

Abstract
CD4+ memory cell development is dependent upon T cell receptor (TCR) signal strength, antigen dose and the cytokine milieu, all of which are altered in type 1 diabetes (T1D). We hypothesized that CD4+ T cell turnover would be greater in type 1 diabetes subjects compared to controls. In vitro studies of T cell function are unable to evaluate dynamic aspects of immune cell homoeostasis. Therefore, we used deuterium oxide (2H2O) to assess in vivo turnover of CD4+ T cell subsets in T1D (n = 10) and control subjects (n = 10). Serial samples of naive, memory and regulatory (Treg) CD4+ T cell subsets were collected and enrichment of deoxyribose was determined by gas chromatography–mass spectrometry (GC–MS). Quantification of T cell turnover was performed using mathematical models to estimate fractional enrichment (f, n = 20), turnover rate (k, n = 20), proliferation (p, n = 10) and disappearance (d*, n = 10). Although turnover of Tregs was greater than memory and naive cells in both controls and T1D subjects, no differences were seen between T1D and controls in Treg or naive kinetics. However, turnover of CD4+ memory T cells was faster in those with T1D compared to control subjects. Measurement and modelling of incorporated deuterium is useful for evaluating the in vivo kinetics of immune cells in T1D and could be incorporated into studies of the natural history of disease or clinical trials designed to alter the disease course. The enhanced CD4+ memory T cell turnover in T1D may be important in understanding the pathophysiology and potential treatments of autoimmune diabetes.
Keywords: deuterium oxide, heavy water, isotope labelling, memory T cell, regulatory T cell, T cell kinetics, type 1 diabetes
Introduction
CD4+ T cells are implicated in the pathogenesis of autoimmune diabetes. In murine models islet antigen-specific CD4+ T cells are required for disease and depletion of CD4+ T cells prevents disease 1. In human type 1 diabetes (T1D), CD4+ T cells are among the cells found infiltrating the pancreatic islets 2 and human leucocyte antigen (HLA) class II alleles are associated strongly with disease risk 3. Because of the importance of T cells in T1D pathogenesis, several studies have looked for differences in the numbers of T cells between T1D subjects and healthy controls. There is no consistent pattern of alterations in naive T cell numbers across studies 4–6. Similarly, both increased as well as decreased numbers of memory T cells have been reported in individuals with T1D compared with controls 7. While lack of CD4+forkhead box protein 3 (FoxP3)+ regulatory T cells (Treg) results unambiguously in the early onset of autoimmune diabetes in both mouse and man 8, most studies have none the less found no difference in the number of Treg present in peripheral blood of humans with and without T1D 9–11.
In contrast to these static measurements of CD4+ T cell numbers, there are indications of altered stability, proliferation and death of CD4+ T cells in autoimmune diabetes. We reported diminished maintenance of FoxP3 expression in CD4+CD25+ Treg of T1D subjects 12 and we and others have found evidence of impaired signalling through interleukin (IL)-2R, required for both Treg and memory T cell homeostasis, in T1D 13–16. Studies in mice suggest Treg turnover 17,18 may have important implications for the durability of immune tolerance 19,20. Signals that promote proliferation may break anergy in potentially autoreactive memory T cells 21, while signals that cause deletion of autoreactive T cells through activation-induced cell death may be impaired in T1D subjects, as they are in non-obese diabetic (NOD) mice 22.
There are also indications that assessments of T cell proliferation and survival in vitro may not correlate with in vivo measures 23, emphasizing the need to study T cell kinetics in vivo. Advances in stable isotope methodologies have created the opportunity to measure the kinetics of cell populations in vivo in humans. DNA labelling with heavy water (deuterium oxide, 2H2O) has been useful in understanding a variety of immunological processes. Others have used these techniques to study T cell kinetics in HIV 24–27, chronic lymphocytic leukaemia 28,29 and ageing 30,31. To our knowledge, the present study is the first to evaluate T cell kinetics in subjects with autoimmune disease in vivo.
Methods and materials
Clinical protocol
All subjects provided written informed consent for the protocol approved by the Benaroya Research Institute Institutional Review Board. Analysis of T cell kinetics from data obtained during the initial protocol required that we assume single exponential disappearance rates of T cell subsets 26. After 10 subjects had been studied, we modified the protocol in order to use models which do not require such an assumption, but rather allow for quantitation of proliferation and disapperance rates from measurements in samples obtained during the post-labelling period 32. The study modifications included measurements of plasma enrichment at each blood draw, extension of the labelling period to 63 days, collection of four samples in the post-labelling period and a minor increase in the amount of heavy water (60–80 ml) consumed to assure an adequate precursor pool of deuterium.
For all participants, the stable isotope deuterium (2H), was administered in the form of deuterium oxide, 70% (Cambridge Isotopes, Andover, MA, USA), as described previously 26,33. Each subject was admitted to the Benaroya Research Institute Clinical Research Center (CRD) on day 0 for 24 h to receive a 480 ml priming dose of heavy water, consumed as 60 ml every 3 h. Participants were observed for dizziness, a possible effect of transient density changes in the fluid of the inner ear, and other adverse reactions; none were observed. Upon discharge from the CRC, subjects were instructed to consume 60 or 80 ml of heavy water daily for 42 or 63 days. Granulocytes and peripheral blood mononuclear cells (PBMCs) were collected at regular intervals during the labelling period in all subjects and after the labelling period in 10 subjects (Fig. 1a). Two subjects did not complete the protocol as directed, stopping heavy water consumption on days 48 and 44 instead of 63 as instructed. Table 1 lists study subjects according to clinical characteristics and study procedures.
Fig. 1.
