

Abstract
MicroRNAs are post-transcriptional regulators that control mRNA stability and the translation efficiency of their target genes. Mature microRNAs are approximately 22-nucleotide in length. They mediate post-transcriptional gene regulation by binding to the imperfect complementary sequences (a.k.a. microRNA regulatory elements, MRE) in the target mRNAs. It is estimated that more than one-third of the protein-coding genes in the human genome are regulated by microRNAs. The experimental methods to examine the interaction between the microRNA and its targeting site(s) in the mRNA are important for understanding microRNA functions. The luciferase reporter gene assay has recently been adapted to test the effect of microRNAs. In this chapter, we use a previously identified miR-138 targeting site in the 3′-untranslated region (3′-UTR) of the RhoC mRNA as an example to describe a quick method for testing the interaction of microRNA and mRNA.
Keywords: MicroRNA, MicroRNA targeting sequence, MicroRNA regulatory element, Luciferase reporter gene assay, miR-138, RhoC
1. Introduction
MicroRNAs (miRNAs) are endogenously expressed, single-stranded noncoding RNAs (approximately 20–24 nucleotides in length) found in almost all eukaryotic cells. miRNAs have been shown to regulate many developmental and physiological processes, and the deregulation of miRNAs has been linked to a number of disease processes (1, 2). miRNAs constitute an important class of fine-tuning gene expression regulators referred to as “dimmer switches” because of their ability to repress gene expression without completely silencing it. They are post-transcriptional regulators that bind to imperfect complementary sequences (a.k.a. miRNA regulatory element, MRE) on the target messenger RNA transcripts (mRNAs) and usually result in translational repression and gene silencing (2). Animal miRNAs usually bind to sites in the 3′-untranslated region (3′-UTR), whereas plant miRNAs usually bind to coding regions of mRNAs. A number of bioinformatics tools are available to predict the miRNA targeting sequences (3). However, to understand the roles of microRNA in complex biological processes, it is important to experimentally assess the functional relevance of the predicted miRNA targeting site(s).
MicroRNA-138 (miR-138) has been shown to regulate a number of essential biological processes, including the development of the mammary gland (4), regulation of dendritic spine morphogenesis (5), modulation of cardiac patterning during embryonic development (6), and thermo-tolerance acquisition (7). The deregulation of miR-138 has been frequently observed in a number of cancer types, including thyroid cancer (8), lung cancer (9), leukemia (10), and head and neck/oral cancers (11–15). Down-regulation of miR-138 is associated with enhanced RhoC expression and cell migration and invasion in oral cancer cells (12, 16). A targeting sequence for miR-138 has recently been identi fied in the 3′-UTR of the RhoC mRNA (Fig. 1) (12). In this chapter, for illustration, we will test the interaction of miR-138 and its targeting sequence in the 3′-UTR of the RhoC mRNA.
Fig. 1.
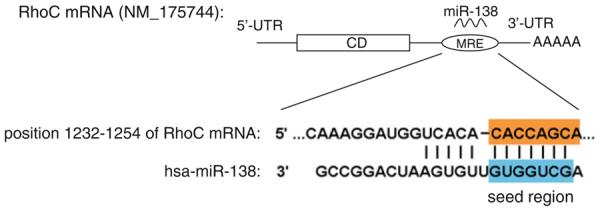
The miR-138 targeting sequence located in the 3′-untranslated region of RhoC mRNA.
Firefly luciferase is commonly used as a reporter to assess the transcriptional activity in intact cells. The most common application of luciferase reporter gene assay is to examine the regulation of transcriptional activities by promoters and transcription factors. Recently, this assay has also been adapted for testing the effect of miRNA-mediated, post-transcriptional regulation on target genes. This is achieved by engineering a luciferase gene construct containing the predicted miRNA targeting sequence from the target gene (often located in the 3′-UTR). For many human genes, luciferase constructs containing the entire 3′-UTR can be obtained from a number of commercial sources (e.g., OriGene Technologies, Inc, GeneCopoeia, Inc, and SwitchGear Genomics). However, the 3′-UTRs may contain multiple targeting sequences and other regulatory elements. Specific assays to test each miRNA targeting sequences are needed. In this chapter, we describe a quick method to test the interaction of miRNA and the specific target sequence. We also present a simple strategy for creating mutant construct as the negative control.
