

Abstract
Glutamic acid decarboxylase (GAD)65 formulated with aluminium hydroxide (GAD-alum) was effective in preserving insulin secretion in a Phase II clinical trial in children and adolescents with recent-onset type 1 diabetes. In addition, GAD-alum treated patients increased CD4+CD25hi forkhead box protein 3+ (FoxP3+) cell numbers in response to in-vitro GAD65 stimulation. We have carried out a 4-year follow-up study of 59 of the original 70 patients to investigate long-term effects on the frequency and function of regulatory T cells after GAD-alum treatment. Peripheral blood mononuclear cells were stimulated in vitro with GAD65 for 7 days and expression of regulatory T cell markers was measured by flow cytometry. Regulatory T cells (CD4+CD25hiCD127lo) and effector T cells (CD4+CD25–CD127+) were further sorted, expanded and used in suppression assays to assess regulatory T cell function after GAD-alum treatment. GAD-alum-treated patients displayed higher frequencies of in-vitro GAD65-induced CD4+CD25+CD127+ as well as CD4+CD25hiCD127lo and CD4+FoxP3+ cells compared to placebo. Moreover, GAD65 stimulation induced a population of CD4hi cells consisting mainly of CD25+CD127+, which was specific of GAD-alum-treated patients (16 of 25 versus one of 25 in placebo). Assessment of suppressive function in expanded regulatory T cells revealed no difference between GAD-alum- and placebo-treated individuals. Regulatory T cell frequency did not correlate with C-peptide secretion throughout the study. In conclusion, GAD-alum treatment induced both GAD65-reactive CD25+CD127+ and CD25hiCD127lo cells, but no difference in regulatory T cell function 4 years after GAD-alum treatment.
Keywords: CD4 T cells (T helper, Th0, Th1, Th2, Th3, Th17), diabetes, immune regulation, regulatory T cells (Treg), therapy/immunotherapy
Introduction
Type 1 diabetes (T1D) is a consequence of an autoimmune reaction towards insulin-producing β cells of the pancreas. Immunomodulatory approaches to prevent or treat T1D have been developed and tested, with variable results 1–4. Autoantigens may be used to induce immunologic tolerance as an alternative to immunosuppression 5. Glutamic acid decarboxylase 65 (GAD65) is one of the main antigens to which patients with T1D mount a destructive immune response, and autoantibodies directed against GAD65 (GADA) and T cells specific for GAD65 epitopes are common in T1D patients 6–8. We have shown previously preservation of residual insulin secretion by GAD-alum treatment in a Phase II clinical trial in children with recent-onset T1D 3. In addition, trial participants treated with GAD-alum up-regulated CD4+CD25hiforkhead box P3+ (FoxP3+) cells in response to GAD65 stimulation in vitro and had a predominant T helper type 2 (Th2) immune response 9,10. Preservation of C-peptide secretion was still detectable after 4 years in patients with <6 months T1D duration at baseline in the same trial 11, and the residual C-peptide secretion was accompanied by sustained high levels of GADA, increased memory T cell frequencies and T cell activation upon in-vitro GAD65 stimulation 12. Recently, additional Phases II and III clinical trials of GAD-alum have been conducted both in Europe and the United States, neither finding an effect on preservation of insulin secretion 13,14. The present Phase II trial included patients with a T1D duration of <18 months, whereas the European Phase III trial included patients with a duration of <3 months, which may contribute to the discrepancy in outcome.
Self-tolerance is maintained physiologically by regulatory T cells (Treg) in the periphery 15, and defects in Treg function have been hypothesized to be involved in the pathogenesis of autoimmune disease 16. Because tolerance in the periphery is maintained by Tregs, induction of active tolerance has long been a proposed mechanism of action of antigen-based therapies such as GAD-alum treatment 17. Tregs typically express high levels of the interleukin (IL)-2 receptor α-chain CD25, the transcription factor FoxP3 and low levels of the IL-7 receptor CD127 18–22. However, both FoxP3 and CD25 can also be expressed by activated non-regulatory T cells. CD39 has also been suggested to be involved in Treg function through the removal of adenosine triphosphate (ATP) and has thus been used to identify subsets of Tregs 23. Tregs can suppress proliferation and cytokine secretion in a broad range of cell types, including CD4+ and CD8+ T cells, and their dysfunction leads to immunopathology 24. It has been reported recently that rather than there being a deficiency in Treg numbers, effector T cells (Teff) from patients with T1D are resistant to Treg-mediated suppression 25,26.