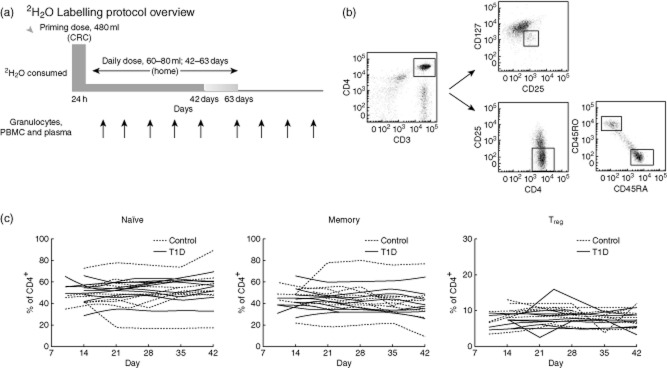
(a) Deuterium-labelling protocol overview. (b) Representative density plots from flow-cytometric isolation of T cell subsets. Regulatory T cells (Treg) were selected as being CD25+127low, CD4+ memory subsets were isolated as CD25-RO+RA− and naive cells as CD25-RO-RA+. (c) T cell subset fractions over common study period (i.e. first 42 days). Solid lines represent Type 1 diabetes (T1D) subjects (n = 10); dashed lines represent control subjects (n = 10).
Table 1.
Summary of study participants
| Subject | Gender | Age (years) | WBC (K/cmm) | Years with T1D | HbA1c (%) | Daily dose of 2H2O (ml) | Labelling period (days) | Samples collected after labelling period? |
|---|---|---|---|---|---|---|---|---|
| C1 | F | 19 | 5·5 | – | – | 60 | 42 | No |
| C2 | M | 18 | 8·3 | – | – | 60 | 42 | No |
| C3 | M | 19 | 5·3 | – | – | 60 | 42 | No |
| C4 | F | 27 | 4·7 | – | – | 60 | 42 | No |
| C5 | M | 38 | 4·4 | – | – | 60 | 42 | No |
| C6 | M | 50 | 6·0 | – | – | 60 | 42 | No |
| C7 | F | 56 | 7·8 | – | – | 80 | 63 | Yes |
| C8 | F | 24 | 9·0 | – | – | 80 | 63 | Yes |
| C9 | M | 28 | 5·5 | – | – | 80 | 63 | Yes |
| C10 | F | 24 | 7·7 | – | – | 80 | 63 | Yes |
| Mean ± s.d. | 30 ± 13 | 6·4 ± 1·6 | ||||||
| DM1 | M | 24 | 4·7 | 21 | 7·8 | 60 | 42 | No |
| DM2 | M | 32 | 6·5 | 15 | 7·2 | 60 | 42 | No |
| DM3 | M | 33 | 4·4 | 27 | 7·1 | 60 | 42 | No |
| DM4 | M | 30 | 10·5 | 3 | 7·8 | 60 | 42 | No |
| DM5 | F | 24 | 6·7 | 15 | 8·3 | 80 | 42 | Yes |
| DM6 | F | 35 | 4·7 | 24 | 7·7 | 80 | 42 | Yes |
| DM7 | F | 27 | 6·0 | 14 | – | 80 | 63 | Yes |
| DM8 | M | 40 | 8·1 | 8 | 7·8 | 80 | 48 | Yes |
| DM9 | M | 34 | 7·5 | 18 | – | 80 | 44 | Yes |
| DM10 | M | 49 | 7·8 | 43 | 8·3 | 80 | 63 | Yes |
| Mean ± s.d. | 33 ± 8 | 6·7 ± 1·9 | 19 ± 11 | 7·8 ± 0·4 |
There were no significant differences between type 1 diabetes and control groups with respect to age (P = 0·59) and white blood cell (WBC) count (P = 0·74). Concurrent medications: C1: oral contraceptive; DM1-10: insulin; DM3: fish oil, aspirin; DM7: fluoxetine, oral contraceptive; DM8: insulin aspirin, paraxetine, bupropion, gabapentin, lisinopril, simvistatin; DM9: lisinopril, claritin, flonase; DM10: zonasamide; F: female; M: male; s.d.: standard deviation.
Cell staining and separation
PBMC were isolated from 50 ml of heparinized fresh blood by Ficoll-Paque (GE Healthcare, Piscataway, NJ, USA) density gradient centrifugation. Cells were stained with either BD (Franklin Lakes, NJ, USA) anti-CD3 allophycocyanin (APC)-cyanin 7 (Cy7) and eBioscience (San Diego, CA, USA) anti-CD45RA fluorescein isothiocyanate (FITC), CD45RO phycoerythrin (PE), CD4 PE-Cy7 and CD25 APC before being sorted for naive (CD4+CD45RA+CD45RO–CD25–) and T memory (CD4+CD45RA–CD45RO+CD25–) populations, or cells were stained with BD anti-CD127 PE and eBioscience anti-CD25 FITC, CD4 PE-Cy7, and CD3 APC, then sorted for Treg cells (CD4+CD25+CD127low). All cells were sorted using a FACS Vantage (BD) with purity greater than 99% and analysed using Diva software (BD). Representative density plots of flow-cytometric isolation of T cell subsets are shown in Fig. 1b. Sorted cells were snap-frozen for analysis of heavy water incorporation. Available residual sorted Treg samples (n = 14) were stained additionally for FoxP3 (Appendix Fig. A1).
Measurement of isotopic enrichment in granulocyte and T cell DNA
DNA enrichment analysis was performed as described previously 33. Briefly, DNA was obtained from all T cell and granulocyte fractions after proteinase K digestion using DNEasy minicolumns (Qiagen Sciences, Valencia, CA, USA). Free nucleotides were prepared by enzymatic hydrolysis with S1 nuclease and acid phosphatase. Deoxyribose from purine nucleotides was derivatized for gas chromatography–mass spectrometry (GC–MS) analysis with pentafluorobenzyl hydroxylamine in acetic acid and acetic anhydride.