2. Materials
2.1. pGL3 Luciferase Reporter Vector
2.1.1. pGL3-Control Firefly Luciferase Control Vector (Promega)
There are a number of available luciferase reporter vectors, including recently introduced ones that are designed for testing miRNA-mediated gene silencing. In this chapter, we use pGL3-Control Vector (Promega), one of the most commonly used luciferase reporter vectors.
2.1.2. pRL-TK Vector (Renilla Luciferase Control Reporter Vectors) (Promega)
The pRL-TK vector provides constitutive expression of Renilla luciferase. pRL-TK vector co-transfected with firefly luciferase vector provides an internal control for normalization of the transfection efficiency.
2.2. pGL3 Luciferase Reporter Constructs
pGL3 luciferase reporter constructs are created by cloning the specific miRNA binding sequence (wild-type/mutants) into the XbaI site located at 3′-UTR of pGL3-Control vector.
2.2.1. Digestion of pGL3-Control Vector
Restriction enzyme XbaI (Fermentas).
Alkaline phosphatase, calf intestinal (CIP, Fermentas).
2.2.2. Oligonucleotides Synthesis (Sigma) and Annealing
Sense and antisense oligos were synthesized with XbaI site at 5′and 3′-end.
10× Annealing buffer: 100 mM Tris-HCl, pH 7.5, 1 M NaCl, 10 mM EDTA.
2.2.3. Agarose Gel Electrophoresis for DNA Analysis
10× TBE buffer (Tris-Borate electrophoresis buffer: 108 mg/mL Tris base, 55 mg/mL Boric acid, 9.3 mg/mL EDTA).
Ethidium bromide (5 mg/mL) (EtBr).
Prepare 1.5% agarose gel: Add 0.75 g agarose to 50 mL 1× TBE. Heat the solution in the microwave to dissolve the agarose. When the solution is cooling down, add 5 μL of EtBr to the dissolved agarose and mix well in a fume hood. Pool the gel to the gel tray and let it cool down completely.
Running buffer: 1× TBE.
Electrophoresis equipment.
Gel loading dye (6×): 50 mM EDTA, 0.2% SDS, 50% glycerol, 0.05% w/v bromophenol blue.
1 kb and 100 bp DNA ladder.
2.2.4. Quick Ligation Kit (New England Biolabs)
-
(a)
Quick T4 DNA ligase; 2× Quick ligation reaction buffer.
2.2.5. Transformation
XL1-blue Supercompetent cells (Stratagene).
SOC media (Cellgro).
LB-agar plates with 100 μg/mL ampicillin: 5.0 g tryptone, 2.5 g yeast extract, 5.0 g NaCl, 7.5 g agar; adjust volume to 500 mL with dH2 O. After autoclaving, add ampicillin to make final concentration 100 μg/mL.
2.2.6. DNA Purification
-
(a)
GeneJet Plasmid Miniprep Kit (Fermentas).
2.3. Transfection Reagent and miRNA Mimic
2.3.1. Hela Cell
HeLa is an immortal cell line used for transfection experiments. Other comparable cell lines can also be used based on experimental design.
2.3.2. Medium and Transfection Reagent
-
(a)
Lipofectamine™ 2000 transfection reagent (Invitrogen).
-
(b)
Opti-MEM I reduced serum medium (Gibco).
2.3.3. miRNA Mimic
hsa-miR-138 mimic: 5′-AGCUGGUGUUGUGAAUCAGGCCG (Thermo scientific-Dharmacon).
Negative control mimic #1: cel-miR-67, 5′-UCACAACCUCCUAGAAAGAGUAGA (Thermo scientific-Dharmacon).
