.029–1.0 nM. Binding affinities for five of the ligands (1l, 1m, 2g, 2h, and 2i), all synthesized at an earlier time, were evaluated in guinea pig brain membranes using established methods.22 Compound 1a was evaluated in both binding assays, providing a reference to enable comparison with the rest of the series. Affinities were lower in the guinea pig brain membrane assay but confirmed the lack of selectivity, with equal affinity for KOR and MOR. Affinities for 1b and 1c at DOR were determined by displacement of [3H]-diprenorphine binding from C6-rat glioma cells expressing recombinant DOR. They bound with high affinity, with Ki values of 0.34 nM and 0.42 nM, respectively.
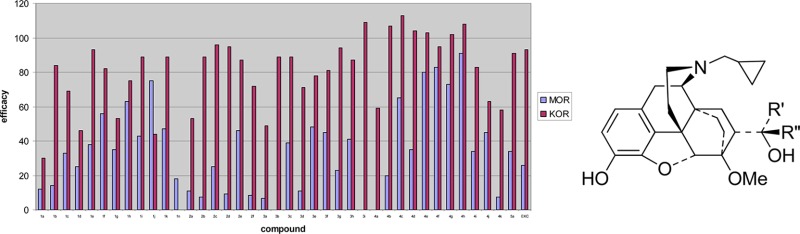
The primary in vitro assay used to determine opioid receptor functional activity was the [35S]GTPγS assay, which, like the binding assays, was performed on recombinant opioid receptors transfected into C6-rat glioma cells (for MOR) and CHO cells (for KOR). Assays were performed as previously described by Traynor and Nahorski.23 Agonist efficacy at these opioid receptors was determined in comparison to the standard selective agonists DAMGO (MOR), and U69593 (KOR) (Table 1). In this assay, buprenorphine (1n) displayed no efficacy at KOR, while M320 (5) was a very potent, full agonist (91% stimulation). The new compounds displayed a range of efficacies at KOR, from low efficacy partial agonists (e.g., 1a, 1d, 1j, and 3a had efficacies from 30–49%) to full agonists of equivalent efficacy to the standard U69593 (e.g., most of series 4 and 2c, 2d, and 3i among others).
The majority of ligands displayed lower efficacy at MOR than at KOR; the most extreme examples being 3b and 3i which are potent, full agonists at KOR, with no efficacy at MOR. They proved to be potent MOR antagonists shifting the concentration effect curve for the standard agonist DAMGO in a parallel fashion giving Ke(MOR) of 0.50 nM and 0.35 nM, respectively. Compound 4a was similarly selective but with partial agonist activity at the KOR. Substantial selectivity in efficacy for KOR was seen in a number of other ligands including 1b, 2b, 2c, 2d, 2f, 3g, 4b, and 4d. Within series 1, 2, and 3, highest MOR efficacy was typically found with the cyclohexylmethyl and closely related benzyl side chains. Compound 4h, having a cyclohexylmethyl side chain, also had high efficacy, although in this series, 4j (having a benzyl group) was only a partial agonist with moderate efficacy. The two ligands, 1b and 1c, evaluated at DOR were both of very low efficacy (2% and 8% stimulation, respectively).
Five ligands (1l, 1m, 2g, 2h, and 2i) were evaluated in the guinea pig ileum (GPI) and mouse vas deferens (MVD) isolated tissue assays, instead of [35S]GTPγS assays (Table 2), using standard procedures.22 Compounds 1a and 1n were also tested in these isolated tissue assays to allow meaningful comparison between results from the different assays. The GPI has both MOR and KOR populations and is sensitive to KOR agonist effects, while the MVD has all three types of opioid receptors and is particularly sensitive to DOR agonists and least sensitive to KOR agonists. The n-propyl analogue (1l) and the diastereomeric n-pentyl analogue (2h) were partial agonists in the GPI, whereas the other ligands evaluated in this assay were all of higher efficacy. Thus, 1l had lower efficacy than its i-propyl isomer 1a and also lower than the bulky t-butyl containing 1n and also 1m, having the longer n-pentyl chain. Compound 2g, the diastereomer of 1l, was also of higher efficacy as was the t-butyl containing 2i. The effects of these ligands in the GPI could not readily be reversed by the standard antagonists CTAP and norBNI. Each of the compounds was found to be an antagonist in the MVD with 1a, 1l, and 1m displaying some selectivity for MOR. Antagonist Ke’s at KOR were not determined for 2g and 2i as they had demonstrated efficacy in the GPI.
Table 2. Agonist Activity (IC50/nM) in the Guinea Pig Ileum (GPI) and Antagonist Activity (Ke/nM) in the Mouse Vas Deferens (MVD).
| GPI | Ke (nM)a |
MVDe |
|||||
|---|---|---|---|---|---|---|---|
| IC50 (nM) | CTAP | norBNI | Ke (MOR) | Ke (KOR) | Ke (DOR) | ||
| 1a | iPr | 0.52 ± 0.49 | N.R.b | N.R. | 0.034 ± 0.004 | 0.37 ± 0.21 | 0.90 ± 0.32 |
| 1l | nPr | P.A.c | N.D.d | N.D. | 0.007 ± 0.0002 | 0.20 ± 0.12 | 0.16 ± 0.03 |
| 1m | nPent | 12.3 ± 2.1 | N.R. | N.R. | 0.004 ± 0.001 | 0.11 ± 0.04 | 0.06 ± 0.02 |
| 1n | tBu | 8.1 ± 3.6 | N.R | N.R | - | - | - |
| 2g | nPr | 0.49 ± 0.31 | N.R. | 0.71 ± 0.07 | 0.028 ± 0.003 | - | 0.31 ± 0.08 |
| 2h | nPent | P.A. | N.D. | N.D. | 0.02 ± 0.006 | 0.01 ± 0.002 | 0.35 ± 0.06 |
| 2i | tBu | 2.0 ± 1.4 | N.R. | 1.8 ± 1.33 | 0.043 ± 0.009 | - | 15 ± 5.9 |
Ke (nM) of the selective antagonists norBNI (KOR) and CTAP (MOR) versus test compound.
N.R.: the antagonists did not reverse the activity of the test compound.