GC–MS analysis was performed using an Agilent model 5973/6890 GC (Agilent Technologies/Quantum Analytics, Foster City, CA, USA) in methane NCI mode, with an Agilent DB-17 column (30 m × 250 μm ID × 25 μm film thickness) under selected ion monitoring of m/z 435–437. The excess M+1 (EM1) mass isotopomer abundance was calculated using enriched DNA standards to correct for abundance sensitivity of mass isotopomer ratios 33. The EM1 value represents the isotope enrichment above natural abundance due to incorporation of 2H2O into newly synthesized DNA (as deoxyribose).
Measurement of plasma deuterium oxide (2H2O) enrichment
Plasma 2H2O enrichment analysis was performed as described previously 34. Briefly, water was distilled from 100 μl aliquots of serum in inverted microvials placed in a 70°C glass bead bath. The distillate was reacted with calcium carbide chips forming acetylene gas, which was trapped and further reacted with bromine in carbon tetrachloride (0·3 M) forming tetrabromomethane. After a 2-h incubation period, excess bromine was sequestered using cyclohexene. A standard curve of % enrichment was prepared from ‘stripped water’ (very low 2H2O content) and 100% enriched 2H2O. GC–MS analysis was performed as above using methane PCI mode, an Agilent DB-225 column, and SIM ions of m/z 264·7–265·7.
Calculations and mathematical modelling
The kinetics of cell turnover can be characterized by determination of fractional enrichment (f), replacement rate (k), average proliferation rate (p) and disappearance rate of labelled cells (d*).
Fractional enrichment (f) and replacement rate (k)
The fractional enrichment, f, represents the fraction of newly synthesized DNA strands or, equivalently, the fraction of newly divided cells 33. The mean f of the study populations at each time-point of the labelling period was compared across T cell subsets in all subjects (n = 10 T1D; n = 10 controls).
To calculate the replacement rate, k, we assumed that our cell subpopulations were kinetically homogeneous and disappearance of cell subsets from the blood follows a single-exponential decay equation 24,35, where 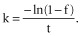 An average of f measured on days 28–42 was used for this calculation.
An average of f measured on days 28–42 was used for this calculation.
Proliferation (p) and disappearance rates (d*)
Obtaining additional samples after the labelling period and measuring the plasma or total body water (TBW), 2H enrichment allowed us to use mathematical models which do not require assumptions about disappearance kinetics in 10 subjects (n = 4 controls; n = 6 T1D). We estimated the average proliferation rate, p, of the subpopulation (including actively proliferating and quiescent subpopulations) and d*, the disappearance rate of labelled cells (the recently proliferated population). Proliferation rates, expressed as doubling time (t2), and disappearance expressed as half-life (t1/2), were calculated as ln2/p and ln2/d*, respectively. Model assumptions and parameter estimates are provided Appendix Tables A1–A3.
Statistical analyses
Unless noted otherwise, simple means were compared using the Mann–Whitney U-test and group means over time were compared using two-way analysis of variance (anova). Differences were considered significant at P ≤ 0·05. These statistical calculations were performed using GraphPad Prism 5 statistical software.
To quantify the variability in T cell fractions over time, we used a linear mixed model including a fixed-group effect (type 1 diabetes versus control) and random effect for each subject to estimate the intraclass correlation coefficient (ICC). The ICC is the proportion of the total variance explained by variability between subjects, while the proportion of variance that is due to within-subject variation is 1-ICC.
Results
Study subject characteristics
As shown in Table 1, study subjects were aged between 18 and 56 years. There were no significant differences between the T1D and control subjects with respect to mean age (33 years versus 30 years, P = 0·59) or baseline white blood cell count (6·7 K/mm3 versus 6·4 K/mm3, P = 0·74). The mean HbA1c in type 1 diabetes subjects was elevated at 7·8% and the mean duration of diabetes was 18·9 years. Mean T cell subset fractions (Treg: 7% T1D versus 10% control, memory: 46% T1D versus control 43%, naive: 50% T1D versus 50% control) were similar between groups.
Composition of CD4+T cell subsets were stable over the study period
The models that we use assume that there is minimal fluctuation in the pool size of the memory, naive and Treg subsets over the time-period of the labelling experiment. As shown in Fig. 1c, the within-subject variation in T cell fractions is small compared to the between-subject variation (ICCmemory = 0·80, ICCnaive = 0·81 and ICCTreg = 0·64), especially for the memory and naive cell populations, indicating that these are reasonably stable cell populations in the periphery with which to calculate proliferation and disappearance rates.
Granulocyte enrichment
Measures of turnover (f, k, p and d*) have been normalized by each individual's granulocyte enrichment to account for differences in deuterium availability across subjects. All subjects, regardless of the amount of deuterated water prescibed by protocol, reached either plasma enrichment levels > 1·0% or granulocyte enrichment levels > 2·5% which have been shown previously to be sufficient for lymphocyte DNA incorporation analyses 33, and there were no differences between T1D (n = 10) and healthy control (n = 10) subjects (two-way anova P = 0·98; data not shown).
Comparisons of T cell subset within each disease group (controls and T1D)
As illustrated in Fig. 2a, among the healthy control subjects (n = 10) the fractional enrichment with deuterium (f) was greater in Treg cells than CD4+ memory cells and both were much greater than the naive cell population, indicating an increased rate of incorporation of deuterium into cellular DNA, thus a more rapid turnover of this T cell compartment. Similar findings were seen among those with T1D (n = 10) (Fig. 2b).
Fig. 2.