The aim of this work was to investigate whether an increase in cells with a Treg phenotype persisted at 4 years after GAD-alum treatment. In addition, we tested whether GAD-alum treatment affected the suppressive capacity of Tregs.
Materials and methods
Ethics statement
This study was approved by the Research Ethics Committee at the Faculty of Health Sciences, Linköping University, Sweden. Written informed consent was obtained from participating individuals, and for those aged <18 years also their parents, in accordance with the Declaration of Helsinki.
Population
The design and characteristics of the Phase II trial have been described elsewhere 3. Briefly, 70 T1D children between 10 and 18 years of age with fewer than 18 months of disease duration were recruited at eight Swedish paediatric centres. Participants had a fasting serum C-peptide level above 0·1 nmol/l and detectable GADA at inclusion. They were randomized to subcutaneous injections of 20 μg GAD-alum (n = 35) or placebo (n = 35) at day 0 and a booster injection 4 weeks later in a double-blind setting. After 4 years, patients and their parents were asked whether they were willing to participate in a follow-up study. Fifty-nine patients, of whom 29 had been treated with GAD-alum and 30 received placebo, agreed to participate.
Antibodies
Fluorescein isothiocyanate (FITC)-conjugated anti-CD39 (clone A1; Biolegend, San Diego, CA, USA), phycoerythrin (PE)-conjugated anti-FoxP3 (clone PCH101), allophycocyanin (APC)-conjugated anti-CD25 (clone BC96) and FITC- and PE-cyanine 7 (PE-Cy7)-conjugated anti-CD127 (clone eBioRDR5; eBioscience, San Diego, CA, USA), Alexa 700- and Pacific Blue-conjugated anti-CD4 (clone RPA-T4), APC-Cy7-conjugated anti-CD25 (clone M-A251; BD Pharmingen, Franklin Lakes, NJ, USA), and relevant isotype- and fluorochrome-matched control antibodies were used in this study. In addition, 7-amino-actinomycin D (7-AAD; BD Pharmingen) was used to measure cell viability.
Flow cytometry
Peripheral blood mononuclear cells (PBMC) from GAD-alum-treated (n = 24) and placebo-treated (n = 25) patients were isolated from whole blood by Ficoll-Paque (Pharmacia Biotech, Piscataway, NJ, USA) density gradient centrifugation within 24 h after drawing. Three of the GAD-alum-treated patients were classified as responders, 14 as intermediate responders and seven were non-responders, while the placebo group contained 10 intermediate responders and 14 non-responders (one patient unclassified due to missing serum sample). PBMC were incubated in AIM-V medium (Invitrogen, Carlsbad, CA, USA) with β-mercaptoethanol at 37°C, 5% CO2 for 7 days, with or without 5 μg/ml recombinant GAD65 (Diamyd Medical, Stockholm, Sweden). One million cells were washed in 2 ml phosphate-buffered saline (PBS) containing 0·1% bovine serum albumin (BSA; Sigma-Aldrich, St Louis, MO, USA) and subsequently stained with anti-CD4, CD39, CD127 and CD25 antibodies. Cells were then fixed and permeabilized using a FoxP3 staining kit (eBioscience), according to the manufacturer's instructions. After washing, cells were stained with PE anti-FoxP3, reconstituted in PBS, acquired on a fluorescence activated cell sorter (FACS) (BD FACSAria) and analysed using Kaluza software version 1·1 (Beckman Coulter, Indianapolis, IN, USA). The FoxP3+ gate was set using the negative population, as the negative population had a higher median fluorescence intensity (MFI) than the isotype control.