2.4. DUAL-Luciferase Reporter Assay System
Dual-luciferase Reporter Assay kit provides an optimized system and all the necessary reagents for the sequential assay of firefly and Renilla luciferase activity.
2.4.1. Dual-Luciferase Reporter Assay Kit (Promega)
1× Passive Lysis Buffer (PLB): Dilute 5× PLB with dH2 O.
LAR II: Resuspend the lyophilized Luciferase Assay Substrate in Luciferase Assay Buffer II. Store at −°C (up to 1 month) or −70°C (up to 1 year).
Stop & Glo Reagent: Add 50× Stop & Glo Substrate to Stop & Glo buffer. Store at −20°C for 15 days.
2.4.2. Glomax 20/20 Luminometer (Promega)
Other comparable luminometer can also be used.
3. Methods
3.1. Cloning Method
3.1.1. Design the Wild-Type and the Mutant miRNA Targeting Sites (Oligonucleotides Synthesis)
Chemical-based oligonucleotide synthesis provides a rapid and inexpensive access to custom-made oligonucleotides of the desired sequence (up to several hundred nucleotide residues). To simplify our cloning strategy, we will take the advantage of vastly available resources for synthesizing oligonucleotides (e.g., Integrated DNA Technologies, Sigma-Genosys). To illustrate, we will design a set of oligonucleotides to test the previously described hsa-miR-138 targeting sequence in the 3′-UTR of the RhoC mRNA (12). Sense and antisense sequences corresponding to a 62-bp fragment from the 3′-UTR of RhoC mRNA (position 1210–1271, NM_175744) will be used. Partial sequences for the XbaI site are appended to the ends of the oligo for creating sticky ends upon annealing. The 5′phosphorylation is also required to facilitate the ligation. The sequences of these oligonucleotides are listed below. The XbaI sites are indicated by bold font, and the seed regions of the hsa-miR-138 targeting site are indicated by underlining.
Wild-type sense: 5′-pCTAGATCTTGCCCCCTTTGACCT T C C C C A A A G G A T G G T C A C A C A C C A G C ACTTTATACACTTCTGGCT-3′.
-
Wild-type antisense: 5′-pCTAGAGCCAGAAGTGTATAAAGT GCTGGTG TGTGACCATCCTTTGGGGAAGGTC AAAGGGGGCAAGAT-3′.
As a negative control, we also designed oligonucleotides containing a mutated hsa-miR-138 targeting site. The mutants vare created by replacing the seed regions of the hsa-miR-138 targeting site with T(7) (see Note 1).
Mutant sense: 5′-pCTAGATCTTGCCCCCTTTGACCTTCCCCAAAGGATGGTCACATTTTTTTACTTTATACACTTCTGGCT-3′.
Mutant antisense: 5′-pCTAGAGCCAGAAGTGTATAAAGTAAAAAAATGTGACCATCCTTTGGGGAAGGTCAAAGGGGGCAAGAT-3′.
3.1.2. Oligonucleotides Annealing
Resuspend both sense and antisense oligos in 1× annealing buffer to make the final concentration 100 μM.
Mix 1 μL of each strand with 1 μL of 10× annealing buffer and 7 μL of H2O.
Incubate the reaction mixture at 95°C for 6 min, and then place at room temperature for 30 min.
Store on ice or at 4°C until ready to use.
Dilute the annealed oligos twice before performing ligation: for the fi rst dilution, dilute 20× with dH2O. For the second dilution, dilute 100× with 1× annealing buffer (final concentration is 5 nM).
3.1.3. pGL3-Control Vector Linearization and Purification
pGL3-Control vector is digested by XbaI (Fermentas) and dephosphorylated by Alkaline phosphatase, and calf intestinal (CIP, Fermentas) simultaneously. To cut the vector, 1 μg of PGL3 DNA is incubated with 1 μL of XbaI, 1 μL of CIP, 1 μL of 10× digestion buffer, and 7 μL of dH2O (total volume 10 μL) at 37°C for 1 h. Then incubate the mixture at 75°C for 5 min to inactivate the enzymes.
