

Abstract
Type 1 diabetes (T1D) results from T helper type 1 (Th1)-mediated autoimmune destruction of insulin-producing β cells. Novel experimental therapies for T1D target immunomodulation, β cell survival and inflammation. We examined combination therapy with the dipeptidyl peptidase-IV inhibitor MK-626 and the histone deacetylase inhibitor vorinostat in the non-obese diabetic (NOD) mouse model of T1D. We hypothesized that combination therapy would ameliorate T1D by providing protection from β cell inflammatory destruction while simultaneously shifting the immune response towards immune-tolerizing regulatory T cells (Tregs). Although neither mono- nor combination therapies with MK-626 and vorinostat caused disease remission in diabetic NOD mice, the combination of MK-626 and vorinostat increased β cell area and reduced the mean insulitis score compared to diabetic control mice. In prediabetic NOD mice, MK-626 monotherapy resulted in improved glucose tolerance, a reduction in mean insulitis score and an increase in pancreatic lymph node Treg percentage, and combination therapy with MK-626 and vorinostat increased pancreatic lymph node Treg percentage. We conclude that neither single nor combination therapies using MK-626 and vorinostat induce diabetes remission in NOD mice, but combination therapy appears to have beneficial effects on β cell area, insulitis and Treg populations. Combinations of vorinostat and MK-626 may serve as beneficial adjunctive therapy in clinical trials for T1D prevention or remission.
Keywords: animal models (mice/rats), autoimmunity, insulitis, lymph nodes, non-obese diabetic (NOD) mouse, regulatory T cells, type 1 diabetes
Introduction
Type 1 diabetes mellitus (T1D) is a chronic autoimmune disease characterized largely by T cell-mediated destruction of insulin-producing pancreatic β cells, resulting in absolute insulin deficiency and hyperglycaemia. A phenotype similar to human T1D is seen in the non-obese diabetic (NOD) mouse, which offers a platform to study both diabetes pathogenesis and therapeutic intervention 1. Unfortunately, despite a number of successful single-agent drug studies in NOD mice that have led to prevention or reversal of diabetes 2–8, none of these drugs have translated into a clinical trial that has induced sustained remission of T1D in humans 9–13. Because of the complexity of diabetes pathogenesis, it is now thought that multi-drug approaches, using drugs that target different aspects of immune pathogenesis and/or β cell destruction, may be engaged for a synergistic benefit. Further, the use of drugs or drug classes with existing Food and Drug Administration (FDA) approval and proven safety in humans provides for the most rapid translation from bench to bedside. To this end, we examined combination therapy with the dipeptidyl-peptidase IV (DPP-IV) inhibitor MK-626 and the histone deacetylase (HDAC) inhibitor vorinostat in the NOD mouse. We employed two models: the first in which diabetic NOD mice were treated with combination therapy to determine effects on glycaemia and insulitis, and the second in which NOD mice were treated at the prediabetic stage to determine effects on incident glycaemic control and insulitis.
MK-626 is a novel inhibitor of DPP-IV 14, a ubiquitous serine-type protease that degrades incretin hormones (such as glucagon-like peptide 1: GLP-1). DPP-IV inhibitors and GLP-1 receptor agonists have proved efficacious and safe in adults with type 2 diabetes. In rodents, incretins have direct β cell effects, including enhancement of insulin synthesis and secretion, as well as enhancement of β cell proliferation and suppression of β cell apoptosis (reviewed in 15). In the NOD mouse, DPP-IV inhibitors have been shown to increase plasma GLP-1 levels 2,16. It is unclear, however, if the increase in GLP-1 is the main mechanism of action of DPP-IV inhibitors in T1D, because they also inhibit the DPP-IV-like cell surface peptidase CD26 17. CD26/DPP-IV inhibition results in suppression of cytotoxic T cells and augmentation of serum TGF-β1, a cytokine critical for the differentiation and expansion of immune-tolerogenic regulatory T cells (Tregs) 18–20.
Vorinostat (Zolinza®) is a class I/II HDAC inhibitor that is FDA-approved for the treatment of refractory cutaneous T cell lymphoma. The orally administered HDAC inhibitor drug class has sparked interest in the field of autoimmunity, owing to its anti-inflammatory and immunomodulatory properties 21,22. With respect to T1D, HDAC inhibitors offer protection from cytokine-induced β cell toxicity in vitro 22–25 and also enhance numbers of Tregs 26,27.
We hypothesized that combination therapy with MK-626 and vorinostat would ameliorate T1D by providing protection from cytokine-induced β cell destruction, while simultaneously shifting the immune response away from cytotoxic T cells and towards beneficial Tregs. These protective benefits could conceivably take effect in either established T1D or in the prediabetic phase of T1D. In this study, we show that combination therapy has promise in increasing endogenous Tregs, reducing insulitis and increasing the β cell area in the NOD mouse model.
