

Abstract
We report a global quantitative phosphoproteomic study of bloodstream and procyclic form Trypanosoma brucei using SILAC labeling of each lifecycle stage. Phosphopeptide enrichment by SCX and TiO2 led to the identification of a total of 10096 phosphorylation sites on 2551 protein groups and quantified the ratios of 8275 phosphorylation sites between the two lifecycle stages. More than 9300 of these sites (92%) have not previously been reported. Model-based gene enrichment analysis identified over representation of Gene Ontology terms relating to the flagella, protein kinase activity, and the regulation of gene expression. The quantitative data reveal that differential protein phosphorylation is widespread between bloodstream and procyclic form trypanosomes, with significant intraprotein differential phosphorylation. Despite a lack of dedicated tyrosine kinases, 234 phosphotyrosine residues were identified, and these were 3–4 fold over-represented among site changing >10-fold between the two lifecycle stages. A significant proportion of the T. brucei kinome was phosphorylated, with evidence that MAPK pathways are functional in both lifecycle stages. Regulation of gene expression in T. brucei is exclusively post-transcriptional, and the extensive phosphorylation of RNA binding proteins observed may be relevant to the control of mRNA stability in this organism.
Keywords: phosphorylation, SILAC, Trypanosoma brucei, quantitative proteomics, phosphoproteomics
Introduction
The protozoan parasite Trypanosoma brucei is the etiological agent of African sleeping sickness, also known as Human African Trypanosomiasis (HAT), which is transmitted by the bite of an infected tsetse fly. The disease is usually fatal if left untreated and is estimated to be responsible for around 10000 deaths per annum in sub-Saharan Africa.1 Current treatments are expensive, toxic and difficult to administer, leaving an urgent unmet need for new therapeutic agents.2T. brucei has a complex digenetic lifecycle between an insect vector and a mammalian host, and its ability to adapt its proteome to these disparate environments is essential to its survival and virulence. During the early stages of an infection, the clinically relevant bloodstream form of the parasite proliferates in the blood and lymph of the human host, then in the second stage enters the cerebral-spinal fluid and brain, resulting in coma and death. When a tsetse fly feeds on an infected host, the parasite is ingested with the blood meal, triggering parasite differentiation into the procyclic form to enable survival in its new environment.
Both the procyclic form and bloodstream form of the parasite may be cultured in vitro. We have recently quantified the changes in protein expression between the procyclic and bloodstream form lifecycle stages using SILAC labeling of procyclic cells, demonstrating that about 10% of the observed proteome is differentially regulated by more than 5-fold.3 The fold-change in protein level showed a strong correlation (Pearson correlation 0.86) to changes in mRNA level recorded by transcriptomic studies,4 despite the atypical mechanism of gene expression in trypanosomes, where genes are transcribed in large polycistronic units that are trans-spliced into mature mRNAs for translation, with mRNA abundance controlled by stability.5
There is evidence that protein phosphorylation may be important in T. brucei; Nett et al. used sequential strong-cation exchange (SCX) and TiO2 enrichment of phosphopeptides derived from the cytosolic fraction of bloodstream form parasites to identify 1204 phosphorylation sites on 491 proteins,6 and immunoprecipitation with antiphosphotyrosine antibodies to identify 34 phosphotyrosine sites induced in response to the treatment of procyclic form cells with hydrogen peroxide.7 However, to date, there has been no quantitative comparison of the phosphoproteomes of the two lifecycle stages. We set out to define the global protein phosphorylation status of the two lifecycle stages to explore the biological processes that might be modulated by phosphorylation, and to highlight stage-specific phosphorylation changes. Here, we report a global quantitative phosphoproteomic study of bloodstream and procyclic form Trypanosoma brucei using SILAC labeling of each lifecycles stage that identifies 9314 previously unidentified phosphorylation sites. The results show that differential phosphorylation is widespread between the procyclic and bloodstream form and that it adds significant complexity to the changes in the proteome.
Experimental Section
Cell Culture Media
HMI9-T, a modification of the original IMDM-based HMI-9,8 uses 56 μM 1-thioglycerol in place of 200 μM 2-mercaptoethanol, 2 mM glutaMAX I (Invitrogen) in place of 4 mM l-glutamine, and contains 10% heat-inactivated fetal bovine serum (PAA). Similarly, HMI11-T is a modification of the original HMI11 (HMI-9 lacking serum plus)8 that use 56 μM 1-thioglycerol and 2 mM glutaMAX I with 10% heat-inactivated fetal bovine serum.
HMI11-SILAC – RK was prepared in the same way as HMI11-T but using IMDM depleted of l-Arginine, l-Lysine (Sigma) and 10% dialyzed heat-inactivated fetal bovine serum (10 kDa molecular weight cutoff, PAA) and was supplemented with 4 mg/L folic acid. The HMI11-SILAC – RK was supplemented with either normal l-Arginine and l-Lysine (HMI11-SILAC + R0K0), or with l-Arginine U–13C6 and l-Lysine 4,4,5,5-2H4 (HMI11-SILAC + R6K4, Cambridge Isotope Laboratories) at 30% of the original HMI11 concentration (120 and 240 μM respectively) unless otherwise stated. Original SDM-79 medium9 and SILAC SDM-79 medium (SDM-79 + R6K6) were prepared as described previously.3
Cell Culture
Procyclic form Trypanosoma brucei Lister 427 clone 29.13.6 cells were grown at 28 °C without CO2 in SDM-79 in the presence of 15 μg/mL G418 and 50 μg/mL hygromycin. Culture adapted strain Lister 427 monomorphic bloodstream form T. brucei (MITat 1.2, expressing VSG221) were cultured in HMI-9T containing 2.5 μg/mL G418 at 37 °C in a 5% CO2 incubator. Both cell lines have been genetically modified to express the T7 RNA polymerase and the tetracycline repressor protein, as described by Wirtz et al.10 Cell dimensions were measured using a CASY cell counter.
For the growth curves, the T. brucei bloodstream form cells were washed twice in 10 mL HMI11-SILAC – RK and resuspended at 5 × 104 cells/mL in either HMI-9T or HMI11-SILAC supplemented with R0K0 at 30% of the original HMI11 concentration. Cells were counted using a Neubauer chamber and phase contrast microscope, and the cultures were diluted 100-fold every two days. After 10 days samples were collected for analysis by light microscopy.
For SILAC labeling, T. brucei bloodstream form log-phase cells were diluted 10000-fold into HMI11-SILAC + R6K4. Cells were harvested after 3 days growth at 2.5 × 106 cells/mL. SILAC labeling of T. brucei procyclic form cells was performed in SDM-79 + R6K6 as described previously.3
Cells were harvested by centrifugation and hypotonically lysed at 5 × 109 cells/mL for 5 min on ice in the presence 0.1 μM 1-chloro-3-tosylamido-7-amino-2-heptone (TLCK), 1 mM benzamidine, 1 mM phenyl-methyl sulfonyl fluoride (PMSF), 1 μg/mL leupeptin, 1 μg/mL aprotinin and Phosphatase Inhibitor Mixture II (Calbiochem). The protein concentration was determined by BCA assay (Pierce) to be ∼5 mg/mL from each cell type. Samples were aliquoted, snap frozen, and stored at −80 °C prior to subsequent processing.
Microscopy
Bloodstream form cells at late log phase grown in either HMI9-T or HMI11-SILAC + R0K0 for 10 days were washed in 10 mL phosphate buffer saline at 800 × g at 4 °C, fixed in 4% paraformaldehyde in phosphate buffered saline at 4 °C for 30 min, and placed on a coverslip. After air-drying the coverslips were washed in phosphate buffered saline and mounted onto slides. The phase contrast images were collected in a Zeiss Axioplan microscope.