Add 6× loading dye to the digested product and load to 1.5% agarose gel.
Electrophorese the sample at 100 V for 20 min. Check the gel frequently while it is running to make sure it is not getting too hot, as this will distort the bands or melt the agarose. If the DNA fragment is ≤200 bp, check the gel every 5 min to make sure the band is not running out of gel.
Following electrophoresis, check the gel under the UV light and take a picture as a record.
Cut the DNA fragment from agarose gel with a clean, sharp scalpel.
Follow the Qiaquick gel extraction kit protocol (Qiagen) to retrieve and purify the DNA fragment (linearized pGL3-Control vector).
Measure the concentration (μg/ μL) of DNA and convert it to molar concentration.
3.1.4. Ligation
Insert synthesized oligos into pGL3-Control vector at XbaI site.
Combine 1 μL XbaI digested, and dephosphorylated pGL3-Control vector (1 nM) with 1 μL of insert (5 nM). The vector and insert ratio is 1:5. Adjust volume to 5 μL with dH2O.
Add 5 μL of 2× quick ligation reaction buffer and 0.5 μL of quick T4 DNA ligase and mix thoroughly.
Centrifuge brie fl y and incubate at room temperature for 30 min.
Chill on ice, the sample is ready for transformation or can be stored at −20°C (see Note 2).
3.1.5. Transformation, Pick up Clones, and Isolation of Plasmid DNA
The ligation products are ready for transformation. Many different transformation methods could be used. We use XL1-blue super-competent cells from Stratagene to perform transformation. We brie fl y describe the procedure here:
Prechill two 14 mL BD falcon polypropylene round-bottom tubes on ice. (One for ligation product transformation and one for control experiment). Preheat SOC medium to 42°C.
Thaw XL1-blue competent cells on ice. Then add 30 μL of cells into each of prechilled tubes and mix gently.
Add 0.5 μL of β-mercaptoethanol to each tube and swirl gently. Incubate tubes on ice for 10 min while gently swirling every 2 min.
Add 1–2 μL of ligation product to one tube and add the same amount of control product to the other tube. Mix gently by pipetting up and down. Incubate the tubes on ice for 30 min.
Heat shock for 45 s in 42°C water bath, then chill on ice for 5 min. The precise duration of the heat shock (45 s) is critical for transformation efficiency.
Add 300 μL of preheated SOC medium and incubate at 37°C for 1 h while shaking at 250–300 rpm.
Spread 50–100 μL of transformation mixture on LB agar plates (100 μg/mL ampicillin).
Incubate the plate overnight (≥ 16 h) at 37°C.
Check the plate for clone growth next day.
Pick up several clones to grow overnight at 37°C with shaking at 250–300 rpm.
Purify plasmid DNA with GeneJet Plasimd Miniprep Kit (Fermentas).
Check the plasmid DNA on 1.5% agarose gel. If the size of the plasmid DNA is as expected, sequence the plasmid to check the orientation of the insert and detecting any mutation (see Note 3).
3.2. Co-transfection of Plasmid DNA and miRNA Mimic Along with pRL-TK Vector into Mammalian Cells
3.2.1. Seed Hela Cells
One day prior to transfection, seed 5–10 × 104 Hela cells to each well of a 24-well plate. Approximately 20–40% confluence of the cells is anticipated at the time of transfection (16 h after seeding).
Incubate cells at 37°C with 5% CO2 overnight.
3.2.2. Lipofectamine 2000 Transfection
All volumes are multiplied by 3.5 to account for the triplicate samples and loss during pipetting.
Prepare stock pGL3 plasmid (250 μg/mL), pRL-TK (5 μg/mL), hsa-miR-138 mimic (25 μM), and negative control mimic (25 μM) in RNase-free water (see Note 4).