Materials and methods
Materials
MK-626 and vorinostat were provided by Merck Research Laboratories (Rahway, NJ, USA) and were dissolved in 0·5% methylcellulose and 60% dimethylsulphoxide (DMSO) in water, respectively.
Animals and procedures
Animal studies were approved by the Indiana University Institutional Animal Care and Use Committees using American Association for Laboratory Animal Care (AALAC) guidelines. NOD/ShiLTJ (NOD) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) at 5 weeks of age and maintained in pathogen-free conditions. For diabetes treatment studies, mice were monitored by twice-weekly tail vein blood glucose levels starting at 10 weeks of age. Diabetes was classified as two consecutive glucose levels above 250 mg/dl. Once diabetes developed, mice were randomized into treatment groups and received treatment 7 days/week for 4 weeks. MK-626 was given at 3 mg/kg/day via oral gavage, as this dose was shown to maximize plasma DPP-IV inhibition and GLP-1 levels in pharmacodynamic studies in mice 14. Vorinostat was administered via intraperitoneal injection at 50 mg/kg/day, a standard dose used widely in the literature that has been determined to provide maximal inflammatory cytokine reduction 28. Control mice received daily intraperitoneal (i.p.) injections of 60% DMSO in water and/or daily oral gavage with 0·5% methylcellulose. To prevent ketosis, sustained-release bovine insulin pellets (LinShin, Ontario, Canada) were implanted subcutaneously as needed during the course of treatment. After 4 weeks of treatment, insulin pellets were removed surgically and mice were monitored for 2 days before final blood glucose measurement and euthanasia. Mice without diabetes by 20 weeks of age were considered as non-diabetic controls. Serum, pancreas, spleen and pancreatic lymph nodes were harvested for analyses. For studies of prediabetic NOD mice, mice were randomized into treatment groups at 6 weeks of age and received treatment 5 days/week for 4 consecutive weeks in the doses and manner described above, and control mice were given carrier by either i.p. injection or oral gavage. At 10 weeks of age, mice were fasted overnight and then subjected to an intraperitoneal glucose tolerance test, as described previously 29. At the time of euthanasia, serum, pancreas, spleen and pancreatic lymph nodes were harvested for analyses.
Insulin, proinsulin, transforming growth factor (TGF)-β1 and GLP-1 measurements
Serum was collected at the time of euthanasia by cardiac puncture. Serum insulin was measured using an Ultra Sensitive Mouse Insulin enzyme-linked immunosorbent assay (ELISA) kit (Crystal Chem, Downers Grove, IL, USA). Serum proinsulin was measured using a Proinsulin Rat/Mouse ELISA kit (Mercodia, Winston-Salem, NC, USA). Serum TGF-β1 was measured using a Mouse/Rat/Porcine/Canine TGF-β1 Quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA). For measurement of GLP-1 levels, serum was drawn 30 min following a oral glucose load of 3 mg/kg body weight, and tested using a multi-species total GLP-1 ELISA kit (Millipore Corporation, Billerica, MA, USA). All ELISA measurements were made according to the manufacturers' instructions.
Immunohistochemistry, β cell area calculation and insulitis scoring
Pancreas isolation, tissue embedding and immunostaining proceeded as described previously 30. Images were acquired on an Axio-Observer Z1 microscope (Zeiss, Thornwood, NY, USA) fitted with an AxioCam high resolution color camera. The β cell area (area of insulin positive staining/total pancreas surface area) was calculated using Zeiss Axiovision software (release 4·8.2). Insulitis was graded as follows using three pancreas sections from each of at least four animals per group: grade 1 = no islet-associated mononuclear cell infiltrates; grade 2 = peri-insulitis affecting less than 50% of the circumference of the islet without evidence of islet invasion; grade 3 = peri-insulitis affecting greater than 50% of the circumference of the islet without evidence of islet invasion; and grade 4 = islet invasion. Insulitis was graded by two observers, one of whom was blinded to sample identity.
Flow cytometric analysis
The spleen and pancreatic lymph nodes were harvested at the time of euthanasia. Spleens were lysed with red blood cell lysis buffer [155 mM NH4Cl, 12 mM NaHCO3, 0·1 mM ethylenediamine tetraacetic acid (EDTA)] before washing. Cells were then stained for Treg analysis using the following fluorochrome-coupled antibodies: anti-CD4-fluorescein isothiocyanate (FITC) (RM4-5; eBioscience, San Jose, CA, USA), anti-CD25-APC (PC61·5; eBioscience) and forkhead box protein 3 (FoxP3)-phycoerythrin (PE) (FJK-16s; eBioscience). Phenotypic analysis of cell populations was performed by multi-parameter flow cytometry. Fluorescence intensities were measured on a fluorescence activated cell sorter (FACSCalibur flow cytometer, BD Biosciences, San Jose, CA, USA) and data were analysed using FlowJo software (Tree Star, Ashland, OR, USA).