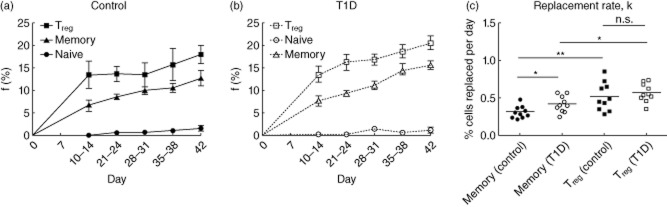
Fractional enrichment (f) and calculated replacement rate (k) in all subjects. (a,b) Fractional enrichment, f, of regulatory T cells (Treg) > memory>>naive in both control subjects (a: n = 10, P < 0·001) and type 1 diabetes (T1D) subjects (b: n = 10, P < 0·001). Group means over time compared using two-way analysis of variance (anova). (c) Mean replacement rate, k. Control subjects (n = 10) T memory cells k = 0·31%/day, T1D subjects (n = 10) memory cells k = 0·42%/day (*P < 0·05). Control subjects (n = 10) Treg cells k = 0·52%/day and T1D subjects (n = 10) Treg cells k = 0·57%/day (P = not significant (n.s.). Control subjects memory < Treg (P < 0·01); T1D subjects T memory < Treg (P < 0·05). Means compared using Mann–Whitney U-test. *P < 0·05; **P < 0·01.
Comparisons of T cell kinetics between healthy controls and those with T1D
Our primary goal was to determine if T cell kinetics were different between healthy controls and those with T1D.
Replacement rate
Significant differences were seen between T1D (n = 10) and control (n = 10) subjects with respect to the calculated replacement rate (k) for CD4+ memory cells, whereas no differences were found in (k) in the Treg subset (Fig. 2c). In control subjects (n = 10) CD4+ memory cell mean k = 0·31% per day, which corresponds to a doubling time (t2) of 222 days. In the T1D subjects (n = 10) CD4+ memory cell population k = 0·42% per day, or t2 = 164 days, which is significantly faster than controls (P = 0·03).
Proliferation and disappearance rates
Quantification of proliferation (p) and disappearance (d*) rates was possible only in subjects who had plasma and other samples obtained before and after the labelling period, as indicated in Table 1. Using the Asquith model described in the Appendix, these data demonstrate a higher (p) and (d*) in the CD4+ memory T cells from T1D (n = 6) compared with control (n = 4) subjects and no difference in (p) and (d*) in the Treg subset (Fig. 3a,b). The mean CD4+ memory T cell proliferation rate was p = 0·36% per day, or equivalently t2 = 195 days in controls versus 0·46% per day or t2 = 153 days in T1D (P = 0·02). The CD4+ memory cell disappearance rate was also greater in T1D subjects (d* = 1·48% per day in controls, which corresponds to t1/2 = 47 days versus 1·92% in T1D, with t1/2 = 37 days, P = 0·002), suggesting that CD4+ memory cells were leaving the memory pool more rapidly in T1D subjects compared to controls.
Fig. 3.
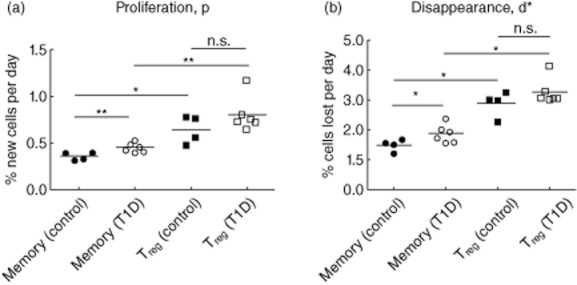
Measures of T cell turnover in control and type 1 diabetes (T1D) subjects who had post-labelling samples collected. (a) Average proliferation rate, p. Control subjects memory cells mean p = 0·36%/day, T1D memory cells p = 0·46%/day (**P < 0·01); control subjects regulatory T cells (Treg) cells mean p = 0·65%/day and T1D Treg cells mean p = 0·91%/day (P = n.s.). (b) Disappearance rate, d*. Control subjects T memory cells mean d* = 1·48%/day, T1D memory cells d* = 1·92%/day (*P < 0·05); control subjects Treg cells mean d* = 2·89%/day and T1D Treg cells d* = 3·56%/day (P = n.s.). Means compared using Mann–Whitney U-test. *P < 0·05; **P < 0·01.
Discussion
In vivo monitoring of the kinetics of immune cells is a potentially important tool for understanding the pathophysiology of autoimmunity and the immune response to various therapies. In vivo labelling of lymphocytes with stable isotopes in humans has been performed in the setting of ageing, infections and cancer but, to our knowledge, has not been performed in the setting of autoimmune disease. Here we have validated an in vivo protocol using deuterium oxide to measure the kinetics of T memory and Treg cell populations in T1D and healthy control subjects. We found that turnover of CD4+ memory cells was significantly faster in T1D subjects compared to control subjects using two different mathematical approaches. Our studies confirmed other reports, that turnover of Treg is greater than memory cells in healthy control subjects 23,39,40, and demonstrate that this is also true in individuals with T1D. Incorporation of deuterium into naive cells was extremely low, although within the detection limits of our methods, suggesting an extremely slow turnover rate.
The significance of increased proliferation and disappearance rates of CD4+ memory in T1D subjects is unknown. It is important to note that increased disappearance of CD4+ memory cells in T1D versus controls reflects the labelled cells leaving the sampling pool. This could be due to cell death, transition to other cell types or migration from peripheral blood into tissues. Using static measurements, we were unable to demonstrate differences in the frequency of proliferating (Ki67+, CD38+) or dying (annexin V+) memory cells between T1D and controls (Appendix Fig. A3). Previous work by Peakman, Roep and others has shown alterations in the frequency of CD4+ memory effector cell populations in peripheral blood of T1D. As CD4+ memory cell development is dependent upon T cell receptor (TCR) signal strength, antigen dose and the cytokine milieu 41–43, altered TCR signalling, persistent antigen and aberrant response to cytokines may all contribute to increased memory T cell turnover in T1D.
While it is possible that metabolic factors including peripheral hyperinsulinaemia and hyperglycaemia affect T cell kinetics, such an effect would be unlikely to explain our findings, which were limited to the CD4+ memory subset. There was also no correlation between baseline HbA1c and turnover measures of T cell subsets within the group of T1D subjects studied. However, additional studies with a larger number of T1D subjects with a wider range of glycaemic control and/or comparison studies with type 2 patients are needed to understand more clearly the impact of metabolic factors on immune cell kinetics. Our finding of increased T memory cell turnover in T1D subjects many years from diagnosis is consistent with the notion suggested previously by others, that there is an ongoing inflammatory state in individuals living with T1D 12,19,44.