Cell sorting
Cells were sorted and expanded when sufficient cell numbers were available. Cryopreserved cells from GAD-alum- (n = 4) and placebo- (n = 3) treated patients were stained with Pacific Blue conjugated anti-CD4, FITC-conjugated anti-CD127 and APC-conjugated anti-CD25 and sorted into Treg and Teff subsets based on CD4+CD25hiCD127lo and CD4+CD25–CD127+ phenotype, respectively. After sorting, cells were pelleted by centrifugation at 400 g for 10 min, resuspended in AIM-V 10% human serum (HS) and allowed to rest for 2 h at 37°C, 5% CO2 before expansion was initiated. Aliquots of sorted cells were re-acquired to assess purity. The average Teff contaminant in sorted Tregs was 0·1%. PBMC from one single freshly drawn healthy donor were stained, sorted as above and stored frozen to serve as interassay control.
Teff and Treg expansion
Tregs were distributed at 4 × 104 cells per well in 125 μl AIM-V 10% HS into 96-well U-bottomed plates, and stimulated with anti-CD3/CD28 Dynabeads (Invitrogen) at a 1:1 bead-to-cell ratio. Teffs were plated at 5 × 105 cells per 500 μl medium, into 96-well flat-bottomed plates precoated overnight with 10 μg/ml anti-CD3 (OKT3; eBioscience) at 4°C. Cultures also contained 1 μg/ml soluble anti-CD28 antibody (CD28·2; eBioscience). Culture volume was doubled the following day, and 30 and 300 U/ml of recombinant human IL-2 (R&D Systems, Abingdon, UK) were added to Teff and Treg cultures, respectively. Tregs were washed and supplemented with fresh IL-2 every 2 days. Tregs and Teffs were restimulated as above on the ninth day of culture, and frozen down after 15 days of expansion. To verify post-expansion phenotype, cryopreserved Tregs and Teffs were cultured for 24 h in AIM-V 10% HS and 5 U/ml IL-2, and subsequently stained and acquired as described above.
Suppression assay
Treg-mediated suppression was assessed using expanded Tregs and Teffs from placebo- (n = 3) and GAD-alum- (n = 4) treated patients. Expanded Tregs and Teffs were thawed and incubated in AIM-V 10% HS at 37°C, 5% CO2 overnight, then resuspended at 0·5 × 105 cells/ml. Teffs were plated into 96-well U-bottomed plates at a density of 5 × 104 cells per well, while Tregs were plated into Teff-containing wells at Treg-to-Teff ratios of 1:1, 1:2, 1:4, 1:8 and 1:16. Treg/Teff cultures were stimulated with 5 μg/ml soluble anti-CD3 and 1 μg/ml soluble anti-CD28 antibodies. Unstimulated wells were included as negative controls, both from patients and interassay control healthy Teffs. IL-2 (1 U/ml) was added to all wells. Supernatants were collected after 3 days of culture and cells were incubated with 0·2 μCi [3H]-thymidine (PerkinElmer, Waltham, MA, USA) for 18 h before harvesting. Thymidine incorporation was measured using a 1450 Wallac MicroBeta counter (PerkinElmer).
C-peptide measurements
C-peptide levels were measured in serum samples with a time-resolved fluoroimmunoassay (AutoDELFIA™ C-peptide kit, Wallac; PerkinElmer), as described 3. Stimulated C-peptide was measured during a mixed meal tolerance test (MMTT) in GAD-alum- (n = 21) and placebo- (n = 10) treated patients who had a maximal C-peptide response of >0·20 nmol/l at the 30-month follow-up. Clinical effect of treatment was defined by changes in stimulated C-peptide measured as area under the curve (AUC) from baseline to 48 months.