Statistics
For comparison between two groups, statistical significance was determined by a two-tailed t-test. For comparison between more than two groups, statistical significance between groups was determined by one-way analysis of variance (anova) with Dunnett's multiple comparison test referenced to the control group using Prism version 5·0 software (GraphPad, La Jolla, CA, USA). All data are shown as mean ± standard error of the mean (s.e.m.). P < 0·05 was considered statistically significant.
Results
Effect of combination therapy on glycaemia, pancreatic β cell area and insulitis in diabetic NOD mice
After development of spontaneous diabetes, female NOD mice were randomized into five treatment groups: (i) vehicle control (‘C’), (ii) MK-626 (‘M’), (iii) vorinostat (‘V’) and (iv) MK-626 and vorinostat (‘M/V’). In an initial cohort, 30 mice per group were followed, but all developed rapid and severe hyperglycaemia and illness, possibly masking potential beneficial effects of drug therapy (not shown). Thus, in a second cohort, insulin pellets were implanted subcutaneously to maintain a blood glucose of <350 mg/dl during a 4-week treatment period. As shown in Fig. 1a, after insulin pellets were removed at the end of the treatment period, the majority of animals in each of the treatment groups exhibited persistent and indistinguishable hyperglycaemia compared to 22-week-old non-diabetic female NOD mice, although it is notable that some animals in each treatment appeared to have near-normal glycaemia. Recognizing that the effects of combination therapy on β cell mass may not be reflected in absolute glucose levels, we next performed morphometric analysis of the β cell area (a parameter that is directly proportional to β cell mass) from fixed pancreatic sections. As shown in Fig. 1b, combination therapy with M/V resulted in significant increases in the β cell area compared to the control diabetic mice and mice receiving monotherapy with M or V. There was a small but significant reduction in the mean insulitis score in mice receiving M/V combination therapy compared to control mice and mice receiving M or V alone (Fig. 1c,d).
Fig. 1.
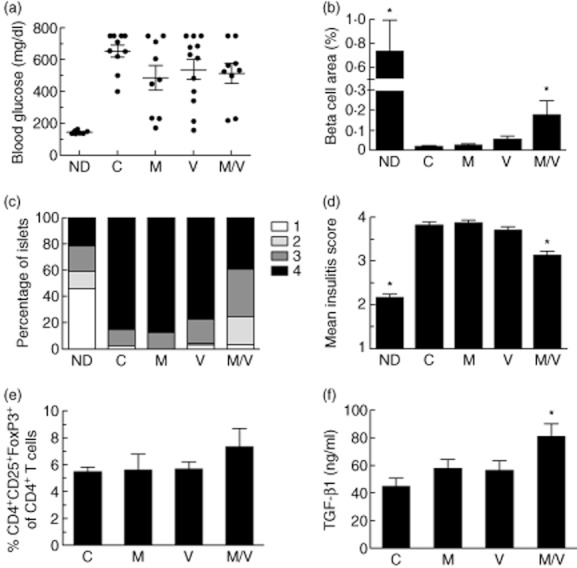
Effects of 4 weeks of single or combination therapy on non-obese diabetic (NOD) mice; 12–16-week-old diabetic female NOD mice were implanted with subcutaneous insulin-releasing pellets and randomized to drug treatments for 4 weeks (C = vehicle controls; M = MK-626; V = vorinostat; M/V = MK-626 and vorinostat) and compared to 22-week-old non-diabetic (ND) control NOD mice (n = 10 mice per group). (a) Random blood glucose levels at the end of the treatment period after removal of insulin pellets; (b) results of β cell area as a percentage of total pancreatic area in mice from each treatment group at the end of the treatment period (n = 4–9 mice per group); (c) insulitis scores at the end of the treatment period as a percentage of total islets for each treatment group (≥100 islets were scored from a total of four to nine mice per group); (d) mean insulitis score for each treatment group; (e) percentage of CD4+CD25+forkhead box protein 3 (Foxp3+) cells among total CD4+ lymphocytes in pancreatic lymph nodes for each treatment group at the end of the treatment period (n = 3–8 mice per group); (f) serum transforming growth factor (TGF)-β1 levels for each treatment group at the end of the treatment period (n = 4–6 per group). *P < 0·05 compared to vehicle controls in all panels.
Effect of combination therapy on splenic and pancreatic lymph node regulatory T cell populations in diabetic NOD mice
Recent data suggest that the preservation of β cell mass and function in T1D may be directly proportional to the relative frequency of Tregs in the CD4+ T cell pool 31. The percentage of CD4+CD25+FoxP3+ Tregs among CD4+ T cells in both the spleens and pancreatic lymph nodes of diabetic NOD mice after 4 weeks of treatment was determined by flow cytometry. Although Tregs in the pancreatic lymph nodes (as a percentage of total CD4+ cells) showed no change in the monotherapy groups versus control, we observed a tendency for increased Tregs in the M/V treatment group versus control (Fig. 1e). Because DPP-IV inhibition stimulates TGF-β1 secretion 18 and TGF-β1 promotes the expansion and maintenance of Tregs 2,26,27,32, we determined TGF-β1 levels in the sera of post-treatment mice as a measure of circulating Treg function. Importantly, we saw a statistically significant increase in TGF-β1 levels in the M/V treatment group versus the control group (Fig. 1f), suggesting that increased TGF-β1 may be contributing to a more favourable population of Tregs in the M/V group. Notably, at the dose of DPP-IV inhibitor used in these studies (3 mg/kg), NOD mice showed increased GLP-1 levels versus controls (4·0 ± 0·73 pM versus 2·3 ± 0·25 pM, P < 0·05).