In our study, although there was a trend towards a greater turnover of Treg in T1D compared to control subjects, we were unable to demonstrate significant differences between groups due potentially to the heterogeneity of our sorted Treg. Although additional staining of available Treg samples with FoxP3 yielded a reasonable purity of 83% (Appendix Fig. A1), Miyara 45, Booth 46 and others reported that within the Treg population, there are naive quiescent Treg (CD4+CD25lowCD45RA+) and rapidly dividing, activated Treg (CD4+CD25highCD45RO+), which we might expect to have differing kinetic profiles. As both cell types were considered together in our study, this may have limited our ability to detect differences in Treg subsets between diabetes and control subjects. Future studies addressing these specific populations as well as central memory and effector memory subtypes may be informative.
The estimated Treg and memory proliferation rates in our control subjects are lower than those published by Vukmanovic-Stejic et al. 23 and Macallan et al. 38. This is attributable both to differences between studies in how the cell populations were defined and the isotope labelling technique chosen. The previous studies included CD25+ cells within the memory pool and 127+ cells within the Treg pool, thus incorporating blasting population and activated memory cells with more rapid turnover. The slower turnover of memory cells in our study was due probably to limiting our memory population to CD25– cells which excludes a ‘blasting’ population. Similarly, by limiting our Treg pool to CD25+127low cells, we excluded activated memory cells which are known to proliferate and die quickly. Choice of isotope for DNA labelling and length of labelling protocol have been shown to impact estimated proliferation and death rates, with higher values being obtained with labelled glucose over labelled water and with shorter labelling times 37. Assuming a model of ‘kinetic heterogeneity’, as we have in our estimation of p and d* (i.e. p ≠ d*), a longer labelling period allows for a greater proportion of cells with a slow turnover to be labelled, thus disappearance rates of labelled cells are expected to be higher in labelling experiments of short duration such as those employed by Vukmanovic-Stejic and Macallan.
A major goal in conducting these studies was to validate the use of heavy isotope labelling in individuals with T1D. Although our sample size was small, we confirmed the stability of T cell pool fractions over time in both T1D and healthy controls. We also confirmed that sufficient enrichment can be achieved in T1D subjects receiving deuterated water to enable adequate labelling of cells of interest. We were able to demonstrate increased turnover in CD4+ memory cells using the calculated replacement rate (k) derived from the fractional enrichment (f), and were able to confirm these analyses using the Asquith model of proliferation (p) and disappearance (d*) rates which do not require assumptions about disappearance kinetics. Moreover, this technique was safe and well-tolerated. Application of heavy isotope labelling to understand T cell kinetics during the period prior to clinical disease or after immunotherapy is likely to enhance our understanding of T1D and the mechanisms involved in response to novel therapuetics.
Appendix
We modelled the measured up- and down-labelling of plasma 2H2O enrichment, P(t), as a marker of deuterium availability for incorporation, where:
after cessation of label intake (t > τ), where t represents time in days, δ represents the turnover rate of body water per day, π is the fraction of 2H2O in drinking water and τ represents length of labelling period in days. Baseline plasma 2H2O enrichment P(0) = β, that is attained after the boost of label by the end of day 0, determined the initial conditions.
Table A1.
Plasma parameter estimates with confidence intervals in subjects who had post-labelling samples collected
| δ | Π | β | |||||
|---|---|---|---|---|---|---|---|
| Subject | τ | Estimate | 95% CI | Estimate | 95% CI | Estimate | 95% CI |
| C7 | 63 | 0·0675 | 0·0296–0·1050 | 0·0156 | 0·0136–0·0176 | 0·0081 | 0·0013–0·0150 |
| C8 | 63 | 0·0547 | 0·0375–0·0712 | 0·0206 | 0·0186–0·0227 | 0·0055 | 0·0000–0·0112 |
| C9 | 63 | 0·0769 | 0·0416–0·1010 | 0·0142 | 0·0134–0·0158 | 0·0117 | 0·0070–0·0154 |
| C10 | 63 | 0·0768 | 0·0601–0·0937 | 0·0170 | 0·0164–0·0177 | 0·0110 | 0·0084–0·0137 |
| DM5 | 42 | 0·1980 | 0·0000–0·5203 | 0·0142 | 0·0126–0·0159 | 0·0078 | 0·0029–0·0126 |
| DM6 | 42 | 0·0971 | 0·0364–0·1580 | 0·0109 | 0·0099–0·0112 | 0·0071 | 0·0054–0·0087 |
| DM7 | 63 | 0·1047 | 0·0017–0·2078 | 0·0125 | 0·0114–0·0137 | 0·0133 | 0·0071–0·0195 |
| DM8 | 48 | 0·0682 | 0·0000–0·1610 | 0·0115 | 0·0091–0·0140 | 0·0098 | 0·0035–0·0162 |
| DM9 | 44 | 0·0344 | 0·0077–0·0611 | 0·0116 | 0·0061–0·0170 | 0·0124 | 0·0064–0·0185 |
| DM10 | 63 | 0·0866 | 0·0559–0·1170 | 0·0139 | 0·0131–0·0147 | 0·0063 | 0·0027–0·0100 |
Parameters estimated with 95% confidence intervals (CI) using fitted excess M+1 (EM1) data for plasma enrichment. δ: turnover rate of body water per day, π: fraction of 2H2O in drinking water, β: baseline body water enrichment attained after priming dose of label by the end of day 1.