Estimating Efficiency of SILAC Labeling
Bloodstream form cells were grown in HMI11-SILAC + R6K4 for 11–12 cell divisions and hypotonically lysed as described above. To reduce the sample complexity the proteins were fractionated by SDS-PAGE, and a band corresponding to 25–50 kDa molecular weight range was excised and subjected to in-gel tryptic digest prior to analysis by LC–MS/MS. Ten peptides were chosen at random and the relative abundance of the major isotopic peak of heavy (arginine-13C6/lysine-2H4) and light (arginine-12C6/lysine-1H4) forms were measured using the extracted ion chromatogram function in Excalibur (Thermo Scientific).
Rodent Infectivity Studies
Bloodstream form cells were grown in HMI9-T or HMI11-SILAC + R0K0 for >14 days prior to inoculation of female Balbc mice (three per condition) by inter peritoneal injection of 1 × 103, 1 × 104 or 1 × 105 cells in the same media. Daily tail bleeds were used to monitor the number of parasites in the bloodstream using a heamocytometer, and animals were euthanized when the parasite burden exceeded 5 × 108 cells/mL prior to overt distress.
Comparative Proteomic Analysis
A 1:1 mix of procyclic form cells grown in light-SILAC media and bloodstream form cells grown in heavy-SILAC media was solubilized with SDS and tryptic peptides were generated by FASP. Peptides were fractionated by SCX prior to analysis by liquid chromatography tandem mass spectrometry as described previously,3 except only 6 SCX fractions were analyzed and technical duplicates were not performed. The previously reported the analysis of a 1:1 mix of procyclic form cells grown in heavy-SILAC media and bloodstream form cells grown in light-SILAC media, consisting of technical duplicates of eight SCX fractions and ten SDS-PAGE fractions,3 was reprocessed to ensure accurate comparison with the current data.
Filter Aided Sample Preparation
Peptide samples for analysis by mass spectrometry were prepared by modification of the filter-aided sample preparation procedure (FASP).11 Briefly, samples were defrosted and combined according to the experiment design to give a total of 2.5 × 109 lysed cells (0.5 mL), solubilized with 4% SDS and then reductively alkylated in a 30000 molecular weight cutoff vertical spin filtration unit (Vivascience) using the FASP procedure adapted for the larger volumes used here. The sample was digested with 1:100 ratio (w/w) of trypsin gold (Promega) in the filtration unit for 18 h at 37 °C, tryptic peptides were eluted by centrifugation, and the filter washed sequentially with 1 mL of 50 mM NH4HCO3 and 1 mL of 0.5 M NaCl. The combined eluent was desalted using a 500 mg C18 cartridge (SepPak, Waters) and lyophilized.
Strong Cation Exchange Chromatography
Strong cation exchange (SCX) was performed on an Agilent 1120 compact LC using a 3.0 × 200 mm 5 μm polysulfethyl aspartamide column (Poly LC) with a flow rate of 350 μL/min and detection at 220 nm. Phosphopeptides were enriched using the method of Beausoleil et al.12 Dried peptides were dissolved in 200 μL of solvent A (5 mM KHPO4 pH 2.7, 30% MeCN) and separated by salt gradient consisting of 5 min at 100% solvent A, a 15 min gradient to 15% solvent B (solvent A + 0.35 M KCl), 1 min gradient to 100% B, 15 min at 100% B, and a 5 min gradient to 100% A. Fractions of 0.7 mL were collected throughout the run and combined into 8 fractions of equal peptide content based on their absorbance at 220 nm. Combined fractions were lyophilized prior to TiO2 enrichment.
TiO2 Enrichment of Phosphopeptides
Phosphopeptides were enriched using TiO2 in batch mode in the presence of 5% TFA and 1 M glycolic acid.13 Briefly, lyophilized peptides were resuspended in 80% MeCN, 5% TFA, 1 M glycolic acid prior to incubation for 15 min with TiO2 beads (GL Sciences) that were prewashed in the same solution. Beads were recovered by centrifugation at 700 × g for 1 min, washed three times in 80% MeCN, 5% TFA and eluted twice in 0.6% NH3 (aq). The combined eluents were lyophilized and stored at −20 °C prior to analysis.
Mass Spectrometry Data Acquisition
Liquid chromatography tandem mass spectrometry was performed by the FingerPrints Proteomic Facility at the University of Dundee. Liquid chromatography was performed on a fully automated Ultimate U3000 Nano LC System (Dionex) fitted with a 1 × 5 mm PepMap C18 trap column and a 75 μm × 15 cm reverse phase PepMap C18 nanocolumn (LC Packings, Dionex). Samples were loaded in 0.1% formic acid (buffer A) and separated using a binary gradient consisting of buffer A (0.1% formic acid) and buffer B (90% MeCN, 0.08% formic acid). Peptides were eluted with a linear gradient from 5 to 40% buffer B over 65 min. The HPLC system was coupled to an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific) equipped with a Proxeon nanospray ion source. For phosphoproteomic analysis, the mass spectrometer was operated in data dependent mode to perform a survey scan over a range 335–1800 m/z in the Orbitrap analyzer (R = 60000), with each MS scan triggering 15 MS2 acquisitions of the 15 most intense ions using multistage activation on the neutral loss of 98 and 49 thomsons in the LTQ ion trap.14 The Orbitrap mass analyzer was internally calibrated on the fly using the lock mass of polydimethylcyclosiloxane at m/z 445.120025.
For proteomic analysis, the HPLC gradient was increased to 145 min and the mass spectrometer was operated in data dependent mode with each MS scan triggering 15 MS2 acquisitions of the 15 most intense ions in the LTQ ion trap.
Data Processing
Data was processed using MaxQuant15 version 1.3.0.5 which incorporates the Andromeda search engine.16 Proteins were identified by searching a protein sequence database containing T. brucei brucei 927 annotated proteins (Version 4.0, downloaded from TriTrypDB,17http://www.tritrypdb.org/) supplemented with the VSG221 sequence and frequently observed contaminants (porcine trypsin, bovine serum albumins and mammalian keratins) that contains a total of 10,081 protein sequences. Search parameters specified an MS tolerance of 6 ppm, an MS/MS tolerance at 0.5 Da and full trypsin specificity, allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionines, N-terminal protein acetylation and N-pyroglutamate were allowed as variable modifications. Phosphoproteomic analysis included phosophorylation of serine, threonine and tyrosine as additional variable modifications. Peptides were required to be at least 7 amino acids in length and a MaxQuant score >5, with false discovery rates (FDRs) of 0.01 calculated at the levels of peptides, proteins and modification sites based on the number of hits against the reversed sequence database. SILAC ratios were calculated using only peptides that could be uniquely mapped to a given protein group, and required a minimum of two SILAC pairs. To account for any errors in the counting of the number of cells mixed, the distribution of SILAC ratios was normalized within MaxQuant at the peptide level so that the median of log2 ratios is zero, as described by Cox et al.15
Prior to statistical analysis, the outputs from MaxQuant were filtered to remove known contaminants and reverse sequences, and phosphorylation sites with a MaxQuant localization probability <0.95 were discarded. This localization probability cut off was chosen following the manual inspection of the tandem mass spectra showing the best localization evidence for phosphorylation sites occurring on protein kinases.