Effect of combination therapy on glucose tolerance, pancreatic β cell area and insulitis in prediabetic NOD mice
In previous studies, we and others have shown that NOD mice exhibit increasing insulitis and progressively worsening glucose tolerance as a result of incident endoplasmic reticulum (ER) stress in β cells in the prediabetic phase between 6 and 10 weeks of age 29,33. To determine if treatment could alter glucose homeostasis and β cell function in the prediabetic phase, we treated 6-week-old prediabetic female NOD mice with a 4-week course of M, V and M/V and compared these to vehicle control (C). Female NOD mice housed in our vivarium do not develop spontaneous diabetes prior to 10 weeks of age 29, so mice were randomized to receive treatment from 6 to 10 weeks of age. As shown in Fig. 2a, control mice exhibited the expected worsening of glucose tolerance at 10 weeks of age compared to 6 weeks of age (but remained diabetes-free). Importantly, the M treatment group exhibited significantly improved glucose tolerance at 10 weeks of age, as assessed by area under the curve (AUC) analysis (Fig. 2b). However, neither the V nor the M/V treatments altered the glucose intolerance seen at 10 weeks of age. To test for incident ER stress in NOD β cells, we measured proinsulin and insulin levels in the sera of 10-week-old mice, as an increased proinsulin : insulin ratio is indicative of β cell stress 29,34. As shown in Fig. 2c–e, there were no statistical differences in insulin levels, proinsulin levels or the ratio of proinsulin : insulin in the sera at the end of treatment period among any of the groups, although all treatment groups showed a tendency to a reduced proinsulin level and a reduced proinsulin : insulin ratio.
Fig. 2.
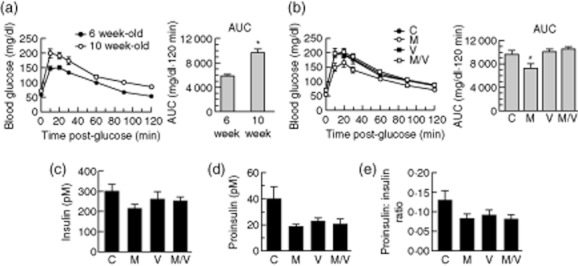
Glucose homeostasis and serum hormone levels after 4 weeks treatment of pre-non-obese diabetic (NOD) mice; 6-week-old prediabetic female NOD mice were randomized to drug treatments for 4 weeks (C = vehicle controls; M = MK-626; V = vorinostat; M/V = MK-626 and vorinostat). (a) Left panel shows results of glucose tolerance tests for 6-week-old mice before randomization (n = 40 mice, closed circles) and in 10-week-old vehicle controls (n = 10 mice, open circles) at the end of the 4-week study period, and the right panel shows area under the curve (AUC) analysis of the glucose tolerance data; (b) left panel shows results of glucose tolerance tests for 10-week-old mice at the end of the study period for each drug, and the right panel shows AUC analysis of the glucose tolerance data (n = 10 mice per group); (c) serum insulin levels in 10-week-old mice at the end of the study period (n = 6–10 mice per group); (d) serum proinsulin levels in 10-week-old mice at end of the study period (n = 6–10 mice per group); (e) serum proinsulin : insulin ratio in 10-week-old mice at the end of the study period (n = 6–10 mice per group). *P < 0·05 compared to either 6-week-old mice (a) or vehicle controls (c).
Pancreata from each group of treated prediabetic NOD mice were harvested and assessed by morphometry for insulitis severity and β cell area. As shown in Fig. 3, the M/V treatment group exhibited a statistically significant increase in β cell area at the end of the 4-week treatment period compared to controls. As Fig. 3b,c demonstrates, the mean insulitis score trended towards being lower in the M/V group and was significantly lower in the M monotherapy group compared to controls. Conversely, V monotherapy treatment resulted in a higher mean insulitis score compared to controls.
Fig. 3.