Prepare transfection reagent: for each transfection, 2 μL of lipofectamine 2000 is incubated with 400 μL of Opti-MEM medium at room temperature for 5 min (Solution 1).
-
Mix plasmid and the appropriate miRNA mimic.
For our experiment testing the hsa-miR-138 targeting site in RhoC mRNA, the following experimental groups are used:
Negative control mimic (1.6 μL) + pGL3-WT (4 μL) + pRL-TK (1 μL).
hsa-miR-138 mimic (1.6 μL) + pGL3-WT (4 μL) + pRL-TK (1 μL).
Negative control mimic (1.6 μL) + pGL3-Mutant (4 μL) + pRL-TK (1 μL).
hsa-miR-138 mimic (1.6 μL) + pGL3-Mutant (4 μL) + pRL-TK (1 μL).
This will make the final working concentrations at 100 nM miRNA mimic, 2.5 ng/ μL pGL3 plasmid, and 12.5 pg/ μL pRL-TK.
-
4.
After 5 min, add Solution 1 into each tube and mix the contents of all tubes gently by pipetting carefully up and down.
-
5.
Incubate tubes for 20 min at room temperature to form the transfection mixture.
-
6.
Remove medium from the wells of the 24-well plate containing cells and wash with PBS twice. Then add the transfection mixture to each well.
-
7.
Incubate cells at 37°C in 5% CO2 for 48 h.
-
8.
If cell toxicity is observed after 24 h, replace the transfection medium with complete medium and continue incubation.
3.3. Luciferase Activity Assay
After 48 h, remove growth medium from wells and rinse cells with PBS twice.
Dispense 200 μL of 1× PLB into each well.
Gently shake the 24-well plate for 15 min at room temperature. Transfer lysate to a 1.5 mL tube. If residual cell debris is presented in the lysate, clear the lysate by centrifuge at 13,500 rpm for 15 min at 4°C and transfer the supernatant to a new tube.
Pre-dispense 100 μL of LAR II into 1.5 mL clear Eppendorf tubes.
Program the luminometer to perform a 2-s pre-measurement delay, followed by a 10-s measurement period for each reporter assay.
Carefully transfer 20 μL of cell lysate into the tube containing LAR II; mix by pipetting 2 or 3 times. Place the tube in the luminometer and initiate reading. Do not vortex, and avoid bubbles which can interfere with the mixing of Stop & Glo Reagent added in the next step, and affect the fi nal reading.
Record the fi re fl y luciferase activity measurement.
Remove the tube from the luminometer, add 50 μL of Stop & Glo Reagent and mix gently. Replace the sample in the luminometer, and initiate reading.
Record the Renilla luciferase activity measurement.
Calculate the ratio of fi re fl y luciferase activity to Renilla luciferase activity. This is the normalized luciferase activity.
The effect of miRNA on the luciferase gene expression can be shown as changes in relative luciferase activity (Fig. 2). This is achieved by setting the normalized luciferase activity of negative mimic + pGL3-WT + pRL-TK sample to 100%, and the reading of miR-138 mimic + pGL3-WT + pRL-TK sample is shown as the percentage of negative mimic + pGL3-WT + pRL-TK. Similarly, for the mutants, relative luciferase activity is calculated by setting the normalized luciferase activity of negative mimic + pGL3- Mutant + pRL-TK sample to 100%, and the reading of miR-138 mimic + pGL3- Mutant + pRL-TK sample is shown as the percentage of negative mimic + pGL3- Mutant + pRL-TK (see Note 5).
Fig. 2.