Quantitation of the changes in abundance are derived from the two SILAC labeled experiments with a 1:1 mix of procyclic form cells grown in heavy-SILAC media and bloodstream cells grown in light-SILAC media, and the label swap experiment with the SILAC labeling reversed. SILAC ratios for phosphorylation sites were calculated using only data from the phosphoproteomic experiments, and SILAC ratios for proteins were calculated using only data from the nonenriched proteomic experiments. When SILAC ratios were reported in both label swap experiments, replicate trimming was applied by calculating the mean and standard deviation σ of the distribution of the log2 (H/L × L/H), and discarding data >2σ from the mean (Figure 3).18 Where SILAC ratios were reported in only one experiment, they were discarded if the percentage variation in the calculated SILAC ratio (calculated by MaxQuant) was >100%.
Figure 3.

Correlation of label-swap biological replicates. (A) Changes in protein abundance. (B) Changes in phosphorylation site abundance. Replicates were trimmed at 2 σ prior to further analysis. Black squares <2 σ, gray square >2 σ.
The Mascot Delta Score was calculated by determining the difference between the best and second best Mascot ion scores for alternative phosphorylation site localizations on an otherwise identical peptide sequence,19 and appended to the MaxQuant outputs. Mascot distiller was used to peak pick mass spectra and read the centroid data from raw tandem mass spectra and submit searches to Mascot (Server version 2.2) using the same sequence database, enzyme specificity, modifications and mass tolerance as described above.
Data was visualized using Perseus 1.3.0.4 (www.perseus-framework.org) and further information on the identified proteins was obtained from TriTrypDB (http://www.tritrypdb.org).17 Gene ontology (GO) term enrichment was carried out using model based gene set analysis or term for term enrichment in Ontologiser20 using a T. brucei specific GO set containing 16765 terms for 4340 genes obtained from GeneDB (http://www.genedb.org).21 To make our data accessible to the scientific community, we uploaded our study to TriTrypDB and deposited the Thermo RAW files and search engine output into ProteomeXchange (http://www.proteomexchange.org) accession number PXD000049, enabling researchers to access the data presented here.
Results
SILAC Labeling of Bloodstream Form Trypanosoma brucei
To enable SILAC labeling of bloodstream form T. brucei cells, a modified medium was created based on HMI11-T (HMI9-T lacking serum plus),8 where l-arginine and l-lysine could be replaced by stable heavy isotope forms (HMI11-SILAC). As bloodstream form cells are usually only cultured to <2 × 106 cells/ml, a 10-fold lower density than procyclic cells, we first set out to establish labeling conditions that would reduce the amount of label required and maximize the number of cells obtained. We found that the growth of bloodstream form T. brucei cells in HMI11-SILAC containing only 30% of the normal concentration of l-arginine and l-lysine was equivalent to cell growth in the HMI9-T medium generally used for bloodstream form cell culture (Figure 1A). As expected, the HMI11-SILAC medium failed to support cell growth in the absence of l-arginine and l-lysine whereas cells were readily able to grow to 5 × 106 cells/ml in the log–linear phase in the presence of l-arginine and l-lysine (Figure 1B). Furthermore, the gross morphology of the cells was unaffected after ten days in culture in HMI11-SILAC + 30% l-arginine and l-lysine, as judged by light microscopy (Figure 1C) and measurement of cell dimensions by light scattering (Supplemental Table 1, Supporting Information). Importantly, cells cultured in HMI11-SILAC + 30% l-arginine and l-lysine for >14 days maintained infectivity in mice (Supplemental Figure 1, Supporting Information). Taking all the above results into consideration, we concluded that culture of bloodstream form T. brucei in HMI11-SILAC + 30% l-arginine and l-lysine is equivalent to culture in HMI9-T media.
Figure 1.

Growth of bloodstream form T. brucei in SILAC labeling media. Cells were grown in conventional HMI9-T medium or in the HMI11-SILAC medium developed in this paper. (A) Cumulative growth over 10 days (with subculturing every 2–3 days). (B) Growth to stationary phase. (C) Light microscopy of cells after 10 days in culture.
To experimentally assess the efficiency of heavy isotope incorporation in HMI11-SILAC, bloodstream form cells were grown in HMI11-SILAC + R6K4 for 11–12 cell divisions and extracted proteins subjected to analysis by LC–MS/MS. If heavy isotope incorporation occurred only by dilution (neglecting protein turnover), then 11–12 cell divisions should produce >99.9% incorporation, but in reality will be limited by the isotopic purity of the labeled amino acids. The heavy isotope incorporation at steady state was estimated to be 98.5 ± 1.5% by comparing the relative abundance of the major isotopic peak of the heavy (arginine-13C6/lysine-2H4) and light forms of ten peptides chosen at random. No significant incorporation of proline-13C5 (by conversion of arginine-13C6) was observed (Supplemental Figure 2, Supporting Information), as noted previously for the procyclic form.3
Comparative Proteomic Analysis
A prerequisite for comparative quantitative phosphoproteomic analyses is access to a relevant comparative quantitative proteomic data with which to distinguish the different types of quantitative phosphorylation differences (i.e., those that simply track changes in protein levels, those that do not correlate with changes in protein level and those that are unique to one or other lifecycle stage). Our previous comparative SILAC proteomic analysis of bloodstream grown in light medium and procyclic form T. brucei grown in heavy medium gave us ratios for 3553 proteins, and demonstrated that large changes in specific protein abundance occur between the lifecycle stages.3 To improve the quality of this comparative quantitative proteome data set, we exploited the development of SILAC labeling of bloodstream form trypanosomes, described above, to perform a “label-swap” experiment using a 1:1 mix of procyclic form cells grown in light-SILAC medium and bloodstream form cells grown in heavy-SILAC medium. The combined cells were solubilized with SDS, and the tryptic peptides generated by FASP were fractionated by SCX prior to analysis as described previously,3 except that six SCX factions were used and a longer gradient was applied for the LC–MS/MS analysis instead of running technical duplicates. A total of 3780 proteins were identified, with SILAC ratios determined for 3313 of them. The new label-swap data set showed very good agreement with our published SILAC data,3 with a comparable depth of coverage and a Pearson correlation of 0.92 (Figure 3A). Given the good correlation between the two data sets, we combined them to provide a definitive comparative quantitative proteome with which to interpret our phosphoproteomic data.
Comparative Phosphoproteomic Strategy
The objective of this study was: (i) To define the phosphoproteome of procyclic form T. brucei for the first time; (ii) To enhance the existing phosphoproteome of bloodstream form T. brucei.6 (iii) To quantify the changes in phosphorylation state which occur between the lifecycle stages using SILAC methodology. Four separate phosphoproteomic experiments were conducted (Figure 2) starting from: (i) Procyclic form cells grown in light-SILAC medium. (ii) A 1:1 mix of procyclic form cells grown in heavy-SILAC medium and bloodstream form cells grown in light-SILAC medium. (iii) A “label swap” experiment consisting of a 1:1 mix of bloodstream form cells grown in light-SILAC medium and procyclic cells grown in heavy-SILAC medium. (iv) Bloodstream form cells grown in light-SILAC medium. To maximize coverage of membrane and structural proteins, total protein extracts were prepared with 4% SDS using the filter-aided sample preparation (FASP) procedure,11 resulting in near-complete solubilization of the parasites. After denaturation and reductive alkylation the proteins were digested with trypsin in solution. Resulting phosphopeptides were enriched and fractionated into eight samples by SCX chromatography12 prior to further enrichment using TiO2 chromatography in batch mode.13 For each phosphoproteomic experiment, the eight fractions were analyzed by liquid chromatography tandem mass spectrometry in technical triplicates on a LTQ Oribtrap Velos using multistage acquisition.14
Figure 2.