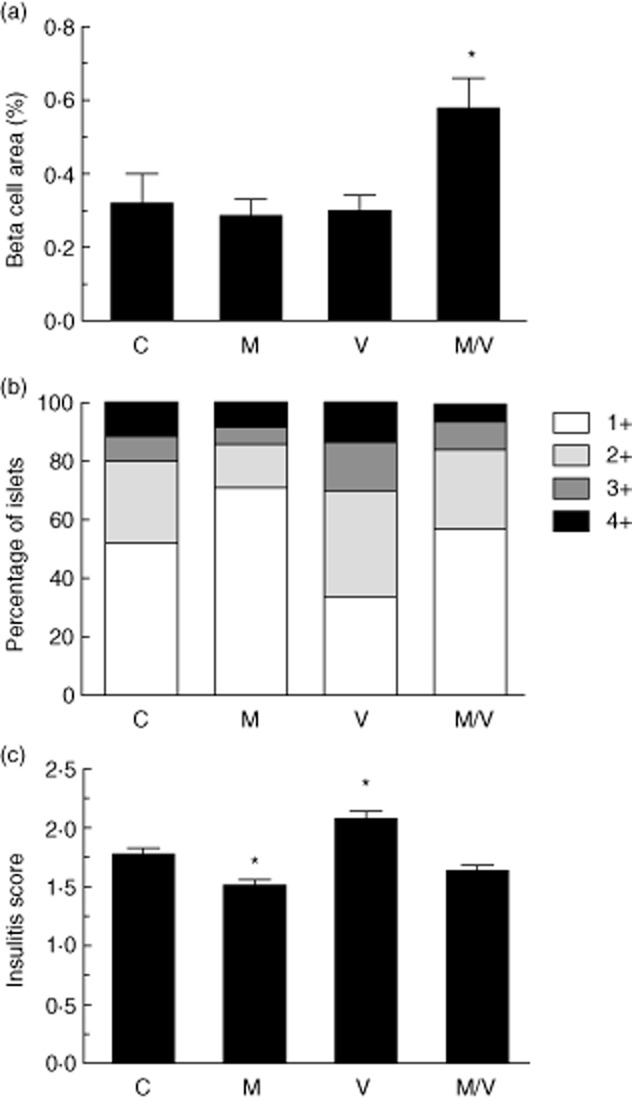
β cell area percentage and insulitis in treated pre-non-obese diabetic (NOD) mice. a, results of β cell area as a percentage of total pancreatic area in prediabetic mice from each treatment group at the end of the treatment period (n = 5 mice per group) (C = vehicle controls; M = MK-626; V = vorinostat; M/V = MK-626 and vorinostat); (b) insulitis scores at the end of the treatment period as a percentage of total islets for each treatment group (≥250 islets were scored from a total of five mice per group); (c) mean insulitis score for each treatment group. *P < 0·05 compared to vehicle controls.
Effect of combination therapy on splenic and pancreatic lymph node regulatory T cell populations in the prediabetic NOD mouse
Because both DPP-IV inhibitors and HDAC inhibitors have been shown to alter T cell differentiation 2,26,27, we sought to determine if early treatment would skew T cell differentiation beneficially towards Tregs, thereby potentially conferring protection from autoimmune diabetes. We measured the percentage of CD4+CD25+FoxP3+ Tregs of total CD4+ cells in the spleens and pancreatic lymph nodes of the prediabetic 10-week-old NOD mice after completing treatment. As shown in Fig. 4a, there was no difference in the mean percentage of Tregs in the spleen after 4 weeks of treatment in any of the groups, but significant increases in percentage of Tregs in pancreatic lymph nodes in the M and M/V groups (Fig. 4b). However, no differences in TGF-β1 levels were observed in the sera of animals from any of the treatment groups (Fig. 4c).
Fig. 4.
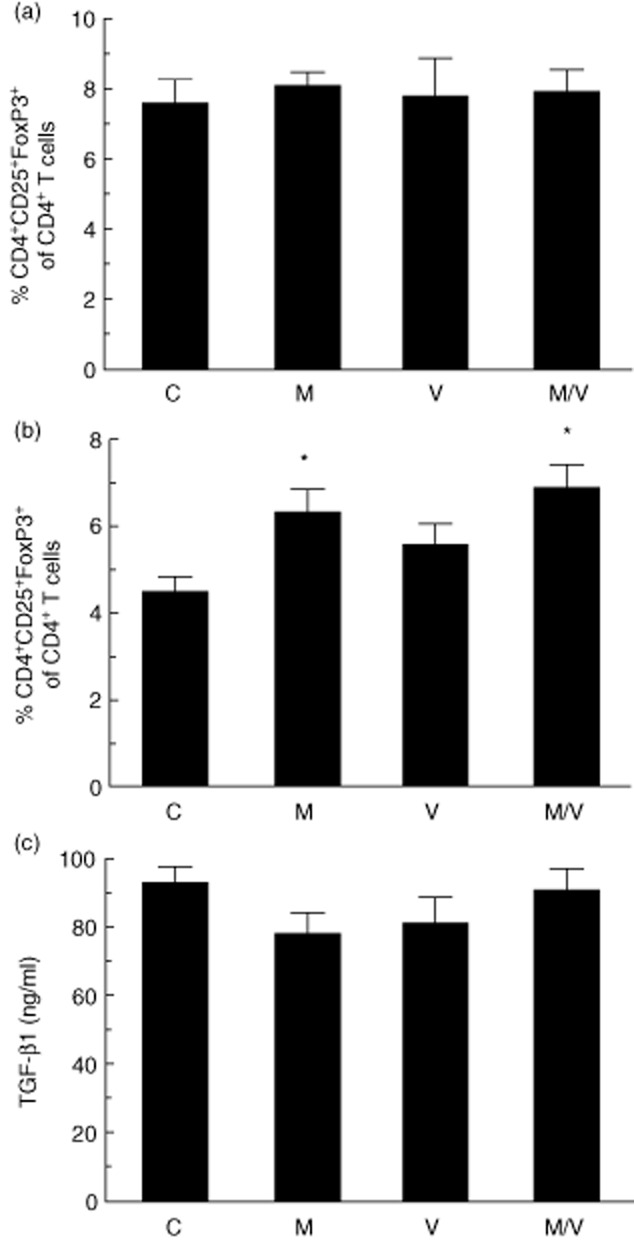
Regulatory T cell populations and transforming growth factor (TGF)-β1 levels in treated pre-non-obese diabetic (NOD) mice. (a) Percentage of CD4+CD25+forkhead box protein 3 (Foxp3+) cells among total CD4+ lymphocytes in splenocytes after 4 weeks' treatment of prediabetic NOD mice with indicated drugs (C = vehicle controls; M = MK-626; V = vorinostat; M/V = MK-626 and vorinostat); (b) percentage of CD4+CD25+FoxP3+ cells among total CD4+ lymphocytes in pancreatic lymph nodes after 4 weeks' treatment of prediabetic NOD mice with indicated drugs; (c) plasma TGF-β1 levels after 4 weeks' treatment of prediabetic NOD mice with indicated drugs. *P < 0·05 compared to vehicle controls.
Discussion
A hallmark feature of T1D is the immune infiltration of pancreatic islets, which leads to β cell dysfunction and eventual loss of β cell mass. The traditional concept, that T1D is a disorder of the immune system in which T helper type 1 (Th1) cells are the major contributor to disease, has been challenged in recent years. For example, from an immunological perspective, the concepts that β cells might contribute to T1D pathogenesis 35 and that immunosuppressive Tregs may mitigate disease severity 31 have both seen increasing attention. From an islet biology perspective, the concept of β cell susceptibility in T1D has been gaining notable traction: that is, there are intrinsic features (gene expression patterns, signalling pathways) that make the β cell more