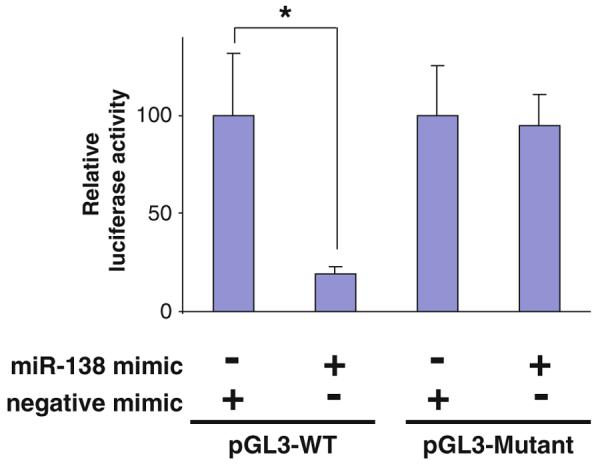
MicroRNA-138 targeting the RhoC mRNA through a targeting sequence located at 3′-UTR. Dual-luciferase reporter assays were performed to test the interaction of hsamiR-138 and its targeting sequence in the RhoC 3′-UTR using constructs containing the predicted targeting sequence (pGL3-WT) and mutated targeting sequence (pGL3-Mutant) cloned into the 3′-UTR of the reporter gene. Data represent three independent experiments with triplicate measurements. * indicates p < 0.05.
4. Notes
As a negative control, we created a reporter construct containing the mutated hsa-miR-138 targeting site by replacing the seed region of the targeting site with T(7). Other strategies for creating mutant reporter constructs are also being used in various studies, including mutating only the bases complementary to the miRNA sequence, mutating the entire miRNA targeting site (e.g., using reversed strand), or deleting the seed region.
Since the XbaI site is the only available restriction enzyme cutting site on 3′ -UTR of pGL3-Control vector, the insert could have two orientations in pGL3-insert plasmid: the forward insert sequence and reversed insert sequence (theoretically a 50/50 chance for each orientation). It may be worth the effort to create additional restriction enzyme cutting site on 3′ -UTR of pGL3-Control vector. Doing so will allow different enzyme sites to be appended to the ends of the synthesized oligos and ensure that the ligation will be in the correct orientation. This can be achieved by simply inserting a short oligo containing multiple restriction enzyme sites into the XbaI site.
Since we only use one restriction enzyme (XbaI) for the cloning, self-ligation is expected. A negative control reaction may be incorporated in the ligation reactions, for controlling the self-ligation. Add all the components in the reaction except insertion DNA. We expect to observe no or very few colonies from the control reaction. The number of colonies to pick from “experimental” plate depends on the number of colonies grown on the control plate.
The negative control of the miRNA mimic used in our experiment is a miRNA from Caenorhabditis elegans (cel-miR-67: UCACAACCUCCUAGAAAGAGUAGA). While this miRNA has been suggested by Thermo scientific-Dharmacon to have minimal sequence identity with known miRNAs in human, mouse, and rat, it is important to verify that there is no potential targeting site for this miRNA in the mRNA fragment of our interest. An alternative control (cel-miR-239b, UUGUACUACACAAAAGUACUG) is available from Thermo scientific-Dharmacon, if additional negative control miRNA mimic is needed.
While most studies have focused on miRNA targeting sites located in the 3′-UTR, a number of miRNA targeting sites have also been found in the coding region (and in the 5′-UTR to a lesser extent). Although our method presented here is for testing the miRNA targeting site located in the 3′-UTR of the targeted mRNA, it can be adapted to test miRNA targeting sites in the coding regions. This can be achieved by simply inserting the miRNA targeting sequence into the XbaI site at the 3′-UTR of the pGL3-Control vector. This approach has been used in a number of studies (17, 18).
Acknowledgment
This work was supported in part by NIH PHS grants (CA135992, CA139596, DE014847) and supplementary funding from UIC CCTS (UL1RR029879). Y.J. is supported by PHS T32DE018381 from NIDCR. We thank Ms. Katherine Long for her editorial assistance.