Partial agonist with maximum inhibition of twitch of 30–50%.
N.D., not determined.
Antagonist Ke (nM) of the test compound versus the standard agonists DAMGO (MOR), U69,593 (KOR), and DPDPE (DOR). Values are from two experiments, each carried out in triplicate.
Comparative molecular field analysis (CoMFA) is a technique used to generate a description of a 3D structure–activity relationship in a quantitative manner, i.e., it is a 3D variant of a quantitative structure–activity relationship (QSAR). The output of a CoMFA is a 3D model showing structural features of the input data that are likely to affect, beneficially or adversely, the activity of the compounds. A CoMFA was used to help identify the regions near C20 that are beneficial or detrimental to KOR efficacy and to help confirm the conformation adopted by the different isomeric series on binding to the KOR. All the compounds evaluated in the [35S]GTPγS assay, including 1n and 5, were used to develop the model. With all compounds overlaid in their lowest energy, C6-OMe to C20-OH hydrogen bonded conformation (as depicted for 1n in Figure 1) and using the % stimulation at KOR data, an R2 of 0.911 and a cross-validated R2 of 0.447 were obtained. As can be seen in Figure 1, the model predicts that for high KOR efficacy there are two areas close to C20 that could beneficially be occupied by lipophilic groups, the first is away from C7 and the second is the region below C8. The model also suggests that lipophilic groups closer to C20 are not well tolerated, leading to lower KOR efficacy. Evaluation of alternative conformations about the C7–C20 bond (e.g., so that the bulky R group of series 1 and 3 occupies the region below C8) led to substantial reductions in R2 and in particular the cross-validated R2.
Figure 1.
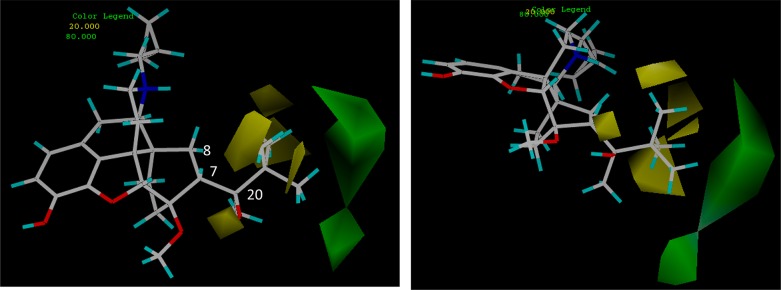
Two views of the areas, predicted by COMFA analysis, where interaction with a lipophilic group would be beneficial for KOR activation (shown in green) and where interaction with a lipophilic group would be detrimental to efficacy (shown in yellow).
The docking of 1n to the recently published crystal structures of the MOR and KOR opioid receptors was examined. The crystal structure24 of the KOR was determined in the presence of the selective KOR antagonist JDTic and presumably represents the antagonist conformation of the receptor. JDTic is enclosed in a largely hydrophobic pocket with only one direct hydrogen bond to the protein. There is a network of water-mediated hydrogen bonds between the ligand and the protein. The docked 1n structure overlays the 7-hydroxy-tetrahydroisoquinoline with the buprenorphine phenolic hydroxy group making the same interactions as the equivalent group in JDTic (Figure 2). There is a hydrogen bond between the protonated nitrogen and the side chain of Asp138. The isopropyl group of JDTic fits into and fills a tight pocket formed by Val108, Asn141, Trp287, and Tyr320: this interaction is replicated by the cyclopropyl moiety of 1n. The t-butyl group makes no specific interactions with the protein but projects into the large pocket that the phenylpiperidine motif of JDTic occupies.
Figure 2.
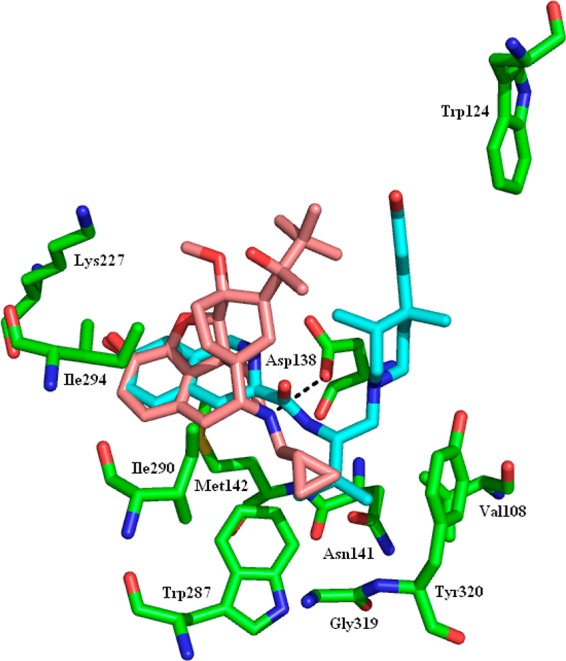
KOR with the protein in green, the crystal structure ligand (JDTic) in cyan, and the docked buprenorphine (1n) in pink. The hydrogen bond between the docked ligand and Asp138 is shown as the black dashed line.
The MOR was crystallized with the irreversible antagonist β-FNA present in the binding site,25 a ligand with significant structural similarity to buprenorphine. β-FNA is covalently bound to the side chain nitrogen of Lys233 (Figure 3). The docked 1n structure approximately overlays β-FNA but, without the covalent restraint, lies a little deeper in the binding site. There is a hydrogen bond between the positively charged nitrogen and the side chain of Asp147. The cyclopropyl moiety overlays that of β-FNA occupying, but not filling, a small pocket formed by Asn150, Trp293, Ile322, and Tyr326. The t-butyl group makes no specific interactions with the protein but projects into the same large pocket occupied by the methyl ester of β-FNA.
Figure 3.