susceptible to death 36. In this context, we were interested in studying the potential for combination therapies that have purported β cell protective effects and Treg modulatory effects, with the potential for translation into human studies. We show here that the combination of the HDAC inhibitor vorinostat and the DPP-IV inhibitor MK-626 result in a statistically greater preservation of β cell area in diabetic NOD mice compared to either drug alone or to vehicle-treated controls. This relative preservation of β cell area correlated with a reduction in overall insulitis, and a trend towards an increase in the percentage of immune-suppressive Tregs in the pancreatic lymph node. It should be noted that these outcomes were seen in the context of exogenous insulin administration, and therefore we must consider the possibility that insulin itself might have had immunomodulatory effects that synergized with the treatments.
In 6-week-old prediabetic mice, MK-626 alone resulted in significant improvements in insulitis and glucose tolerance after 4 weeks of treatment, accompanied by an increase in pancreatic lymph node Tregs. The combination of MK-626 plus vorinostat increased the β cell area and also enhanced pancreatic lymph node Tregs in these prediabetic animals. Interestingly, the increased β cell area seen after combination treatment with MK-626 and vorinostat in prediabetic mice was not accompanied by an increase in serum insulin levels, suggesting that the β cell area and serum insulin may not always correlate directly. It remains possible, however, that differences in serum insulin were below the detection threshold of our ELISA.
Our results in NOD mice offer important insights in the context of the literature, and suggest possible avenues and expectations in future clinical trials. A recent study by Lundh et al. 24 suggested that class I HDAC inhibitors provide intrinsic β cell protection against cytokine-induced toxicity in vitro, possibly by reducing expression of inducible nitric oxide synthase. In our studies in diabetic NOD mice, however, vorinostat (a class I/II HDAC inhibitor, ref. 37) alone had no effect on β cell area or reversal of diabetes, suggesting that use of the drug at a late stage after hyperglycaemia is established provided little/no benefit. Even in prediabetic NOD mice, vorinostat resulted in no differences in glucose tolerance or insulitis, although our studies did not extend for sufficiently long enough to determine if use of vorinostat in the prediabetic phase offered protection from incipient diabetes. Our vorinostat findings parallel studies by Patel et al. 38, who observed that trichostatin A (a class I/II HDAC inhibitor) did not reverse or improve hyperglycaemia in diabetic NOD mice. Thus, the use of HDAC inhibitors alone may not represent a viable therapeutic approach in fully established T1D, but the potential beneficial effects of such inhibitors may become more obvious when combined with other therapeutic agents. In this respect, our studies suggest that use of vorinostat with the DPP-IV inhibitor MK-626 may have therapeutic value.