To model the enrichment of the deoxyribose moiety of adenosine in the DNA of our subpopulations, we assume identical reaction kinetics of hydrogen and deuterium and of labelled and unlabelled adenosines. Because the adenosine deoxyribose (dR) moiety contains seven hydrogen atoms that may be replaced with deuterium, we expect an amplification factor, c > 1, in the enrichment of dR in DNA relative to the plasma enrichment. To estimate c more accurately we measured enrichment of dR in a cell population that is known to have a rapid turnover with no evidence of kinetically distinct subpopulations, i.e. granulocytes, such that p, the average production rate of the cell population is assumed to equal d, the disappearance rate of labelled cells. Amplification factor has been characterized previously 26 1 < c < 7 and is typically between 2·5–4·0 when plasma enrichment levels are 1·0–1·5%.
Label enrichment of adenosine in the DNA of a population of cells was modelled by:
where l is the total amount of labelled adenosine in DNA and A is the total amount of adenosine in the DNA of that population. Normalizing the equation by the total amount of adenosine in the DNA, L = l/A yields:
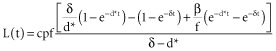 |
during the label intake (t ≤ τ), and
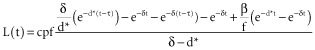 |
after labelling stopped (t > τ). In this model, the constancy of pool size has been assumed but no assumption of equality between p and d* has been made. Estimates for δ, π and β derived from plasma enrichment measures are shown in Appendix Table A1. Parameter estimates of d* and cp are shown with confidence intervals for granulocytes in Appendix Table A2and memory and regulatory T cells (Treg) in Appendix Table A3. The measured deuterium enrichment and fitted curves are graphed in Appendix Fig. A2.
Table A2.
Granulocyte parameter estimates for subjects with post-labelling samples
| cp | d* = p | ||||
|---|---|---|---|---|---|
| Subject | Estimate | 95% CI | Estimate | 95% CI | c |
| C7 | 0·3169 | 0·265–0·369 | 0·0910 | 0·074–0·108 | 3·4813 |
| C8 | 0·3810 | 0·297–0·465 | 0·1080 | 0·080–0·136 | 2·9227 |
| C9 | 0·2608 | 0·168–0·367 | 0·0746 | 0·044–0·112 | 3·4946 |
| C10 | 0·2112 | 0·160–0·262 | 0·0689 | 0·049–0·089 | 3·0653 |
| DM5 | 0·2659 | 0·209–0·032 | 0·0682 | 0·048–0·088 | 3·8971 |
| DM6 | 0·3596 | 0·203–0·517 | 0·1020 | 0·047–0·157 | 3·5255 |
| DM7 | 0·2838 | 0·196–0·371 | 0·0875 | 0·056–0·120 | 3·2438 |
| DM8 | 0·3012 | 0·176–0·426 | 0·0741 | 0·034–0·114 | 4·0670 |
| DM9 | 0·2399 | 0·119–0·361 | 0·0695 | 0·026–0·112 | 3·4538 |
| DM10 | 0·2882 | 0·206–0·370 | 0·0930 | 0·062–0·124 | 3·1006 |
Parameters estimated with 95% confidence intervals (CI) using fitted excess M+1 (EM1) data for granulocyte enrichment. d*: disappearance rate of labelled cells, c: amplification factor or likelihood of deuterium incorporation, p: average turnover rate or rate at which each adenosine residue replicates. cp and d* are estimated in the granulocyte model allowing for calculation of c for each subject. d* is assumed to equal p for granulocyte population known to have a very rapid turnover rate.
Table A3.
CD4+ T memory and regulatory T cells (Treg) parameter estimates for cp and d*
| CD4+ T memory | CD4+ Treg | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cp | d* | cp | d* | |||||||
| Subject | Estimate | 95% CI | p | Estimate | 95% CI | Estimate | 95% CI | p | Estimate | 95% CI |
| C7 | 0·0114 | 0·0079–0·0148 | 0·0033 | 0·0155 | 0·0077–0·0233 | 0·0268 | 0·0171–0·0364 | 0·0077 | 0·0326 | 0·0179–0·0473 |
| C8 | 0·0093 | 0·0075–0·0111 | 0·0026 | 0·0120 | 0·0075–0·0166 | 0·0164 | 0·0111–0·0217 | 0·0047 | 0·0301 | 0·0180–0·0422 |
| C9 | 0·0137 | 0·0093–0·0183 | 0·0039 | 0·0151 | 0·0070–0·0241 | 0·0274 | 0·0212–0·0339 | 0·0078 | 0·0299 | 0·0215–0·0397 |
| C10 | 0·0123 | 0·0114–0·0131 | 0·0040 | 0·0168 | 0·0148–0·0187 | 0·0147 | 0·0128–0·0165 | 0·0048 | 0·0228 | 0·0187–0·0267 |
| DM5 | 0·0189 | 0·0167–0·0210 | 0·0048 | 0·0194 | 0·0155–0·0234 | 0·0253 | 0·0197–0·0309 | 0·0065 | 0·0306 | 0·020–0·0412 |
| DM6 | 0·0161 | 0·0117–0·0205 | 0·0046 | 0·0158 | 0·0063–0·0253 | 0·0412 | 0·0314–0·0510 | 0·0117 | 0·0412 | 0·0272–0·0574 |
| DM7 | 0·0109 | 0·0115–0·0315 | 0·0034 | 0·0204 | 0·0098–0·0311 | 0·0189 | 0·0140–0·0275 | 0·0058 | 0·0305 | 0·0125–0·0486 |
| DM8 | 0·0215 | 0·0071–0·0148 | 0·0053 | 0·0237 | 0·0094–0·0379 | 0·0297 | 0·0166–0·0429 | 0·0073 | 0·0328 | 0·0143–0·0513 |
| DM9 | 0·0152 | 0·0072–0·0232 | 0·0044 | 0·0188 | 0·0019–0·0358 | 0·0495 | 0·0190–0·0800 | 0·0143 | 0·0484 | 0·0093–0·0874 |
| DM10 | 0·0128 | 0·0099–0·0156 | 0·0041 | 0·0173 | 0·0112–0·0233 | 0·0232 | 0·0145–0·0318 | 0·0075 | 0·0300 | 0·0158–0·0442 |
Parameters estimated with 95% confidence intervals (CI) using fitted excess M+1 (EM1) data for CD4+ memory and Treg enrichment. d*: disappearance rate of labelled cells, c: the amplification factor or likelihood of deuterium incorporation, p: average rate at which each adenosine residue replicates, are estimated together as ‘cp’ in our model. cp is divided by c from Appendix Table 2 to obtain p, which is plotted in Fig. 3 of the main text.