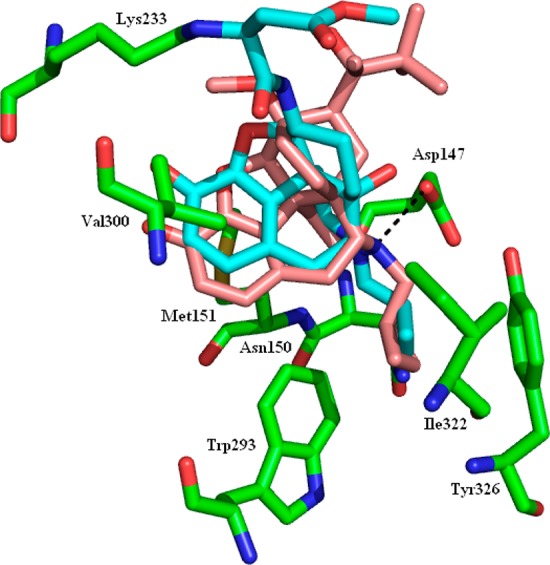
MOR with the protein in green, the crystal structure ligand (β-FNA) in cyan, and the docked buprenorphine (1n) in pink. The hydrogen bond between the docked ligand and Asp147 is shown as the black dashed line.
Two ligands, the i-butyl (1b) and i-pentyl (1c) analogues of buprenorphine (1n), were selected for preliminary in vivo evaluation in the para-phenylquinone (PPQ) abdominal stretch assay (Table 3).26 In this assay, PPQ acts as a fairly low intensity nociceptive stimulus such that even partial opioid agonists are active as antinociceptives. These two ligands were chosen as they had the desired lower efficacy partial agonist activity at MOR in the [35S]GTPγS assay coupled with higher efficacy at KOR. As it is difficult to accurately predict the in vivo profile of mixed-action ligands, one ligand (1b) was chosen that was substantially biased toward KOR, while the other (1c) had a more balanced profile. In the PPQ assay, 1b was a potent agonist (ED50 0.02 mg/kg s.c.) that could be reversed by norBNI (AD50 6.5 mg/kg s.c.) but only partly reversed by β-FNA (a maximum reversal of 27% at the highest β-FNA dose) (Table 3). Compound 1c was equally potent (0.02 mg/kg s.c.) and again could be reversed by norBNI (AD50 2.45 mg/kg s.c.) and partially reversed by β-FNA (59% reversed at 10 μg/brain β-FNA).
Table 3. Antinociceptive Activity in the PPQ Induced Abdominal Stretch Assaya.
| reversal by selective opioid antagonists:
AD50 or % reversal |
|||
|---|---|---|---|
| ED50, mg/kg s.c. | β-FNA, μg/brain i.c.v (MOR) | norBNI, mg/kg s.c. (KOR) | |
| 1b | 0.02 (0.01–0.03) | 15% at 1, 5% at 10, 27% at 30 | 6.5 (2.6–16) |
| 1c | 0.02 (0.01–0.04) | 14% at 3, 59% at 10, 12% at 30 | 2.5 (0.53–11.4) |
Methods were as described previously (ref (26)).
Discussion
As predicted, based on earlier work by ourselves and others,16,17,27 the N-cyclopropylmethylnororvinols synthesized and evaluated in this study had very high affinity but little to no MOR/KOR selectivity in binding assays. Also as predicted, in the [35S]GTPγS assay used to determine the level of agonist activity at each receptor, the majority of the ligands were KOR agonists with lower efficacy at the MOR. Importantly, the [35S]GTPγS data for the known compounds buprenorphine (1n) and M320 (5) were in agreement with previously reported isolated tissue data and findings from in vivo studies, with 1n displaying no efficacy at the KOR, while 5 was a very potent, full agonist at this receptor, and both displayed partial agonist activity at the MOR such that 1n profiled as a MOR partial agonist, KOR antagonist and 5 as a high efficacy KOR agonist with some MOR activity.14,28 Similarly, evaluation of 1a in both [35S]GTPγS and isolated tissue assays (Tables 1 and 2) gave results in good agreement, indicating low efficacy at each receptor.
We have previously proposed that KOR agonist activity can be achieved in orvinol and related series through interaction with either a region above and away from C7 (relating to the site occupied by the R-group in series 1 and 3) or a region below C8 (relating to the site occupied by the R-group in series 2 and 4).16,17 The results of the CoMFA analysis performed on the compounds prepared for this study are in agreement with this hypothesis. With all compounds held in their low energy H-bonding conformation, an excellent correlation was found between predicted and actual KOR efficacy and two regions, corresponding to the two lipophilic sites just described, were highlighted as being beneficial to KOR efficacy (Figure 1). Importantly, this appears to confirm that the hydrogen bonded conformation of the orvinols is preserved on interaction with the KOR.
In general, the 2° and 3° alcohols having opposite relative stereochemistry to buprenorphine (series 2 and 4) display higher KOR efficacy than series 1 and 3, which have the same relative stereochemistry to buprenorphine. Substantial differences in efficacy between series was also found at the MOR, with series 4 > series 1 > series 2 ≈ series 3. Comparing the buprenorphine-like 3° alcohols (1) with their diastereoisomers (2), the results from the [35S]GTPγS assays indicate that those having the same relative stereochemistry as buprenorphine have equal or higher efficacy at MOR than their diastereoisomers (2) but that 2 have higher efficacy at KOR, often substantially so. This finding was replicated in the GPI assay where buprenorphine (1n) was found to be a MOR partial agonist, while its isomer (2i) was a potent KOR agonist. Within the 2° alcohols, those with the opposite relative stereochemistry (4) to buprenorphine mostly had higher efficacy for both MOR and KOR than their diastereoisomers (3). The 2° alcohols (3,4) were in general of higher efficacy at both MOR and KOR than the 3° alcohols (1,2).