DPP-IV inhibitors improve glycaemic control in type 2 diabetes, presumably by increasing endogenous incretin levels 39. Recently, a study of sitagliptin in subjects with long-standing T1D also suggested potential glycaemic benefit, although it was unclear if that benefit resulted from effects on incretin levels or T cell populations 40. Preclinical studies also suggest the potential for beneficial effects of DPP-IV inhibitors in established T1D, an effect that is thought to be secondary to inhibition of a DPP-IV-like protease CD26 TGF-β1 2. Although remission of T1D was not observed in our studies, the use of MK-626 in combination with vorinostat increased serum TGF-β1, pancreatic lymph node Tregs and β cell area in diabetic NOD mice, and MK-626 alone improved glucose tolerance and insulitis in prediabetic mice.
Taken together, our results provide preclinical information that may be useful in potential future clinical trials. Based on clinical trials of multiple drugs and drug classes in T1D, it appears that single drug-based therapies are unlikely to have substantial effects on prevention or amelioration of hyperglycaemia. It is possible that the effects on Treg populations observed in our studies could have a more robust effect on glycaemia if treatment were continued for longer than the 4 weeks employed here. Nevertheless, a consensus is emerging from the clinical literature that, independently of effects on glycaemia, therapies that preserve β cell function and/or mass should have important beneficial effects, such as reductions in the rates of microvascular complications and hypoglycaemia 41,42. Future multiple drug regimen trials for T1D that include vorinostat and MK-626, along with other disease modifying agents that target T cell stimulation or co-stimulation, may provide therapeutic benefit by increasing Tregs and preserving β cell mass.
Acknowledgments
The authors wish to acknowledge Ms N. Stull for technical assistance in harvesting tissues. This work was supported by an investigator-initiated study from Merck & Co., Inc. (to R.G.M.) and by grants R01 DK083583 (to R.G.M.), T32 DK065549 (to S.M.C.) and F32 DK094489 (to S.C.C.) from the National Institutes of Health, and by an institutional Clinical and Translational Sciences Institute KL2 award (to S.A.T.).
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 2.Tian L, Gao J, Hao J, et al. Reversal of new-onset diabetes through modulating inflammation and stimulating beta-cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology. 2010;151:3049–3060. doi: 10.1210/en.2010-0068. [DOI] [PubMed] [Google Scholar]
- 3.Menard V, Jacobs H, Jun HS, Yoon JW, Kim SW. Anti-GAD monoclonal antibody delays the onset of diabetes mellitus in NOD mice. Pharm Res. 1999;16:1059–1066. doi: 10.1023/a:1018939900961. [DOI] [PubMed] [Google Scholar]
- 4.Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- 5.Yamada K, Nonaka K, Hanafusa T, Miyazaki A, Toyoshima H, Tarui S. Preventive and therapeutic effects of large-dose nicotinamide injections on diabetes associated with insulitis. An observation in nonobese diabetic (NOD) mice. Diabetes. 1982;31:749–753. doi: 10.2337/diab.31.9.749. [DOI] [PubMed] [Google Scholar]
- 6.Baeder WL, Sredy J, Sehgal SN, Chang JY, Adams LM. Rapamycin prevents the onset of insulin-dependent diabetes mellitus (IDDM) in NOD mice. Clin Exp Immunol. 1992;89:174–178. doi: 10.1111/j.1365-2249.1992.tb06928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkinson MA, Maclaren NK, Luchetta R. Insulitis and diabetes in NOD mice reduced by prophylactic insulin therapy. Diabetes. 1990;39:933–937. doi: 10.2337/diab.39.8.933. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZJ, Davidson L, Eisenbarth G, Weiner HL. Suppression of diabetes in nonobese diabetic mice by oral administration of porcine insulin. Proc Natl Acad Sci USA. 1991;88:10252–10256. doi: 10.1073/pnas.88.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gale EAM, Bingley PJ, Emmett CL, Collier T European Nicotinamide Diabetes Intervention Trial (ENDIT) Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- 10.Diabetes Prevention Trial – Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346:1685–1691. doi: 10.1056/NEJMoa012350. [DOI] [PubMed] [Google Scholar]
- 11.Sherry N, Hagopian W, Ludvigsson J, et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherrett DK, Bundy B, Becker DJ, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. 2011;378:319–327. doi: 10.1016/S0140-6736(11)60895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skyler JS, Krischer JP, Wolfsdorf J, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial – type 1. Diabetes Care. 2005;28:1068–1076. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 14.Edmondson SD, Mastracchio A, Mathvink RJ, et al. (2 S,3 S)-3-Amino-4-(3,3-difluoropyrrolidin-1-yl)- N,N-dimethyl-4-oxo-2-(4-[1,2,4]triazolo[1,5- a]- pyridin-6-ylphenyl)butanamide: a aelective α-amino amide dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2006;49:3614–3627. doi: 10.1021/jm060015t. [DOI] [PubMed] [Google Scholar]