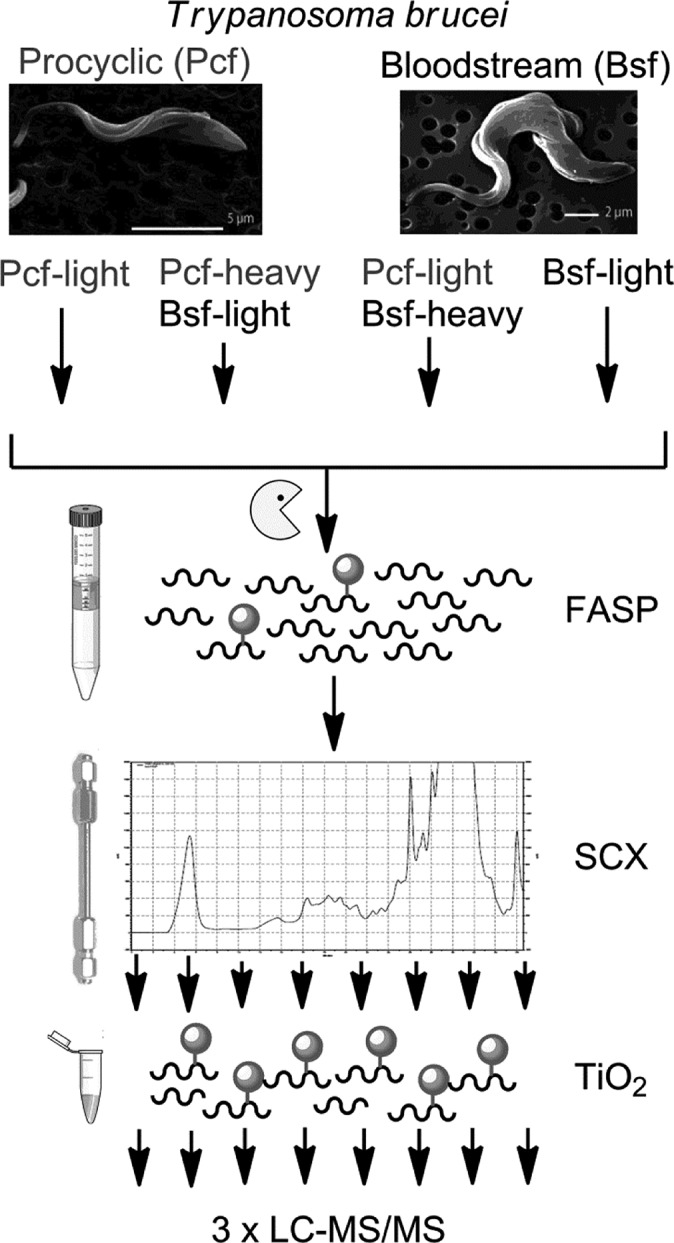
Phosphoproteomic workflow. Differentially labeled (heavy and light) bloodstream and procyclic form trypanosomes were lysed in SDS, and total proteins S-alkylated and digested with trypsin using the FASP method. Phosphopeptides were separated into eight fractions by SCX HPLC, enriched with TiO2, and analyzed by LC–MS/MS using MSA.
Overview of Data
The combined data were from a total of 138 LC–MS/MS runs, with 96 runs for samples enriched for phosphopeptides and 88 for samples with SILAC labeling. Altogether 589101 MS/MS spectra were identified, corresponding to 58087 nonredundant peptide sequences belonging to 5478 protein groups with a false discovery rate of 1% (Supplementary Tables S2 and S3, Supporting Information). A total of 10096 phosphorylation sites were identified on 2551 proteins (Supplementary Tables S4 and S5, Supporting Information), of which SILAC ratios could be determined for 8275 phosphorylation sites on 2233 protein groups, with a Pearson correlation of 0.78 between the label-swap experiments. A total of 8120 phosphorylation sites on 2137 protein groups were also identified using Mascot software, of which 6196 phosphorylation sites on 1933 protein groups had a Mascot delta score ≥10, reported to give a 1% false localization rate (FLR) for Orbitrap-MSA data.19 Of the 10096 phosphorylation sites identified by MaxQuant, 9314 (92%) have not previously been reported. The data presented here therefore represents a major advance in the depth of the trypanosome phosphoproteome.
The distribution of phosphorylation between residues was found to be 8087 phosphoserine (pS) sites (80%), 1,720 phosphothreonine (pT) sites (17%), and 289 phosphotyrosine (pY) sites (3%), in reasonable agreement with the T. brucei bloodstream form cytosolic phosphoproteome observations of 75% pS, 21.5% pT, 3.5% pY.6 Of the 10096 phosphorylation sites observed, 6493 occurred on 1491 proteins annotated as hypothetical or hypothetical conserved, reflecting the high proportion of genes of unknown function in the trypanosome genome. Gene Ontology terms are associated with only 1310 out of the 2552 phosphoproteins we observed, limiting the power of GO term enrichment analysis. Performing model based gene set analysis,20 a method designed to give a high-level summarized view of the data, identified only three terms: cilliary or flagella motility (GO:001539, PEP 1), protein tyrosine kinase activity (GO:004713, PEP 0.7), and regulation of gene expression (GO:0010468, PEP 0.5). This is an intriguing outcome as the T. brucei genome lacks any identifiable dedicated tyrosine kinases,6,22 yet has the associated GO terms, illustrating the pitfalls of such analyses. Performing term-for-term enrichment generated 45 significantly enriched GO terms (p < 0.01, Figure 4) confirming the predominance of terms associated with flagella motility, protein kinase activity and regulation of gene expression, and revealing additional terms for RNA, zinc and calmodulin binding (GO:0005488, GO:0008270, GO:0005516), and peptidase activity (GO:0004181, GO:0004197). Greater biological insight from the data is likely to come from the community inspecting the phosphorylation patterns, and their changes between lifecycle stages, of proteins of specific interest to them, a few examples of which are provided below.
Figure 4.

Gene Ontology term enrichment for all observed phosphoproteins. Term-for-term enrichment, P < 0.01; white box, Biological process; light gray box, Cellular component; dark gray box, Molecular function. For clarity parental terms are omitted; the complete graph can be found in the Supporting Information (Figure S3).
Comparison of the Phosphoproteomes of Bloodstream and Procyclic Form T. brucei
SILAC labeling allows the fold-changes that occur between the bloodstream and procyclic form of T. brucei (log2 Pcf/Bsf) to be quantified at both the protein and phosphorylation site level. As the phosphoproteomic workflow includes selective enrichment for phosphorylated peptides, this data cannot be used in the calculation of protein SILAC ratios, which are instead derived from separate proteomic experiments. We were able to determine fold-changes for a total of 8275 phosphorylation sites on 2233 protein groups, with 5768 (70%) of the phosphorylation sites on 1385 protein groups having ratios at both the protein- and site-level. In general, the changes in phosphorylation site occupancy between the lifecycle stages are greater than the changes in protein levels; for example, 318 (5.5%) of the phosphorylation sites change ≥10-fold (log2 ≥|3.32|) between lifecycle stages, while only 33 (0.87%) of protein groups change ≥10-fold. Indeed, plotting the fold-changes in protein ratio against the fold-changes in phosphorylation site ratio (Figure 5A) reveals that the observed changes are relatively weakly correlated (Pearson correlation 0.49). However, there are numerous examples where: (i) The change in phosphorylation correlates with the change in protein abundance, suggesting that phosphorylation stoichiometry is maintained between lifecycle stages in these cases. (ii) The change in phosphorylation correlates negatively with the change in protein abundance. (iii) Protein abundance is maintained but phosphorylation stoichiometry changes significantly (both up and down). (iv) Different phosphorylation sites on the same protein show a wide range of fold-changes (Figure 5B and Figure 6). These data demonstrate that changes in protein phosphorylation status between the lifecycle stages of T. brucei add significant complexity to the differential expression of the proteome. Some individual cases are discussed in more detail below.