Within tertiary alcohols (1), KOR efficacy was lowest in those ligands having branching at C21, while insertion of one or two methylene units increased KOR efficacy. Therefore, for example, 1a having an isopropyl group was a low efficacy partial agonist (30% relative to U69,593), whereas 1b and 1c (isobutyl and isopentyl containing) were higher efficacy agonists (84% and 69% relative to U69,593). A similar increase in efficacy on going from isopropyl to isobutyl and isopentyl was seen in the diastereomeric series of tertiary alcohols (2a versus 2b and 2c), but efficacy at the KOR was generally higher for this series than for their diastereomers. The isopropyl group also led to lowest KOR efficacy in both series of secondary alcohols (3a and 4a), though in these series other ligands having branching at C21, for example, the cyclopentyl in 3d and 4d had significantly higher efficacy. These results are consistent with previous studies in isolated tissue assays with 17-NMe orvinols (series 1, but N-cyclopropylmethyl replaced by N-Me) where it was found that branching of the alkyl group at the point of attachment to the carbinol function (i.e., at C21) resulted in lower potency and efficacy at the MOR, while in general, MOR and KOR potency and efficacy increased with increasing chain length.17,29 The results of the CoMFA study provide further support for these conclusions with the region adjacent to C21 found to be detrimental to efficacy at the KOR (Figure 1), appearing to confirm our hypothesis that the loss of KOR efficacy in 1n relative to closely related orvinols is due to a methyl group of the t-butyl moiety interacting favorably with the antagonist conformation of KOR receptor and/or disfavoring the agonist conformation.17,27 Docking of 1n to the recently published crystal structures of the KOR confirms that the t-butyl group accesses a large lipophilic region when the receptor is in an antagonist-bound conformation but does not provide any more detailed information on why buprenorphine is not an agonist at KOR. The modeling does shed some light on why the N-cyclopropylmethyl group, and related moieties such as allyl, helps confer substantial antagonist character to the orvinols and other series; groups of this size appear to bind tightly to a pocket formed by Val108, Asn141, Trp287, and Tyr320 in the KOR and Asn150, Trp293, Ile322, and Tyr326 in the MOR.
Comparison of 5 and 1c suggests that reducing the 6,14-bridge from etheno to ethano has little or no effect on efficacy at MOR but substantially decreases efficacy at KOR. Previously, it has been suggested that reduction of the bridge leads to some attenuation of MOR intrinsic activity,14 though this conclusion was based on in vivo antinociceptive activity and without the benefit of selective antagonists or of data from isolated tissue assays.
The in vivo evaluation of 1b and 1c gave results entirely in keeping with the in vitro analysis. Both had substantial KOR agonism with 1c having a greater MOR component than 1b. While there is no clear dose dependency found in the experiments using β-FNA pretreatment, this may simply be indicative of the low partial agonist character of these ligands at the MOR, meaning we are looking at inhibition in vivo of a group of receptors responsible for only part of the activity (analgesia) that is the measured end-point. The current in vitro analysis also helps rationalize the in vivo data that has been reported for a limited number of these compounds previously.30 The in vivo data was collected on compounds of type 1 with R groups from methyl to butyl (including branched isomers) and suggested that compounds with R = n-propyl or larger (up to t-butyl) were MOR partial agonists that in some cases had high efficacy KOR agonist activity.30 The in vitro data generated for the compounds common to these previous studies and the current work (1a, 1b, 1l, 1n; R = i-propyl, i-butyl, n-propyl, and t-butyl) is mostly consistent with these findings with low MOR efficacy but variable KOR efficacy that appears to initially increase with the size of the R group (e.g., i-propyl → i-butyl, 30 → 84%) before then decreasing again (i-butyl → t-butyl, 84 → 0%). The n-propyl analogue (1l) was only evaluated in isolated tissue assays where it was found to be of low efficacy agonist in guinea pig ileum (GPI). The difficulty in reversing the agonist effects of the ligands in the GPI by selective antagonists for MOR and KOR (CTAP and norBNI) makes commenting on the receptor selectivity of this activity difficult but is typical of the orvinol series and results from their tight, long-lived binding to the receptors and can be indicative of an extended duration of action.17,31 Differences between the GPI and the cell based [35S]GTPγS assays may be related to the easier access to receptors in the cell homogenates. The activity of these compounds as antagonists in the mouse vas deferens (MVD) suggested that all were partial, rather than full, agonists. However, in vivo 1l, having a n-propyl group, appeared to have higher efficacy than the C20 branched chain analogues 1n (t-butyl) and 1a (i-propyl),30 contrary to the GPI derived data just described.
Conclusions
The present study confirms the orvinols’ lack of selectivity in binding to opioid receptors and that KOR/MOR agonists of varying efficacy can be obtained from this series. As predicted, efficacy can be controlled by the chain length of the C20 R group and by the introduction of branching or ring systems into the chain; a full range of profiles from EKC-like KOR agonists/MOR partial agonists (e.g., 1c, 1e), selective KOR agonists (e.g., 3b, 3i, and 4a), nonselective agonists (e.g., 1h and 4f), and even a ligand with predominant MOR activity (1j) were seen. Preliminary in vivo evaluation of 1b and 1c, coupled with the known profiles of 1n and 5a, suggests that the [35S]GTPγS data is predictive of the in vivo activity of these ligands. Compounds have successfully been obtained with the targeted profile of potent moderate to high efficacy KOR agonism combined with potent partial agonist activity at the MOR.
Experimental Section
Reagents and solvents were purchased from Sigma-Aldrich or Alfa Aesar and used as received. Buprenorphine (1n) was supplied by the National Institute on Drug Abuse, Bethesda, Maryland. 1H and 13C NMR spectra were obtained with a Bruker-400-MHz instrument (1H at 400 MHz, 13C at 100 MHz); δ in ppm, J in Hz with TMS as an internal standard. ESIMS: microTOF (BRUKER). Microanalysis: Perkin-Elmer 240C analyzer. Column Chromatography was performed using RediSep prepacked columns with a Teledyne Isco CombiFlash instrument. Ligands were tested as their hydrochloride salts, prepared by adding 5 equivalents of HCl (1 N solution in diethyl ether) to a solution of compound in anhydrous methanol. All reactions were carried out under an inert atmosphere of nitrogen unless otherwise indicated. All compounds were >95% pure as determined by microanalysis. A representative synthesis for each series is reported here.
General Procedure A: Grignard Addition
The Grignard reagents were prepared from the corresponding bromides (5 mmol) by reaction with magnesium (182 mg, 7.5 mmol) in anhydrous THF (5 mL) containing a crystal of iodine. The Grignard reagents were titrated prior to use by adding 1 mL of the Grignard solution to a flask containing 1,10-phenanthroline (∼2 mg) in anhydrous THF (2 mL) (purple solution) and titrating with 1 M 2-butanol (anhydrous) in THF (end point pale yellow solution).