Figure A1.
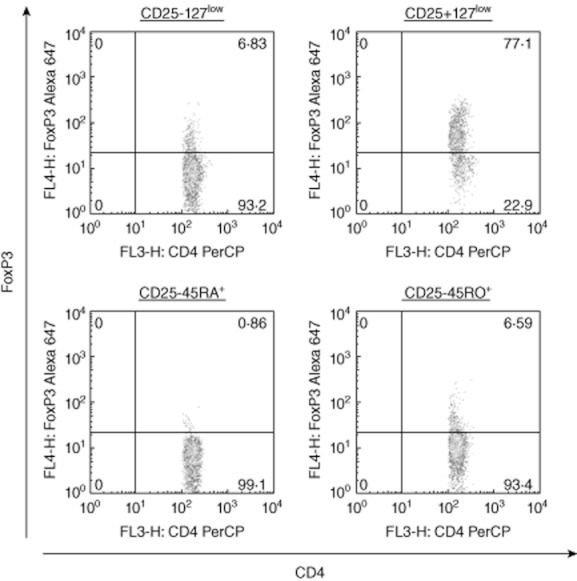
Representative fluorescence activated cell sorter (FACS) plot with forkhead box protein 3 (FoxP3) staining.
Figure A2.

Measured deuterium enrichment [excess M+1 (EM1)] with fitted curves for subjects from whom post-labelling samples were collected. To correct for the fraction of heavy water available in the body water, we fitted the measured label enrichment of plasma during up- and down-labelling, as described in the Methods. Measured enrichment (EM1) of plasma, granulocytes, T memory and regulatory T cells (Treg) is plotted along with fitted curves for each subject. Vertical line represents last day of oral 2H2O consumption.
Figure A3.
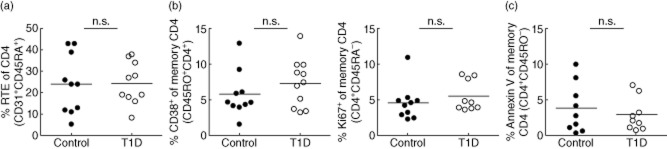
Static measures of CD4 T cell composition and death. Age- and gender-matched peripheral blood mononuclear cells (PBMC) from control (n = 10) and type 1 diabetes (T1D) (n = 10) subjects were thawed, stained and analysed by flow cytometry for expression of markers of maturation, activation and death including (a) CD31 and CD45RA for recent thymic emigrants, (b) CD38 and Ki67 for proliferation and (c) annexin V for apoptosis. Black circles: control subjects; white circles: T1D subjects. Statistical differences were determined using a Student's t-test.
Disclosure
This work was funded by the JDRF. The authors have no conflicts to report related to this work.
References
- 1.Makhlouf L, Grey ST, Dong V, et al. Depleting anti-CD4 monoclonal antibody cures new-onset diabetes, prevents recurrent autoimmune diabetes, and delays allograft rejection in nonobese diabetic mice. Transplantation. 2004;77:990–997. doi: 10.1097/01.tp.0000118410.61419.59. [DOI] [PubMed] [Google Scholar]
- 2.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segurado OG, Arnaiz-Villena A, Wank R, Schendel DJ. The multifactorial nature of MHC-linked susceptibility to insulin-dependent diabetes. Autoimmunity. 1993;15:85–89. doi: 10.3109/08916939309004844. [DOI] [PubMed] [Google Scholar]
- 4.Mikulkova Z, Praksova P, Stourac P, et al. Numerical defects in CD8+ Cell Immunol. 2010;262:75–79. doi: 10.1016/j.cellimm.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Petersen LD, Duinkerken G, Bruining GJ, van Lier RA, de Vries RR, Roep BO. Increased numbers of in vivo activated T cells in patients with recent onset insulin-dependent diabetes mellitus. J Autoimmun. 1996;9:731–737. doi: 10.1006/jaut.1996.0095. [DOI] [PubMed] [Google Scholar]
- 6.Hedman M, Faresjo M, Axelsson S, Ludvigsson J, Casas R. Impaired CD4 and CD8 T cell phenotype and reduced chemokine secretion in recent-onset type 1 diabetic children. Clin Exp Immunol. 2008;153:360–368. doi: 10.1111/j.1365-2249.2008.03720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peakman M, Mahalingam M, Leslie RD, Vergani D. Co-expression of CD45RA (naive) and CD45R0 (memory) T-cell markers. Lancet. 1994;343:424. doi: 10.1016/s0140-6736(94)91262-9. [DOI] [PubMed] [Google Scholar]
- 8.Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005;25(Suppl):56–62. doi: 10.1016/j.jaut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 9.Brusko T, Wasserfall C, McGrail K, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 2007;56:604–612. doi: 10.2337/db06-1248. [DOI] [PubMed] [Google Scholar]
- 10.Tree TI, Roep BO, Peakman M. A mini meta-analysis of studies on CD4+CD25+ T cells in human type 1 diabetes: report of the Immunology of Diabetes Society T Cell Workshop. Ann NY Acad Sci. 2006;1079:9–18. doi: 10.1196/annals.1375.002. [DOI] [PubMed] [Google Scholar]
- 11.Kukreja A, Cost G, Marker J, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. 2002;109:131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long SA, Cerosaletti K, Bollyky PL, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes. 2010;59:407–415. doi: 10.2337/db09-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcina A, Fedetz M, Ndagire D, et al. IL2RA/CD25 gene polymorphisms: uneven association with multiple sclerosis (MS) and type 1 diabetes (T1D) PloS ONE. 2009;4:e4137. doi: 10.1371/journal.pone.0004137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dendrou CA, Wicker LS. The IL-2/CD25 pathway determines susceptibility to T1D in humans and NOD mice. J Clin Immunol. 2008;28:685–696. doi: 10.1007/s10875-008-9237-9. [DOI] [PubMed] [Google Scholar]
- 15.Hakonarson H, Qu HQ, Bradfield JP, et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes. 2008;57:1143–1146. doi: 10.2337/db07-1305. [DOI] [PubMed] [Google Scholar]
- 16.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol. 2011;186:6329–6337. doi: 10.4049/jimmunol.1100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koenecke C, Czeloth N, Bubke A, et al. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol. 2009;39:3091–3096. doi: 10.1002/eji.200939432. [DOI] [PubMed] [Google Scholar]