Statistical analysis
Statistically significant differences were determined using the Mann–Whitney two-tailed U-test for unpaired observations, as data were determined to be significantly different from a Gaussian distribution. Wilcoxon's signed-rank test was used to compare paired samples. Linear regression was used to compare slope and Y-intercept of suppression curves, and correlations were determined with Spearman's rank correlation coefficient test. A probability level of <0·05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism software, version 5·04 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
GAD65 stimulation increases the proportion of both CD25hiCD127lo and CD25+CD127+/hi cells in GAD-alum-treated patients
We have demonstrated previously that in-vitro stimulation with GAD65 induced CD4+CD25hi FoxP3+ cells in PBMC from GAD-alum-treated patients 9. To determine whether this effect persisted 4 years after treatment, we analysed CD25hiCD127lo cells and used FoxP3 and CD39 as additional markers to discriminate Tregs from activated T cells more accurately. Thus, the expression of CD25, CD127, FoxP3 and CD39 on CD4+ lymphocytes was analysed in PBMC after 7 days of incubation with or without GAD65. Gates used for analysis and representative PBMC samples describing the expression of CD4, CD25 and CD127 are shown in Fig. 1a,b.
Fig. 1.
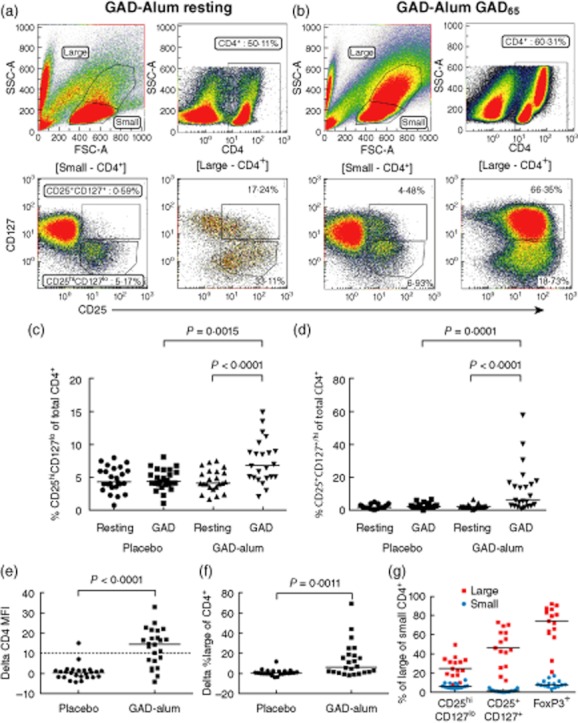
Gating strategy and glutamic acid decarboxylase (GAD65)-induced T cell populations. (a,b) Gating strategy and representative peripheral blood mononuclear cell (PBMC) samples from a GAD-alum-treated subject at resting conditions and after in-vitro GAD65 stimulation, respectively. Upper left panel describes the division of lymphocytes into small and large. Upper right panel describes CD4 expression. The lower panels describe the distribution of CD4+ cells between the CD25hiCD127lo and the CD25+CD127+ populations, gates indicated above plots. (c,d) Expression of CD25 and CD127 on CD4+ cells (placebo n = 25, GAD-alum n = 23). (e) Changes in CD4 median fluorescence intensity (MFI) and (f) frequency of forward-scatter (FSC)hiside-scatter (SSC)hi cells were calculated by subtracting MFI and frequencies at resting conditions from those obtained after GAD65 stimulation and are represented as delta values (placebo n = 25, GAD-alum n = 24). The cut-off for high CD4 expression was defined as an increase of 10 units of CD4 MFI in CD4+ cells following GAD65 stimulation and is indicated by the dashed line in (e). (g) Frequencies of CD25hiCD127lo, CD25+CD127+ and forkhead box protein 3+ (FoxP3+) cells among FSChiSSChi (red squares) and small (blue circles) CD4+ cells from GAD-alum-treated patients (n = 15) are shown as %. Each point represents an individual and median values are indicated with horizontal lines. Statistical significances are shown as P-values.
The frequency of CD25hiCD127lo cells in the CD4+ population was increased significantly upon GAD65 stimulation in GAD-alum-treated patients compared to unstimulated cells (7·4% and 4·5%, respectively), but not in the placebo group (Fig. 1c). The frequency of CD4+CD25+CD127+ cells was also increased following GAD65 stimulation in the GAD-alum-treated patients compared to resting cells (13·1% and 2·6%, respectively; Fig. 1d). In contrast, GAD65 stimulation did not induce expression of CD25hiCD127lo or CD25+CD127+ compared to resting cells in the placebo group (Fig. 1c,d). The frequencies of CD4+CD25hiCD127lo and CD4+CD25+CD127+ cells were also significantly higher in the GAD-alum-treated group compared to placebo individuals after stimulation with GAD65 (Fig. 1c,d).