Figure 5.

Correlation of fold-changes in protein and fold-changes in phosphorylation sites. (A) Correlation of all phosphorylation sites. (B) Correlation of average fold-change in phosphorylation sites for each phosphoprotein, with bars indicating the upper and lower limits of the range.
Figure 6.

Examples of the assignments of phosphorylation site location from tandem mass spectral data. (A) STE11 family protein kinase (Tb11.46.003) with pS357 and pS358 with MaxQuant localization score of 1.0 and Mascot delta score of 57.8. (B) CAMK family protein kinase Tb11.01.2290 with pS5 with MaxQuant localization score of 1.0 and Mascot delta score of 27.1, and pS8 with MaxQuant localization of 1.0 but which was not identified by Mascot. Data acquired on a LTQ Orbitrap Velos using MSA, * designates loss of phosphate (−98 Da), ** loss of two phosphates (−196 Da).
Manual validation of protein kinase phosphorylation sites
In order to judge the quality of the phosphorylation site assignments, and to empirically assess the false localization rate, a subset of the automated phosphorylation site assignments were manually inspected (Figure 6 and Supporting Information). The subset chosen was the T. brucei kinome consisting of 176 protein kinases22b as they are expected to occur at relatively low abundance and to show physiologically relevant phosphorylation. Using a MaxQuant localization score of ≥0.75 as an initial cutoff, we identified 641 specific phosphorylation sites on 127 protein kinases (for comparison a cutoff of ≥0.95 gave 493 sites on 124 kinases). Of these 641 automatically annotated sites on 127 kinases, 549 sites on 122 kinases were validated by manual inspection (Supplemental Table S6, Supporting Information). Overall, the manual validation correlated well with the sites chosen using the MaxQuant localization score and the Mascot delta score (MDS) methods.19 However, we noted that only 42% of sites in the MaxQuant localization score range 0.75–0.95 were manually validated, whereas over 94% of the sites MaxQuant localization score >0.95 were manually validated, increasing to over 98% at a localization score of >0.99. Further, at an MDS of 10, reported to give a global 1% false localization rate for Orbitrap Velos – MSA data,19 we were able to manually confirm 98% of the automatically annotated sites. However, it is worth noting that 81% of sites with MDS < 10 and 66% of sitesidentified by MaxQuant but not by Mascot were manually validated. Based on this manual survey of a significant number of phosphorylation site assignments, we have chosen to report all automatically annotated phosphorylation sites with MaxQuant localization scores >0.95 (with an estimated false localization rate of <6%).
Interestingly, the variation in the phosphorylation of the kinome between the two lifecycles stages is lower than the general data set, with only 15 phosphorylation sites showing a 10-fold or greater change. The protein kinase A catalytic subunit isoform 1 (Tb09.211.2410) showed strongly up-regulated phosphorylation in the bloodstream form parasite at three sites (log2 range −5.0 to −4.3), although the assignment of increased phosphorylation in the putative activation loop at T179 (Log2 −2.6) is slightly ambiguous as an identical (phospho)peptide is shared by protein kinase A catalytic subunit isoform 2 (Tb09.211.2360). Similarly, the protein kinase A regulatory subunit (Tb11.02.2210) has 5 sites which are also up-regulated in the bloodstream form, though to a lesser extent (log2 range −4.4 to −0.8). Protein kinases with phosphorylation up-regulated in the procyclic form include a NEK kinase (Tb11.01.6650) with two sites with a log2 range 5.6 to 4.7, and two CLK dual specificity kinases Tb927.2.4200 (5 sites, log2 range 3.3 to 0.6) and Tb927.10.15020 (4 sites, log2 range 3.2 to 2.4). Two “Other” group protein kinases have phosphorylation sites that are up-regulated in the procyclic and bloodstream form, respectively, (Tb927.7.3210, 5 sites, range log2 −3.7 to 2.8; Tb927.7.3220, 5 sites, range log2 −3.2 to 1.6).
Of the 15 MAPKs present in the T. brucei kinome, 9 (ECK1, KFR1, MOK, RCK, MAPK2, MAPK9, MAPK10, MAPK12, and an unnamed MAPK) are shown by the data presented here to have dual phosphorylation in the TxY motif required for activation. In addition, multiple phosphorylation sites are observed on three potential upstream STE7 MAP2K and nine STE11 MAP3K protein kinases, suggesting the presence of active MAPK signaling cascades. The observation of differential phosphorylation on certain MAPKs, MAP2Ks and MAP3Ks between the two lifecycle stages suggests that MAPK signaling may play distinct roles in each lifecycle stage (Figure 7).
Figure 7.
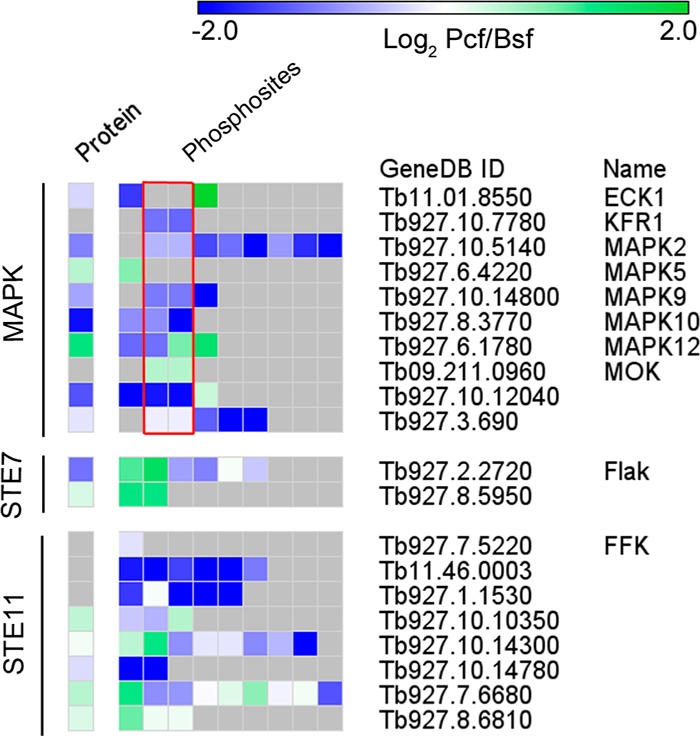
Differential phosphorylation of MAPK signaling components. Heatmap showing the change in abundance between the procyclic to bloodstream (Log2) at the protein and phosphorylation site level for the MAPK, STE7 and STE11 kinase quantified in this study. Red box, TxY motif; Gray shading, not observed. The Heatmap is generated with GENEE (http://www.broadinstitute.org/cancer/software/GENE-E/).