A solution of the appropriate Grignard reagent (1 M in THF, 1.2 mL, 1.2 mmol) was treated dropwise at room temperature with a solution of N-cyclopropylmethyl-6,14-endo-ethanonorthevinone (8) (500 mg, 1.18 mmol) or N-cyclopropylmethyl-6,14-endo-ethanonorthevinal (10) (500 mg, 1.22 mmol) in anhydrous toluene (12 mL). After stirring at room temperature for 20 h, the reaction was quenched by the addition of saturated aqueous ammonium chloride solution (20 mL). The phases were separated and the aqueous phase extracted with EtOAc. The combined organic phases were washed with saturated aqueous sodium bicarbonate, dried over MgSO4, filtered, and evaporated in vacuo. The residue was purified by column chromatography over silica gel eluting with a gradient from 10% to 30% ethyl acetate in hexane. Rf values are recorded from TLC eluted with 30:1:69 ethyl acetate/ammonia solution/hexane.
General Procedure B: 3-O-Demethylation with Propane Thiolate and HCl Salt Formation
A solution of the appropriate thevinol (0.25 mmol) in anhydrous HMPA (1 mL) under an inert atmosphere was treated with sodium hydride (21 mg, 0.875 mmol) followed by 1-propanethiol (79 μL, 0.875 mmol). After the addition was complete, the reaction mixture was heated to 120 °C and stirred for 3 h. On cooling to room temperature, NH4Cl (sat, aq) was added and the mixture extracted with diethyl ether. The organic extracts were washed with water (3×) and brine. The organic phase was dried (MgSO4), filtered, and evaporated to dryness. The residue was purified by column chromatography over silica gel.
The HCl salts were prepared by the addition of 2 M HCl in diethyl ether (1.2 equiv) to a solution of the orvinol in diethyl ether. The white precipitate that formed was collected by filtration, washed with ether, and dried under high vacuum.
General Procedure C: Reduction with LiAlH4
A solution of the ketone (0.215 mmol) in anhydrous THF (7 mL) was added dropwise to a suspension of LiAlH4 (20.5 mg, 0.54 mmol) in THF at room temperature. The resulting mixture was allowed to stir for 1 h and then treated carefully with saturated aqueous sodium sulfate sulfate solution until all aluminum salts had precipitated. The aluminum salts were removed by filtration and washed with Et2O. The filtrate was dried over MgSO4, evaporated to dryness, and the residue purified by column chromatography over silica gel (30% ethyl acetate and 0.5% NH4OH in hexane).
General Procedure D: Swern Oxidation
A solution of DMSO (170 μL, 2.4 mmol) in anhydrous DCM (5 mL) was added dropwise to a solution of oxalyl chloride (93 μL, 1.1 mmol) in DCM (2 mL) at −78 °C. The resulting solution was stirred at −78 °C for 10 min and then treated dropwise with a solution of the alcohol (1 mmol) in DCM (5 mL). The reaction was stirred for a further 15 min before triethylamine (0.7 mL, 5 mmol) was added slowly and the resulting solution allowed to warm to room temperature. Water (10 mL) was added and the resulting mixture stirred for 10 min. The phases were separated and the aqueous phase extracted with DCM. The combined organic extracts were washed with saturated ammonium chloride, dried over MgSO4, filtered, and the solvent removed under reduced pressure. The residue was purified by column chromatography over silica gel (0.5% NH4OH, 30% ethyl acetate in hexane) to afford the product.
(2′R,5α,6R,7R,14α)-2′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-5′-methyl-hexan-2′-ol (9c)
Using general procedure A on 8, with isopentyl magnesium bromide, 9c was isolated as a white solid (216 mg, 37%). 1H NMR (400 MHz, CDCl3) δ 0.08–0.12 (2H, m), 0.44–0.52 (2H, m), 0.70–0.83 (2H, m), 0.91 (3H, d), 0.93 (3H, d), 1.01–1.08 (2H, m), 1.19–1.56 (5H, m), 1.34 (3H, s), 1.66 (1H, dd), 1.74–1.85 (2H, m), 1.90–2.03 (2H, m), 2.19–2.38 (4H, m), 2.64 (1H, dd), 2.79–2.86 (1H, m), 2.95–3.01 (2H, m), 3.54 (3H, s), 3.88 (3H, s), 4.42 (1H, s), 5.10 (1H, s), 6.54 (1H, d), 6.70 (1H, d). HRMS (ESI+) calcd for C31H46NO4 (MH+), 496.3421; found, 496.3414 (100%).
(2′R,5α,6R,7R,14α)-2′-(4,5-Epoxy-7,8-dihydro-3-hydroxy-6-methoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-5′-methyl-hexan-2′-ol (1c)
Using general procedure B with 9c (50 mg) gave 1c as a yellow solid (47 mg, 98%) 1H NMR (400 MHz, CDCl3) δ 0.08–0.12 (2H, m), 0.46–0.50 (2H, m), 0.70–0.83 (2H, m), 0.91 (6H, d), 1.00–1.08 (2H, m), 1.19–1.26 (1H, m), 1.34 (3H, s), 1.34–1.54 (4H, m), 1.65–1.69 (1H, m), 1.77–1.81 (2H, m), 1.90–2.03 (2H, m), 2.18–2.38 (4H, m), 2.66 (1H, dd), 2.79–2.86 (1H, m), 2.95–3.00 (2H, m), 3.52 (3H, s), 4.42 (1H, s), 5.16 (1H br s), 6.50 (1H, d, J = 8.0 Hz), 6.69 (1H, d). HRMS (ESI+) calcd for C30H44NO4 (MH+), 482.3265; found, 482.3241 (100%); Anal. (C30H43NO4·HCl·1.5H2O) C, H, N.