- 15.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 16.Jelsing J, Vrang N, van Witteloostuijn SB, Mark M, Klein T. The DPP4 inhibitor linagliptin delays the onset of diabetes and preserves β-cell mass in non-obese diabetic mice. J Endocrinol. 2012;214:381–387. doi: 10.1530/JOE-11-0479. [DOI] [PubMed] [Google Scholar]
- 17.Morimoto C, Schlossman SF. The structure and function of CD26 in the T-cell immune response. Immunol Rev. 1998;161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 18.Reinhold D, Bank U, Bühling F, et al. Inhibitors of dipeptidyl peptidase IV (DP IV, CD26) induces secretion of transforming growth factor-beta 1 (TGF-beta 1) in stimulated mouse splenocytes and thymocytes. Immunol Lett. 1997;58:29–35. doi: 10.1016/s0165-2478(97)02716-8. [DOI] [PubMed] [Google Scholar]
- 19.Reinhold D, Bank U, Bühling F, et al. Inhibitors of dipeptidyl peptidase IV induce secretion of transforming growth factor-beta 1 in PWM-stimulated PBMC and T cells. Immunology. 1997;91:354–360. doi: 10.1046/j.1365-2567.1997.d01-2258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preller V, Gerber A, Wrenger S, et al. TGF-beta1-mediated control of central nervous system inflammation and autoimmunity through the inhibitory receptor CD26. J Immunol. 2007;178:4632–4640. doi: 10.4049/jimmunol.178.7.4632. [DOI] [PubMed] [Google Scholar]
- 21.Leoni F, Fossati G, Lewis EC, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis EC, Blaabjerg L, Storling J, et al. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet β cells in vivo and in vitro. Mol Med. 2011;17:369–377. doi: 10.2119/molmed.2010.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larsen L, Tonnesen M, Ronn SG, et al. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia. 2007;50:779–789. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 24.Lundh M, Christensen DP, Damgaard Nielsen M, et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia. 2012;55:2421–2431. doi: 10.1007/s00125-012-2615-0. [DOI] [PubMed] [Google Scholar]
- 25.Chou DH-C, Holson EB, Wagner FF, et al. Inhibition of histone deacetylase 3 protects Beta cells from cytokine-induced apoptosis. Chem Biol. 2012;19:669–673. doi: 10.1016/j.chembiol.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 27.Reilly CM, Thomas M, Gogal R, et al. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J Autoimmun. 2008;31:123–130. doi: 10.1016/j.jaut.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 28.Leoni F, Zaliani A, Bertolini G, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans-Molina C, Robbins RD, Kono T, et al. PPAR-{gamma} activation restores islet function in diabetic mice through reduction of ER stress and maintenance of euchromatin structure. Mol Cell Biol. 2009;29:2053–2067. doi: 10.1128/MCB.01179-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabrera SM, Rigby MR, Mirmira RG. Targeting regulatory T cells in the treatment of type 1 diabetes mellitus. Curr Mol Med. 2012;12:1261–1272. doi: 10.2174/156652412803833634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes A-MB, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 34.Robbins RD, Tersey SA, Ogihara T, et al. Inhibition of deoxyhypusine synthase enhances islet {beta} cell function and survival in the setting of endoplasmic reticulum stress and type 2 diabetes. J Biol Chem. 2010;285:39943–39952. doi: 10.1074/jbc.M110.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atkinson MA, Bluestone JA, Eisenbarth GS, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes. 2011;60:1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel T, Patel V, Singh R, Jayaraman S. Chromatin remodeling resets the immune system to protect against autoimmune diabetes in mice. Immunol Cell Biol. 2011;89:640–649. doi: 10.1038/icb.2010.144. [DOI] [PubMed] [Google Scholar]
- 39.Mest HJ, Mentlein R. Dipeptidyl peptidase inhibitors as new drugs for the treatment of type 2 diabetes. Diabetologia. 2005;48:616–620. doi: 10.1007/s00125-005-1707-5. [DOI] [PubMed] [Google Scholar]
- 40.Ellis SL, Moser EG, Snell-Bergeon JK, Rodionova AS, Hazenfield RM, Garg SK. Effect of sitagliptin on glucose control in adult patients with Type 1 diabetes: a pilot, double-blind, randomized, crossover trial. Diabet Med. 2011;28:1176–1181. doi: 10.1111/j.1464-5491.2011.03331.x. [DOI] [PubMed] [Google Scholar]
- 41.Ryan EA, Paty BW, Senior PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 42.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–836. doi: 10.2337/diacare.26.3.832. [DOI] [PubMed] [Google Scholar]