Phosphorylation Sites Up-regulated in Bloodstream Form T.brucei
A total of 203 phosphorylation sites on 119 protein groups were up-regulated ≥10-fold (Log2 ≤ −3.32) in bloodstream form T. brucei, with a distribution of these up-regulated phosphorylation sites of 74% pS, 15% pT, 11% pY, showing a significant enrichment in pY compared to the total observations. An additional 429 sites with no SILAC ratio (on 319 protein groups) were observed exclusively in the bloodstream form, with the distribution of 64% pS, 27.5% pT, 8.5% pY showing a similar trend. These data suggest that tyrosine phosphorylation is significantly over represented in sites differentially regulated between the lifecycle stages.
In total, 439 proteins have 631 individual phosphorylation sites that are either ≥10-fold up-regulated in the bloodstream form or are bloodstream form specific (Supplemental Table S7, Supporting Information). This group contains a large number of proteins annotated as hypothetical or hypothetical conserved, including 13 of the 15 proteins with >40-fold change (Log2 < −5.32) at the site level. GO term enrichment of the bloodstream form up-regulated group (of the 191 annotated proteins out this total 439) produced a very similar set of enriched terms to the global phosphoproteome (flagella, protein kinase, and gene expression), with the addition of terms for glycolysis and energy metabolism, although this may be due, at least in part, to large changes at the protein level in some of these proteins.3 Nevertheless, researchers interested in the changes in parasite metabolism between the procyclic and bloodstream form parasites are likely to find clues to physiologically relevant phosphorylation changes in the data provided. Significantly, there are examples where protein up-regulation in bloodstream form parasites may be further “amplified” by the coordinate up-regulation of phosphorylation. Notable examples of this include the RNA recognition motif protein RBP10 (Tb927.8.2780) that has been reported to promote a bloodstream form mRNA expression pattern,23 which has 4 phosphorylation sites changing by an average of log2 −4.8 (from log2 −5.4 to −4.2), potentially amplifying a change in protein abundance of Log2 −3.3, and the receptor-type adenylate cyclase GRESAG4 (Tb927.8.7940) which has 6 phosphorylation sites changing by an average of log2 −3.9 (from log2 −4.7 to −3.1), potentially amplifying a change in protein abundance of log2 −2.4.
Phosphorylation Sites Up-regulated in Procyclic Form T. brucei
A total of 115 phosphorylation sites on 83 protein groups were ≥10-fold up-regulated (log2 ≥ 3.32) in procyclic form T. brucei with a residue distribution of 70% pS, 22% pT, 9% pY, and an additional 499 sites with no SILAC ratio on 376 protein groups were observed exclusively in the procyclic form with a residue distribution of 77% pS, 15.5% pT, 7.5% pY. The significant enrichment in pY compared to the total observations mirrors that seen in the bloodstream form up-regulated phosphorylation sites, reinforcing the importance of changes in the phosphorylation of tyrosine between the lifecycle stages.
In total, 459 proteins have 614 individual phosphorylation sites that are either ≥10-fold up-regulated in the procyclic form or are procyclic form specific (Supplemental Table S8, Supporting Information). This procyclic up-regulated group contains a large number of proteins annotated as hypothetical or hypothetical conserved, as observed for the bloodstream form up-regulated group. GO term enrichment of the procyclic up-regulated group (of 207/460 proteins) again produced a very similar set of enriched terms to the global and bloodstream form up-regulated phosphoproteome (flagella, protein kinase, and gene expression) with no significant new terms. A total of 9 proteins annotated as transporters have >10-fold change (log2 > 3.32) at the site level, potentially amplifying the corresponding changes in protein abundance. Interesting specific examples include: The PAD2 carboxylic acid transporter (Tb927.7.5940), associated with slender to stumpy bloodstream form transformation in pleomorphic cell lines,24 where 3 sites increase in phosphorylation with an average log2 of 3.3 (range log2 1.5 to 6.2) in addition to a protein level increase of log2 2.9. The ubiquitin carboxy-terminal hydrolase (Tb927.10.2210), which has 10 phosphorylation sites with an average log2 of 3.6 (range log2 1.6 to 5.6), while the protein has almost no change in level (log2 −0.41). The C-14 sterol reductase (Tb11.01.7170), involved in the biosynthesis of ergosterol and its isomers that are the principal sterols in the procyclic parasites,25 has 2 phosphorylation sites with an average log2 of 3.8 (range log2 3.7 to 3.9), potentially amplifying the protein fold change of log2 2.2.
Phosphoproteins with Differentially Regulated Sites
A significant number of proteins that are multiply phosphorylated exhibit phosphorylation sites that are differentially regulated within the protein between the lifecycle stages (Figure 8). There are 234 proteins (Supplementary Table S9, Supporting Information) where the range of fold-changes of phosphorylation sites within the protein is greater than 10-fold (range Log2 > |3.32|), with an average of 10 phosphorylation sites per protein. Interestingly, of the 18 proteins where the change in site occupancy is greater than 80-fold (range Log2 > |6.32|), 16 are annotated as hypothetical or hypothetical conserved proteins. GO term enrichment analysis (of 113/234 proteins) produced significantly enriched terms (p < 0.01) including flagellum, gene regulation, and protein phosphatase activity, but that did not include protein kinase activity. Three protein phosphatases are found in the intraprotein differentially regulated group: (i) A kinetoplastid-specific protein phosphatase (Tb11.01.4320) with a differential range log2 of |6.6| (10 sites, range log2 −4.8 to 1.8). (ii) An atypical dual specificity phosphatase (Tb11.01.5870) with a differential range log2 of |5.6| (3 sites, range log2 −3.8 to 1.7). (iii) The TFIIF-Stimulated CTD phosphatase (Tb927.10.11450) with a differential range log2 of |4.1| (4 sites, range log2 −2.1 to 2.0).
Figure 8.

Distribution of intraprotein differentially regulated phosphorylation sites. Only proteins with more than one phosphorylation site are shown.
Discussion
We have previously developed SILAC labeling in procyclic form T. brucei,(3) and here we report the development of SILAC labeling in the clinically relevant bloodstream form and demonstrate that growth, morphology and infectivity are unchanged. Both lifecycle stages are rapidly dividing, enabling efficient isotope incorporation >98%, and show no propensity to convert arginine to proline. Two recent proteomic analysis using SILAC labeling in either procyclic26 or procyclic and bloodstream form27T. brucei also observed a similar lack of arginine to proline conversion.
The quantitation of changes in the global phosphoproteome between procyclic and bloodstream form lifecycle stages of Trypanosoma brucei, described here, reveals that differential phosphorylation is widespread, is not strongly correlated to change in protein abundance and adds significant complexity to the differential expression of the proteome. The observation of intraprotein differential phosphorylation suggests that phosphorylation may play distinct functional roles in each lifecycle stage, although how this effect is mediated is at present unclear and, because of the lack of comparative phosphorylation data to date, there are few existing hypotheses to examine. However, one process where protein phosphorylation is suggested to play a central role is the differentiation of the parasite from bloodstream to procyclic form in a coordinated, programmed event.28 While we have not studied the actual process of differentiation in this study, we have examined the phosphoproteomes of the initial and final states of the process, and observe phosphorylation changes in many of the proteins involved in differentiation. For example, TOR4 (Tb927.1.1930)29 has 10 sites with log2 changes in the range 0.4–2.7, MAPK5 (Tb927.6.4220)30 has a protein change log2 of 0.5 and pT207 site change log2 of 0.8; ZFK (Tb11.01.1030)31 has a pS520 log2 of −1.3; NOPP44/46 (Tb927.8.760)32 has a protein log2 of 1.7 and pY5 observed uniquely in procyclic parasites; PAD2 (Tb927.7.5940)24 has a protein log2 of 2.9 and 3 sites with log2 in the range 1.5 to 6.2. The absence of detected phosphorylation on PIP39 (Tb09.160.4480, protein log2 of 0.9), and PTP1 (Tb927.10.6690, not observed in our data set) may be due to up-regulation or transient phosphorylation during differentiation, such as in the division arrested stumpy form that was not studied here. Alternatively, the cocktail of phosphatase inhibitors used in this study may have limited effect against the activity of one or more trypanosome phosphatase. Clearly, further detailed studies of the process are required before firm conclusions can be drawn on the precise rules(s) of protein phosphorylation in trypanosome differentiation.