(1′S,5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)- 2′-cyclohexyl-ethan-1′-ol (11a: R = Cyclohexylmethyl)
Using general procedure A on 10 with cyclohexylmethylmagnesium bromide gave 11a, isolated as a white solid, 89%. 1H NMR (400 MHz, CDCl3) δ 0.09–0.10 (2H, m), 0.46–0.50 (2H, m), 0.68–0.72 (1H, m), 0.78–0.89 (3H, m), 0.95–0.98 (1H, m), 1.13–1.28 (5H, m), 1.47–1.53 (4H, m), 1.63–1.71 (8H, m), 1.83–1.86 (1H, m), 2.00–2.02 (2H, m), 2.21–2.29 (3H, m), 2.32–2.42 (1H, m), 2.58–2.66 (2H, m), 2.95–3.00 (1H, d), 3.06–3.08 (1H, d), 3.41 (1H, s), 3.86 (3H, s), 4.41 (1H, s), 6.53 (1H, d), 6.68 (1H, d); HRMS (ESI+) calcd for C32H46NO4 (MH+), 508.34; found, 508.34.
(5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-2′-cyclohexylethanone (12: R = Cyclohexylmethyl)
Using general procedure D on 11a (R = cyclohexylmethyl) gave 12 (R = cyclohexylmethyl) as a white solid (80 mg, 32%). 1H NMR (400 MHz, CDCl3) δ 0.07–0.08 (2H, m), 0.45–0.49 (2H, m), 0.68–0.78 (2H, m), 0.84–0.95 (2H, m), 1.12–1.16 (1H, m), 1.22–1.35 (4H, m), 1.50–1.51 (1H, m), 1.54 (3H, s), 1.58–1.68 (5H, m), 1.83–1.85 (1H, m), 1.90–2.00 (1H, m), 2.26–2.35 (3H, m), 2.37–2.38 (1H, d), 2.47–2.53 (1H, dd), 2.59–2.63 (1H, m), 2.66–2.73 (1H, m), 2.94–2.99 (2H, m), 3.04–3.06 (1H, d), 3.39 (1H, s), 3.86 (3H, s), 4.44 (1H, s), 6.54 (1H, d), 6.69 (1H, d); HRMS (ESI+) calcd for C32H44NO4 (MH+), 506.33; found, 506.33.
(2′R,5α,6R,7R,14α)-2′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-cyclohexyl-propan-2′-ol (13: R = Cyclohexylmethyl)
Using procedure A for the addition of methylmagnesium bromide to 12 (R = cyclohexylmethyl), 13 (R = cyclohexylmethyl) was isolated as a white solid (34 mg, 89%). 1H NMR (400 MHz, CDCl3) δ 0.08–0.10 (2H, m), 0.47–0.50 (2H, m), 0.69–0.72 (2H, m), 0.90–0.99 (2H, m), 1.04–1.10 (2H, m), 1.15 (3H, s), 1.20–1.28 (3H, m), 1.56–1–68 (8H, m), 1.74–1.77 (2H, m), 1.85 (1H, m), 1.99–2.03 (2H, m), 2.18–2.24 (2H, m), 2.26–2.36 (1H, m), 2.58–2.63 (1H, m), 2.78–2.86 (1H, m), 2.96–2.99 (1H, d), 2.99–2.03 (1H, d), 3.51 (1H, s), 3.87 (3H, s), 4.38 (1H, s), 4.68 (1H, s), 6.53 (1H, d), 6.69 (1H, d); HRMS (ESI+) calcd for C33H48NO4 (MH+), 522.36; found, 522.36.
(2′S,5α,6R,7R,14α)-2′-(4,5-Epoxy-7,8-dihydro-3-hydroxy-6-methoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-cyclohexyl-propan-2′-ol (2e)
Using General procedure B on 13 (R = cyclohexylmethyl) gave 2e as a white solid. 1H NMR, 270 MHz (CDCl3) δ 0.07–0.11 (2H, m), 0.46–0.50 (2H, m), 0.67–0.81 (2H, m), 0.86–1.30 (10H, m), 1.55–1.89 (12H, m), 1.94–2.08 (2H, m), 2.14–2.26 (3H, m), 2.34–2.41 (1H, m), 2.55 (1H, dd), 2.76–2.86 (1H, m), 2.92 (1H, d), 2.99 (1H, d), 3.50 (3H, s), 4.40 (1H, s), 4.71 (1H, s), 6.48 (1H, d), 6.66 (1H, d); HRMS, m/z for (C32H46NO4) [MH]+, calcd, 508.3427; found, 508.3424. Anal. (C32H45NO4·HCl·1.5H2O) C, H, N.
(1′R,5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-phenyl-propan-1′-ol (11a: R = Phenethyl)
Phenethylmagnesium bromide addition to 10 using general procedure A gave 11a, isolated as a white solid (218 mg, 58%). Rf 0.48: 1H NMR (400 MHz, CDCl3) δ 0.08–0.10 (2H, m), 0.46–0.49 (2H, m), 0.72–0.96 (2H, m), 1.15–1.25 (3H, m), 1.51–1.66 (2H, m), 1.72–1.79 (4H, m), 1.90–2.03 (2H, m), 2.21–2.35 (4H, m), 2.62–2.69 (3H, m), 2.95–3.06 (2H, dd), 3.38 (3H, s), 3.86 (3H, s), 4.18 (1H, m), 4.40 (1H, s), 6.53 (1H, d), 6.68 (1H, d), 7.16–7.30 (5H, m); HRMS (ESI+) calcd for C33H42NO4 (MH+), 516.31; found, 516.31.
(5α,6R,7R,14α)-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3-phenyl-propanone (12: R = Phenethyl)
Compound 11a, R = phenylethyl (0.36 mmmol) was treated as described in general procedure D. The residue was purified by column chromatography using a combi flash machine (30% ethyl acetate in hexane) to afford 12 (R = phenethyl). Isolated as a white solid (100 mg, 54%). Rf 0.55: 1H NMR (400 MHz, CDCl3) δ 0.07–0.09 (2H, d), 0.44–0.49 (2H, m), 0.73–0.76 (1H, m), 1.15–1.32 (2H, m), 1.53–1.57 (1H, m), 1.56–1.71 (2H, m), 1.99–2.03 (2H, m), 2.23–2.32 (4H, m), 2.59–2.60 (1H, m), 2.70–2.78 (2H, m), 2.89–3.08 (5H, m), 3.37 (3H, s), 3.86 (3H, s), 4.43 (1H, s), 6.54 (1H, d), 6.68 (1H, d), 7.14–7.24 (5H, m); HRMS (ESI+) calcd for C33H40NO4 (MH+), 514.30; found, 514.30.