GAD65 stimulation induces a population of activated T cells in GAD-alum-treated subjects
Stimulation with GAD65 in GAD-alum-treated patients induced a population of forward-scatter (FSC)hiside-scatter (SSC)hi cells, consisting mainly of CD4+ memory T cells, as we have reported previously 12. These FSChiSSChi cells are illustrated in Fig. 1a,b and are characterized by high CD4 expression (Fig. 1e). The FSChiSSChi population was observed in 16/24 GAD-alum patients and in one of 25 placebo individuals. In line with the GAD65 recall response induced in GAD-immunized individuals, GAD65 stimulation induced higher CD4 MFI (Fig. 1e) and higher percentages of FSChiSSChi cells (Fig. 1f) among CD4+ cells from GAD-alum patients compared to the placebo group. Next, we analysed the expression of Treg-associated markers among FSChiSSChi CD4+ cells from the GAD-alum group, and found that 25% were CD25hiCD127lo, 46·2% were CD25+CD127+/hi and 74% were FoxP3+ (Fig. 1g).
GAD65 stimulation induces cells with a Treg phenotype in GAD-alum-treated subjects
FoxP3 expression on CD4+ and CD4+FSChiSSChi cells was enhanced significantly by GAD65 stimulation in the GAD-alum group (Fig. 2a–c), while GAD65 stimulation did not induce any change compared to resting cells in the placebo group (Fig. 2c). To define further whether the increased CD25+CD127lo population in GAD65 stimulated PBMC from GAD-alum-treated patients corresponded to a Treg population, CD39 and FoxP3 were added as additional Treg markers. Indeed, CD4+CD25hiCD127lo FoxP3+CD39+ cells were also found to be increased selectively in these patients following in-vitro GAD65 stimulation (Fig. 2d). Thus, in-vitro GAD recall leads to expansion of both Tregs and activated CD25+CD127+ T effector cells, which is observed only in patients treated previously with GAD-alum. There were no significant differences in expression of any measured marker on resting cells between the two treatment arms (Figs 1 and 2).
Fig. 2.
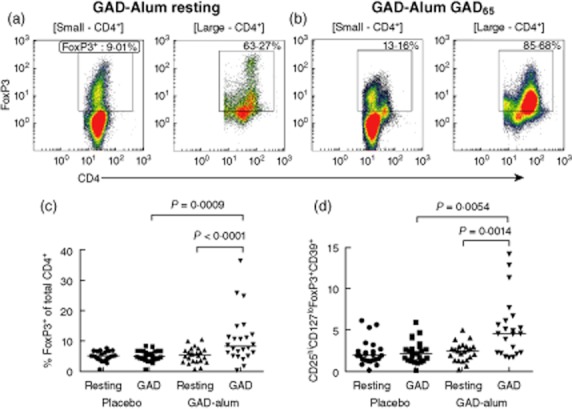
Expression of regulatory T cell (Treg)-associated markers. (a) Expression of forkhead box protein 3 (FoxP3) on CD4+ resting cells from a representative glutamic acid decarboxylase (GAD)-alum-treated subject; (b) expression of FoxP3 in GAD-stimulated cells, gates indicated above plots. (c) Expression of FoxP3 on CD4+ cells (placebo n = 25, GAD-alum n = 24); (d) the fraction of CD4+ cells co-expressing CD25hiCD127loFoxP3+CD39+ from placebo (n = 24) and GAD-alum (n = 22) patients. Each point represents an individual and median values are indicated with horizontal lines. Statistical significances are shown as P-values.