- 19.Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–859. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz RH. Natural regulatory T cells and self-tolerance. Nat Immunol. 2005;6:327–330. doi: 10.1038/ni1184. [DOI] [PubMed] [Google Scholar]
- 22.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 23.Vukmanovic-Stejic M, Zhang Y, Cook JE, et al. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest. 2006;116:2423–2433. doi: 10.1172/JCI28941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellerstein M, Hanley MB, Cesar D, et al. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- 25.Hellerstein MK, Hoh RA, Hanley MB, et al. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J Clin Invest. 2003;112:956–966. doi: 10.1172/JCI17533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neese RA, Misell LM, Turner S, et al. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci USA. 2002;99:15345–15350. doi: 10.1073/pnas.232551499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohri H, Perelson AS, Tung K, et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J Exp Med. 2001;194:1277–1287. doi: 10.1084/jem.194.9.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Defoiche J, Debacq C, Asquith B, et al. Reduction of B cell turnover in chronic lymphocytic leukaemia. Br J Haematol. 2008;143:240–247. doi: 10.1111/j.1365-2141.2008.07348.x. [DOI] [PubMed] [Google Scholar]
- 29.Defoiche J, Zhang Y, Lagneaux L, Willems L, Macallan DC. In vivo ribosomal RNA turnover is down-regulated in leukaemic cells in chronic lymphocytic leukaemia. Br J Haematol. 2010;151:192–195. doi: 10.1111/j.1365-2141.2010.08334.x. [DOI] [PubMed] [Google Scholar]
- 30.Akbar AN, Vukmanovic-Stejic M, Taams LS, Macallan DC. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat Rev Immunol. 2007;7:231–237. doi: 10.1038/nri2037. [DOI] [PubMed] [Google Scholar]
- 31.Vukmanovic-Stejic M, Agius E, Booth N, et al. The kinetics of CD4+Foxp3+ T cell accumulation during a human cutaneous antigen-specific memory response in vivo. J Clin Invest. 2008;118:3639–3650. doi: 10.1172/JCI35834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asquith B, Debacq C, Macallan DC, Willems L, Bangham CR. Lymphocyte kinetics: the interpretation of labelling data. Trends Immunol. 2002;23:596–601. doi: 10.1016/s1471-4906(02)02337-2. [DOI] [PubMed] [Google Scholar]
- 33.Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc. 2007;2:3045–3057. doi: 10.1038/nprot.2007.420. [DOI] [PubMed] [Google Scholar]
- 34.Collins ML, Eng S, Hoh R, Hellerstein MK. Measurement of mitochondrial DNA synthesis in vivo using a stable isotope-mass spectrometric technique. J Appl Physiol. 2003;94:2203–2211. doi: 10.1152/japplphysiol.00691.2002. [DOI] [PubMed] [Google Scholar]
- 35.Macallan DC, Fullerton CA, Neese RA, Haddock K, Park SS, Hellerstein MK. Measurement of cell proliferation by labeling of DNA with stable isotope-labeled glucose: studies in vitro, in animals, and in humans. Proc Natl Acad Sci USA. 1998;95:708–713. doi: 10.1073/pnas.95.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vrisekoop N, den Braber I, de Boer AB, et al. Sparse production but preferential incorporation of recently produced naive T cells in the human peripheral pool. Proc Natl Acad Sci USA. 2008;105:6115–6120. doi: 10.1073/pnas.0709713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asquith B, Borghans JA, Ganusov VV, Macallan DC. Lymphocyte kinetics in health and disease. Trends Immunol. 2009;30:182–189. doi: 10.1016/j.it.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Macallan DC, Asquith B, Zhang Y, et al. Measurement of proliferation and disappearance of rapid turnover cell populations in human studies using deuterium-labeled glucose. Nat Protoc. 2009;4:1313–1327. doi: 10.1038/nprot.2009.117. [DOI] [PubMed] [Google Scholar]
- 39.Macallan DC, Wallace D, Zhang Y, et al. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J Exp Med. 2004;200:255–260. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace DL, Zhang Y, Ghattas H, et al. Direct measurement of T cell subset kinetics in vivo in elderly men and women. J Immunol. 2004;173:1787–1794. doi: 10.4049/jimmunol.173.3.1787. [DOI] [PubMed] [Google Scholar]
- 41.Park SO, Han YW, Aleyas AG, et al. Low-dose antigen-experienced CD4+ T cells display reduced clonal expansion but facilitate an effective memory pool in response to secondary exposure. Immunology. 2008;123:426–437. doi: 10.1111/j.1365-2567.2007.02707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swain SL, Bradley LM, Croft M, et al. Helper T-cell subsets: phenotype, function and the role of lymphokines in regulating their development. Immunol Rev. 1991;123:115–144. doi: 10.1111/j.1600-065x.1991.tb00608.x. [DOI] [PubMed] [Google Scholar]
- 44.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–7355. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 46.Booth NJ, McQuaid AJ, Sobande T, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol. 2010;184:4317–4326. doi: 10.4049/jimmunol.0903781. [DOI] [PubMed] [Google Scholar]