In this study we have used culture adapted Lister 427 bloodstream form (MITat 1.2) and procyclic form (29.13.6) cell lines that have been made amenable to genetic manipulation by Wirtz et al. by the insertion of exogenous genes coding for the T7 RNA polymerase and tetracyclic repressor protein using either one (bloodstream form) or two (procyclic form) selectable drug resistance markers.10 The two cell lines used are independent clones that are not genetically identical, and this may lead to some differences in protein expression and phosphorylation status, although such differences are likely to be minor in comparison to the developmentally regulated changes between the lifecycle stages. In addition, the culture adapted Lister 427 strains are neither genetically nor morphologically identical to field isolates, and it is not possible to account for the impact this will have on protein expression and phosphorylation status.
A significant proportion of the T. brucei kinome of 176 protein kinases are phosphorylated, with individual examples of phosphorylation sites up-regulated in either procyclic or bloodstream form, and examples of intraprotein differential phosphorylation. The abundance of active MAP kinases, as judged by the presence of dual TxY motif phosphorylation, suggests that MAPK pathways are functional in both lifecycle stages of T. brucei, although how or whether the pathways are regulated remains unclear. Efforts to determine the detailed biological role of T. brucei protein kinases are ongoing and knock-down by RNA interference has provided evidence of the essential nature of a significant number of protein kinases.33 We have recently used chemical proteomic profiling to establish that T. brucei protein kinases are sensitive to typical kinase inhibitors with nanomolar potency, demonstrating the potential for the development of species-specific inhibitors.22b Further analysis of protein kinase mediated phosphorylation cascades by stimulus–response studies, using the data published here to anchor them, may reveal the pinch-points that are most suitable for therapeutic intervention.
Often, eukaryotic phosphorylation signaling cascades terminate in the modulation of transcription factors to mediate changes in gene expression. However, classical transcription factors are missing in T. brucei, where the processing and stability of polycistronically transcribed mRNA precursors appears to regulate gene expression.5 One potential mechanism to modulate gene expression in T. brucei is the use of RNA binding zinc finger proteins to influence mRNA processing and stability, such as such as ZFP3 (Tb927.3.720)34 and RBP10 (Tb927.8.2780).23 A significant number of ZFPs (31 out of 49) and RBPs (13 out of 47) are present in the phosphoproteomic data reported here, with a subset of these (11 and 5, respectively) showing lifecycle specific regulation of phosphorylation status (≥5-fold change). Since the dynamic phosphorylation/dephosphorylation of these proteins has the potential to modulate gene expression, they are reasonable candidates to be the ultimate effector molecules of at least some of the trypanosome signaling cascades.
Despite the lack of identifiable dedicated tyrosine kinases in T. brucei, 234 phosphotyrosine residues were identified, and tyrosine phosphorylation was 3–4 fold over-represented in the phosphorylation sites that change between the two lifecycle stages. Tyrosine phosphorylation is proposed to arise from putative dual-specificity protein kinases such as DYRK, MEK and CLK, but there has been no experimental verification of the dual specificities of the trypanosome kinases to date.
GO term enrichment analysis has revealed that the flagellum is consistently over-represented among the phosphorylated proteins. Flagella can play both motility and sensory roles, and while it is clear that the trypanosome flagellum is highly motile, its potential sensory role is less well-defined.35 The dynamic phosphorylation of dynein motors is proposed to control flagella motility in other organisms, and in trypanosomes flagella motility is dispensable in the procyclic form but essential for cytokinesis in the bloodstream form.36 Interestingly, a number of flagella proteins show extensive intraprotein differential phosphorylation, including: (i) dynein heavy chain (Tb927.7.920) with 7 sites with log2 changes in the range −2.8 to 3.2 and protein log2 changes of −0.5; (ii) a paraflagella rod component (Tb927.8.1550) with 14 sites with log2 changes in the range −3.1 to 2.0 and a protein log2 change of −0.4; and (iii) an outer dynein docking complex (Tb11.01.7750) with 3 sites with log2 changes in the range −3.6 to 1.8 and a protein log2 change of 0.6.
Conclusions
In summary, we have presented a global quantitative phosphoproteomic analysis of the bloodstream and procyclic form of Trypanosoma brucei, which represents both the largest set of phosphorylation sites and the first report of genome wide quantitative phosphorylation data for any kinetoplastid. The establishment of SILAC labeling in both culture-amenable lifecycle stages of the parasite provides the community with the tools required to examine signaling events at the molecular level. This data set reveals the breadth of protein expression and phosphorylation in T. brucei, and provides a resource for the community to begin to examine the effect of phosphorylation upon biological processes. The present study of rapidly dividing nonsynchronized cells provides a static, averaged snapshot of the basal level of protein phosphorylation, and valuable information will be gained from future studies of the dynamics of stimulus-response events such as differentiation.
Acknowledgments
We thank the FingerPrints Proteomic Facility at the University of Dundee for acquisition of MS data and helpful discussions, Lucia Guther for helpful discussions on trypanosome cell culture, and Laste Stojanovski and Frederick Simeons for assistance with the animal work. This work was funded by the Wellcome Trust (Programme Grant 085622 and Strategic Awards 083481 and 100476) and Tenovus Tayside (T10/34).