(1′R,5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3,6-dimethoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-phenyl-propan-1′-ol (11b: R = Phenethyl)
Compound 12 (R = phenylethyl) (100.0 mg, 0.19 mmol) was treated as described in general procedure C to give 11b (R = phenethyl). Isolated as a white solid (86 mg, 86%). Rf 0.51. 1H NMR (400 MHz, CDCl3) δ 0.07–0.09 (2H, m), 0.47–0.49 (2H, m), 0.72–0.80 (2H, m), 0.87–0.99 (2H, m), 1.59–1.82 (5H, m), 1.96–2.04 (2H, m), 2.18–2.34 (5H, m), 2.61–2.72 (2H, m), 2.74–2.82 (1H, m), 2.92–2.99 (3H, m), 3.54 (3H, s), 3.87 (3H, s), 4.47–4.48 (1H, d), 5.46 (1H, s), 6.54 (1H, d), 6.70 (1H, d), 7.15–7.18 (1H, m), 7.24–7.28 (4H, m); HRMS (ESI+) calcd for C33H42NO4 (MH+), 516.31; found, 516.31.
(1′R,5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3-hydroxy-6-methoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-phenyl-propan-1′-ol (3i)
Compound 11b (R = phenylethyl) (54 0.0 mg, 0.10 mmol) was treated as described in general procedure B to give 3i. It was isolated as a white solid (48 mg, 96%). Rf 0.14; 1H NMR, 270 MHz (CDCl3) δ 0.06–0.09 (2H, m), 0.45–0.50 (2H, m), 0.64–0.80 (2H, m), 0.82–0.98 (2H, m), 1.58–1.86 (5H, m), 1.91–2.08 (2H, m), 2.13–2.32 (4H, m), 2.60–2.84 (3H, m), 2.91–3.03 (3H, m), 3.50 (3H, s), 3.81 (1H, t), 4.49 (1H, s), 5.45 (1H, s), 6.48 (1H, d), 6.67 (1H, d), 7.13–7.32 (5H, m); HRMS, m/z for (C32H40NO4) [MH]+, calcd, 502.2957; found, 502.2956. Anal. (C32H39NO4·HCl·1.5H2O) C, H, N.
(1′S,5α,6R,7R,14α)-1′-(4,5-Epoxy-7,8-dihydro-3-hydroxy-6-methoxy-17-cyclopropylmethyl-6,14-ethano-morphinan-7-yl)-3′-phenyl-propan-1′-ol (4k)
Compound 11a (R = phenethyl) was treated as described in general procedure B to give 4k. 1H NMR (400 MHz, CDCl3) δ 0.08–0.09 (2H, m), 0.45–0.50 (2H, m), 0.66 (1H, m), 079 (1H, m), 1.12–1.13 (1H, m), 1.39–1.48 (1H, m), 1.52–1.57 (2H, m), 1.63–1.67 (1H, d), 1.70–1.80 (4H, m), 1.90–1.99 (3H, m), 2.19–2.38 (5H, m), 2.60–2.70 (3H, m), 2.85 (1H, m), 2.93–2.98 (1H, d), 3.04–3.05 (1H, d), 3.35 (3H, s), 4.41 (1H, s), 6.49 (1H, d), 6.67 (1H, d), 7.16 (1H, m), 7.16–7.30 (5H, m); HRMS (ESI+) calcd for C32H40NO4 (MH+), 502.2957; found, 502.3002. Anal. (C32H39NO4.HCl.0.5H2O) C, H, N.
Molecular Modeling
CoMFA: all of the modeling was carried out using the Sybyl-X 1.0 molecular modeling environment from Tripos Inc. The molecules were built, assigned with Gasteiger–Huckel charges, and minimized using the MMFF94s force field. The CoMFA analysis was carried out as per the instructions in the Sybyl-X 1.0 documentation. Docking: the 4DJH crystal structure of the KOR and the 4DKL crystal structure of the MOR were prepared for docking work by running them through the Protein Preparation Wizard tool of the Schrödinger software. Compound 1n was built using the Schrödinger software. GOLD was used to dock 1n into both prepared protein structures. The binding site was defined as a sphere of 5 Å radius centered on the centroid of the crystal structure ligand with the requirement that the centroid of the docked ligand lies within this sphere. Water molecules in the crystal structure were left in place.
Acknowledgments
This work was funded by the National Institutes of Health, National Institute on Drug Abuse grant DA07315 (to S.M.H.). M.P.T. was supported by the Wellcome Trust (Programme Grant 082837 to BVL Potter, University of Bath).