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai Y, Zhou X. Computational methods for the identification of microRNA targets. Open Access Bioinform. 2010;2:29–39. doi: 10.2147/OAB.S6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Li Q. Identification of differentially expressed microRNAs during the development of Chinese murine mammary gland. J Genet Genomics. 2007;34:966–973. doi: 10.1016/S1673-8527(07)60109-X. [DOI] [PubMed] [Google Scholar]
- 5.Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, Hubel K, Dekker F, Hedberg C, Rengarajan B, Drepper C, Waldmann H, Kauppinen S, Greenberg ME, Draguhn A, Rehmsmeier M, Martinez J, Schratt GM. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morton SU, Scherz PJ, Cordes KR, Ivey KN, Stainier DY, Srivastava D. microRNA-138 modulates cardiac patterning during embryonic development. Proc Natl Acad Sci USA. 2008;105:17830–17835. doi: 10.1073/pnas.0804673105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kisliouk T, Yosefi S, Meiri N. MiR-138 inhibits EZH2 methyltransferase expression and methylation of histone H3 at lysine 27, and affects thermotolerance acquisition. Eur J Neurosci. 2011;33:224–235. doi: 10.1111/j.1460-9568.2010.07493.x. [DOI] [PubMed] [Google Scholar]
- 8.Mitomo S, Maesawa C, Ogasawara S, Iwaya T, Shibazaki M, Yashima-Abo A, Kotani K, Oikawa H, Sakurai E, Izutsu N, Kato K, Komatsu H, Ikeda K, Wakabayashi G, Masuda T. Downregulation of miR-138 is associated with overexpression of human telom-erase reverse transcriptase protein in human anaplastic thyroid carcinoma cell lines. Cancer Sci. 2008;99:280–286. doi: 10.1111/j.1349-7006.2007.00666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seike M, Goto A, Okano T, Bowman ED, Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, Gemma A, Kudoh S, Croce CM, Harris CC. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci USA. 2009;106:12085–12090. doi: 10.1073/pnas.0905234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Yang L, Hu J, Ruan J. miR-138 might reverse multidrug resistance of leukemia cells. Leuk Res. 2010;34:1078–1082. doi: 10.1016/j.leukres.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, Dai Y, Liu X, Wang C, Wang A, Chen Z, Heidbreder CE, Kolokythas A, Zhou X. Identification and experimental validation of G protein alpha inhibiting activity polypeptide 2 (GNAI2) as a microRNA-138 target in tongue squamous cell carcinoma. Hum Genet. 2011;129:189–197. doi: 10.1007/s00439-010-0915-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L, Liu X, Kolokythas A, Yu J, Wang A, Heidbreder CE, Shi F, Zhou X. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138 mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int J Cancer. 2010;127:505–512. doi: 10.1002/ijc.25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong TS, Liu XB, Chung-Wai Ho A, Po-Wing Yuen A, Wai-Man Ng R, Ignace Wei W. Identification of pyruvate kinase type M2 as potential oncoprotein in squamous cell carcinoma of tongue through microRNA pro fi ling. Int J Cancer. 2008;123:251–257. doi: 10.1002/ijc.23583. [DOI] [PubMed] [Google Scholar]
- 14.Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin Cancer Res. 2008;14:2588–2592. doi: 10.1158/1078-0432.CCR-07-0666. [DOI] [PubMed] [Google Scholar]
- 15.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Jiang L, Wang A, Yu J, Shi F, Zhou X. MicroRNA-138 suppresses invasion and promotes apoptosis in head and neck squamous cell carcinoma cell lines. Cancer Lett. 2009;286:217–222. doi: 10.1016/j.canlet.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawasaki H, Taira K. Hes1 is a target of microRNA-23 during retinoic-acid-induced neuronal differentiation of NT2 cells. Nature. 2003;423:838–842. doi: 10.1038/nature01730. [DOI] [PubMed] [Google Scholar]
- 18.Qin W, Shi Y, Zhao B, Yao C, Jin L, Ma J, Jin Y. miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS One. 2010;5:e9429. doi: 10.1371/journal.pone.0009429. [DOI] [PMC free article] [PubMed] [Google Scholar]