Glossary
Abbreviations
- FASP
filter aided sample preparation
- GO
gene ontology
- SCX
strong cation exchange
- SILAC
stable isotope labeling by amino acids in cell culture
- T. brucei
Trypanosoma brucei
Supporting Information Available
Assignment of MS2 spectra for protein kinase phosphorylation sites, as pdfs contained in a zipped file. Supplementary Figures S1. Rodent infectivity of bloodstream form T. brucei cultured in HMI9-T and HI11-SILAC media. Figure S2. Efficiency of isotope incorporation in bloodstream form T. brucei. Figure S3. Gene Ontology term enrichment for all phosphoproteins. Table S1. Cell dimensions. Table S2. Protein groups including SILAC ratios. Table S3. Peptide including SILAC ratios. Table S4. Phosphorylation sites including SILAC ratios. Table S5. Phosphopeptides including SILAC ratios. Table S6. Manual validation of protein kinase phosphorylation sites. Table S7. Phosphorylation sites up-regulated in the bloodstream form. Table S8. Phosphorylation sites up-regulated in the Procyclic form. Table S9. Phosphoproteins with phosphorylation sites differentially regulated between lifecycle stages. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Simarro P.; Diarra A.; Ruiz Postigo J.; Franco J.; Jannin J. The Human African Trypanosomiasis control and Surveillance Programme of the WHO 2000–2009: The Way Forward. PLoS Negl. Trop. Dis. 2011, 52e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 2312589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D.; Guther M. L. S.; Ferguson M. A. J. Comparative SILAC Proteomic Analysis of Trypanosoma brucei Bloodstream and Procyclic Lifecycle Stages. PLoS One 2012, 75e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen B. C.; Sivam D.; Kifer C. T.; Myler P. J.; Parsons M. Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics 2009, 10, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton C.; Shapira M. Post-translational regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [DOI] [PubMed] [Google Scholar]
- Nett I. R. E.; Martin D. M. A.; Miranda-Saavedra D.; Lamont D.; Barber J. D.; Mehlert A.; Ferguson M. A. J. The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol. Cell. Proteomics 2009, 8.7, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nett I. R. E.; Davidson L.; Lamont D.; Ferguson M. A. J. Identification and specific localization of tyrosine-phosphorylated proteins in Trypanosoma brucei. Eukaryot. Cell 2009, 84617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H.; Hirumi K. Continous cultivation of Trypanosoma brucei bloodstream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [PubMed] [Google Scholar]
- Brun R.; Schonenberger M. Cultivation and in vivo cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979, 36, 289–292. [PubMed] [Google Scholar]
- Wirtz E.; Leal S.; Ochatt C.; Cross G. A. M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universial sample preparation method for proteome analysis. Nat. Methods 2009, 65359–362. [DOI] [PubMed] [Google Scholar]
- Beausoleil S. A.; Jedrychowski M.; Schwartz D.; Elias J. E.; Villen J.; Li J.; Cohn M. A.; Cantley L. C.; Gygi S. P. Large-scale charactersation of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thingholm T. E.; Jorgensen T. J. D.; Jensen O. N.; Larsen M. R. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protocols 2006, 1, 1929–1935. [DOI] [PubMed] [Google Scholar]; b Jensen S. S.; Larsen M. R. Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun. Mass Spectrom. 2007, 21, 3635–3645. [DOI] [PubMed] [Google Scholar]
- Schroeder M. J.; Shabanowitz J.; Schwartz J. C.; Hunt D. F.; Coon J. J. A neutral loss activation method for improved phosphopeptide sequence analysis by quadropole ion trap mass spectrometry. Anal. Chem. 2004, 76, 3590. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualised p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhausert N.; Michalskit A.; Scheltemat R. A.; Olsen J. V.; Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Aslett M.; Aurrecoechea C.; Berriman M.; Brestelli J.; Brunk B. P.; Carrington M.; Depledge D. P.; Fischer S.; Gajria B.; Gao X.; Gardner M. J.; Gingle A.; Grant G.; Harb O. S.; Heiges M.; Hertz-Fowler C.; Houston R.; Innamorato F.; Iodice J.; Kissinger J. C.; Kraemer E.; Li W.; Logan F. J.; Miller J. A.; Mitra S.; Myler P. J.; Nayak V.; Pennington C.; Phan I.; Pinney D. F.; Ramasamy G.; Rogers M. B.; Roos D. S.; Ross C.; Sivam D.; Smith D. F.; Srinivasamoorthy G.; Stoeckert C. J.; Subramanian S.; Thibodeau R.; Tivey A.; Treatman C.; Velarde G.; Wang H. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acid Res. 2010, 38, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quakenbush J. Microarray data normalisation and transformation. Nat. Genet. 2002, 32, 496–501. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Lemeer S.; Boesche M.; Lang M.; Mathieson T.; Bantscheff M.; Kuster B. Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell. Proteomics 2011, 102M110 003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S.; Gagneur J.; Robinson P. N. GOing Bayesian: model-based gene set analysis of genome-scale data. Nucleic Acid Res. 2010, 38113523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C.; Peacock C. S.; Wood V.; Aslett M.; Kerhornou A.; Mooney P.; Tivey A.; Berriman M.; Hall N.; Rutherford K.; Parkhill J.; Ivens A. C.; Rajandream M.-A.; Barrell B. GeneDB: a resource for prokaryotic and eukaryotic organisms. Nucleic Acid Res. 2004, 32, 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parsons M.; Worthey E. A.; Ward P. N.; Mottram J. C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 2005, 6, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Urbaniak M. D.; Mathieson T.; Bantcheff M.; Eberhard D.; Grimaldi R.; Miranda-Saavedra D.; Wyatt P. G.; Ferguson M. A. J.; Frearson J. A.; Drewes G. Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem. Biol. 2012, 7, 1858–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurst M.; Seliger B.; Anand Jha B.; Klein C.; Quiroz R.; Clayton C. Expression of the RNA recognition motif protein RBP10 promotes a bloodstream-form transcript pattern in Trypanosoma brucei. Mol. Microbiol. 2012, 85, 1048–1063. [DOI] [PubMed] [Google Scholar]
- Dean S.; Marchetti R.; Kirk K.; Matthews K. R. A surface transporter family conveys the trypanosome differentiation signal. Nature 2009, 459, 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Cross G. A.; Nes W. D. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol syntehsis and cell proliferation of Trypanosma brucei. J. Lipid Res. 2007, 48, 665–73. [DOI] [PubMed] [Google Scholar]
- Gunasekera K.; Wuthrich D.; Braga-Lagache S.; Heller M.; Ochsenreiter T. Proteome remodelling during development from bloodstream to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Geneomics 2012, 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butter F.; Bucerius F.; Michel M.; Cicova Z.; Mann M.; Janzen C. J. Comparative proteomics of two life cycle stages of stable isotope-labelled Trypanosoma brucei reveals novel components of the parasite’s host adaption machinery. Mol. Cell. Proteomics 2013, 121172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn K.; Matthews K. R. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 2007, 10, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barquilla A.; Saldivia M.; Diaz R.; Bart J.-M.; Vadal I.; Calvo E.; Hall M. N.; Navarro M. Third target of rapamycin complex negatively regulates development of quiescence in Trypanosma brucei. Proc. Natl. Acad. Sci. U.S.A. 2012, 1093614399–14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister D. D.; Burklard G.; Morand S.; Renggli C. K.; Roditi I.; Vassella E. A mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryotic Cell 2006, 571126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassella E.; Kramer R.; Turner C. M. R.; Wankell M.; Modes C.; van de Bogaard M.; Boshart M. Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol. Microbiol. 2001, 47133–46. [DOI] [PubMed] [Google Scholar]
- Chou S.; Jensen B. C.; Parsons M.; Alber T.; Grundner C. The Trypanosoma brucei lifecycle switch TbPTP1 is structurally conserved and dephosphorylates the nucleolar protein NOPP44/46. J. Biol. Chem. 2010, 2852922075–22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Urbaniak M. D. Casein kinase 1 isoform 2 is essential for bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 2009, 166, 183–185. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Alsford S.; Turner D. J.; Obado S. O.; Sanchez-Flores A.; Glover L.; Berriman M.; Hertz-Fowler C.; Horn D. High-Throughput phenotyping using parallel sequencing of RNA interference targets in the African Trypanosome. Genome Res. 2011, 21, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mackey Z. B.; Koupparis K.; Nishino M.; McKerrow J. H. High-Throughput Analysis of an RNAi Library Identifies Novel Kinase Targets in Trypanosoma brucei. Chem. Biol. Drug Des. 2011, 78, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walrad P. B.; Capewell P.; Fenn K.; Matthews K. R. The post-transcriptional trans-acting regulator, TbZFP3, co-ordinates transmission-stage enriched mRNAs in Trypanosoma brucei. Nucleic Acid Res. 2011, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginger M. L.; Portman N.; McKean P. G. Swimming with protists: perception, motility and flagellum assembly. Nat. Rev. Microbiol. 2008, 6, 838–850. [DOI] [PubMed] [Google Scholar]
- Broadhead R.; Dawe H. R.; Farr H.; Griffiths S.; Hart S. R.; Portman N.; Shaw M. K.; Ginger M. L.; Gaskell S. J.; Paul G. M.; Gull K. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 2006, 440, 224–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.