Glossary
Abbreviations Used
- MOR
mu opioid receptor
- DOR
delta opioid receptor
- KOR
kappa opioid receptor
- LAAM
levo-α-acetylmethadol (or (3S,6S)-(6-dimethylamino-4,4-diphenyl-heptan-3-yl)acetate)
Supporting Information Available
Full experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newman A. H. Novel Pharmacotherapies for cocaine abuse 1997–2000. Expert Opin. Ther. Pat. 2000, 10, 1095–1122. [Google Scholar]
- Negus S. S.; Mello N. K.; Portoghese P. S.; Lin C. E. Effects of κ-opioids on cocaine self-administration by rhesus monkeys. J. Pharmacol. Exp. Ther. 1997, 282, 44–55. [PubMed] [Google Scholar]
- Mello N. K.; Negus S. S. Effects of κ opioid agonists on cocaine- and food-maintained responding by rhesus monkeys. J. Pharmacol. Exp. Ther. 1998, 286, 812–824. [PubMed] [Google Scholar]
- Mello N. K.; Negus S. S. Interactions between kappa opioid agonists and cocaine – preclinical studies. New medications for drug abuse. Ann. N.Y. Acad. Sci. 2000, 909, 104–132. [DOI] [PubMed] [Google Scholar]
- Glick S. D.; Maissoneuve I. M.; Raucci J.; Archer S. κ-Opioid inhibition of morphine and cocaine self-administration in rats. Brain Res. 1995, 681, 147–152. [DOI] [PubMed] [Google Scholar]
- Bidlack J. M.; Cohen D. J.; McLaughlin J. P.; Lou R.; Ye Y.; Wentland M. P. 8-Carboxamidocyclazocine: A long-acting, novel benzomorphan. J. Pharmacol. Exp. Ther. 2002, 302, 374–380. [DOI] [PubMed] [Google Scholar]
- Tzaferis J. A.; McGinty J. F. Kappa opioid receptor stimulation decreases amphetamine-induced behavior and neuropeptide mRNA expression in the striatum. Mol. Brain Res. 2001, 93, 27–35. [DOI] [PubMed] [Google Scholar]
- Heidbreder C.; Shippenberg T. S. U69,593 prevents cocaine sensitization by normalizing basal accumbens dopamine. NeuroReport 1994, 5, 1797–1800. [DOI] [PubMed] [Google Scholar]
- Crawford C. A.; McDougall S. A.; Bolanos C. A.; Hall S.; Berger S. P. The effects of the κ agonist U-50,488 on cocaine-induced conditioned and unconditioned behaviours and Fos immunoreactivity. Psychopharmacology 1995, 120, 392–399. [DOI] [PubMed] [Google Scholar]
- Shippenberg T. S.; LeFevour A.; Heidbreder C. H. κ-Opioid receptor agonists prevent sensitization to the conditioned rewarding effects of cocaine. J. Pharmacol. Exp. Ther. 1996, 276, 545–554. [PubMed] [Google Scholar]
- Walsh S. L.; Strain E. C.; Abreu M. E.; Bigelow G. E. Enadoline, a selective kappa opioid agonist: Comparison with butorphanol and hydromorphone in humans. Psychopharmacology 2001, 157, 151–162. [DOI] [PubMed] [Google Scholar]
- Negus S. S.; Mello N. K. Effects of kappa opioid agonists on cocaine self-administration under a progressive ratio schedule in rhesus monkeys. Drug Alcohol Depend. 2001b, 63Suppl. 1S113. [Google Scholar]
- Johnson R. E.; Chutuape M. A.; Strain E. C.; Walsh S. L.; Stitzer M. L.; Bigelow G. E. A comparison of levomethadyl acetate, buprenorphine and methadone for opioid dependence. N. Engl. J. Med. 2000, 343, 1290–1297. [DOI] [PubMed] [Google Scholar]
- Cowan A.; Lewis J. W.. Buprenorphine: Combatting Drug Abuse with a Unique Opioid; Wiley-Liss: New York, 1995. [Google Scholar]
- Boura A. L. A.; Fitzgerald A. E. The pharmacology of N-(cyclopropylmethyl)-19-isopentyl-nororvinol hydrochloride. A potent and long lasting central depressant. Br. J. Pharmacol. 1966, 26, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop A.; Berzetei-Gurske I.; Burnside J.; Toll L.; Traynor J. R.; Husbands S. M.; Lewis J. W. Structural Determinants of Opioid Activity in the Orvinols and Related Structures. Ethers of 7,8-Cyclopenta-analogs of Buprenorphine. Helv. Chim. Acta 2000, 83, 687–693. [Google Scholar]
- Lewis J. W.; Husbands S. M. The orvinols and related opioids – high affinity ligands with diverse efficacy profiles. Curr. Pharm. Des. 2004, 10, 717–732. [DOI] [PubMed] [Google Scholar]
- Grundt P.; Martinez-Bermejo F.; Lewis J. W.; Husbands S. M. Formic acid catalysed rearrangement of thevinols and their vinylogous analogues. Effects of 5β-methyl substitution. Helv. Chim. Acta 2003, 86, 2287–2298. [Google Scholar]
- Bentley K. W.; Hardy D. G. Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6,14-endoethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and −norcodeine. J. Am. Chem. Soc. 1967, 89, 3281–3292. [DOI] [PubMed] [Google Scholar]
- Bentley K. W.; Bower J. D.; Lewis J. W. Novel analgesics and molecular rearrangements in morphine-thebaine group 0.16. Some derivatives of 6,14-endo-etheno-7,8-dihydromorphine. J. Chem. Soc., C 1969, 18, 2569–2572. [DOI] [PubMed] [Google Scholar]
- Lee K. H.; Woods J. H.; Traynor J. R. Differential binding properties of oripavines at cloned mu- and delta-opioid receptors. Eur. J. Pharmacol. 1999, 378, 323–330. [DOI] [PubMed] [Google Scholar]
- Berzetei-Gurske I. P.; White A.; Polgar W.; DeCosta B. R.; Pasternak G. W.; Toll L. The in vitro pharmacological characterization of naloxone benzoylhydrazone. Eur. J. Pharmacol. 1995, 277, 257–263. [DOI] [PubMed] [Google Scholar]
- Traynor J. R.; Nahorski S. R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-[S-35]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [DOI] [PubMed] [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X. P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aceto M. D.; Harris L. S.; Bowman E. R. Etorphines: Mu-opioid receptor-selective antinociception and low physical dependence capacity. Eur. J. Pharmacol. 1997, 338, 215–223. [DOI] [PubMed] [Google Scholar]
- Husbands S. M.; Lewis J. W. Structural determinants of efficacy for κ-opioid receptors in the orvinol series: 7,7-Spiro analogues of buprenorphine. J. Med. Chem. 2000, 43, 139–141. [DOI] [PubMed] [Google Scholar]
- Katz J. L.; Woods J. H.; Winger G. D.; Jacobson A. E. Compounds of novel structure having kappa agonist behavioral effects in rhesus monkeys. Life Sci. 1982, 31, 2375–2378. [DOI] [PubMed] [Google Scholar]
- Lewis J. W.Structure-Activity Relationships of Opioids – A Current Perspective; Proceedings of the VIIIth International Symposium on Medicinal Chemistry I; Swedish Pharmaceutical Press: Stockholm, Sweden, 1985; p 123. [Google Scholar]
- Lewis J. W.Ring C-Bridged Derivatives of Thebaine and Oripavine; Advances in Biochemical Psychopharmacology; Harris B., Smith M.; Raven Press: New York, 1974; Vol 8, pp p123–136. [PubMed] [Google Scholar]
- Husbands S. M.; Lewis J. W. Opioid ligands having delayed long-term antagonist activity: Potential pharmacotherapies for opioid abuse. Mini-Rev. Med. Chem. 2003, 3, 137–144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.