The post-expansion phenotype of Tregs and Teffs remains lineage-specific
Tregs (CD4+CD25hiCD127lo) from GAD-alum-treated patients expanded approximately 900-fold, to a similar extent as Tregs from placebo-treated patients (800-fold; Table 1). Teffs (CD4+CD25–CD127+) from both GAD-alum- and placebo-treated patients expanded approximately 100-fold. To verify the phenotype of sorted and expanded Tregs and Teffs after cryopreservation, we analysed the expression of Treg markers on thawed cells by flow cytometry. Tregs maintained predominant expression of CD25, FoxP3, cytotoxic T lymphocyte antigen-4 (CTLA-4) and low expression of CD127 and CD45RA, and roughly 50% were CD39+. Meanwhile, expression of FoxP3, CTLA-4, CD45RA and CD25 was scarce on Teff, but the majority expressed CD127 and expressed CD39 in the same proportion as Tregs. Measurement of cell viability by 7-AAD staining 24 h after thawing demonstrated that Teffs had a viability of 90%, whereas 70% of Tregs were viable (data not shown).
Table 1.
Characteristics of expanded cells after freeze/thaw.
| Treg | Teff | |
|---|---|---|
| CD25hiCD127lo | CD25−CD127+ | |
| In-vitro expansion | Fold increase (range) | Fold increase (range) |
| GAD-alum | 896 (706–1389) | 117 (75–291) |
| Placebo | 800 (667–1039) | 80 (45–180) |
| Protein expression phenotype | % (range) | % (range) |
|---|---|---|
| CD4+ out of total CD3+ | 98·8 (92·3–99·3) | 97·9 (67·6–99·3) |
| CD25hiCD127lo out of total CD4+ | 94·5 (88·8–98·5) | 5·3 (2·7–32·3) |
| FoxP3+ out of total CD4+ | 81·4 (51·7–89·7) | 9·6 (4·9–16·2) |
| CTLA-4+ out of total CD4+ | 74·7 (30·7–96·5) | 14·4 (3·8–26·4) |
Expression of regulatory T cell (Treg) markers was measured by flow cytometry 24 h after thawing. Data are pooled from both placebo and glutamic acid decarboxylase (GAD)-alum-treated type 1 diabetes (T1D) patients. CTLA-4: cytotoxic T lymphocyte antigen-4; FoxP3: forkhead box protein 3; Teff: effector T cells.
Expression of Treg markers is not related to clinical outcome
We tested whether the expression of any of the markers affected by GAD65 stimulation was related to clinical outcome of treatment. We found no significant correlation between expression of Treg markers used in this study and changes in stimulated C-peptide measured as ΔAUC or AUC 4 years after treatment. C-peptide secretion was not significantly different in patients where an FSChiSSChi population was induced by GAD65 stimulation compared to those who did not respond in this way (data not shown).
Suppression of polyclonally activated Teffs is not affected by GAD-alum treatment
To test whether the function of Tregs in T1D children included in the Phase II trial was affected by GAD-alum or placebo administration, suppression assays using sorted and expanded Tregs (CD4+CD25hiCD127lo) and Teffs (CD4+CD25–CD127+) were performed. Gates used to sort Tregs and Teffs are illustrated in Fig. 3a. Expanded Tregs from patients treated with GAD-alum suppressed proliferation of autologous Teffs to the same extent as Tregs from placebo patients (Fig. 3b). Tregs from both groups of patients displayed dose-dependent suppression of proliferation. As reported previously 25,27,28, we further found that suppression in autologous cultures of Tregs and Teffs was reduced in all patients (n = 7, placebo and GAD-alum combined) compared to a healthy control (seven repeated measurements, Fig. 3c, P < 0·0001). To determine whether this attenuated suppression was intrinsic to Tregs or Teffs, we tested the suppression of Tregs from T1D patients (either GAD-alum- or placebo-treated, with similar results; Fig. 3b,d), and from a healthy control in autologous and cross-over culture suppression assays. As shown in Fig. 3c, T1D Tregs exerted the same level of suppression on Teffs coming from either T1D or healthy subjects. In the reverse experiment, healthy Tregs were able to suppress Teffs from healthy or T1D subjects to a similar degree. Taken together, these results suggest that attenuated suppression from Tregs of T1D patients is due to reduced Treg efficacy rather than to increased Teff resistance to suppression.
Fig. 3.
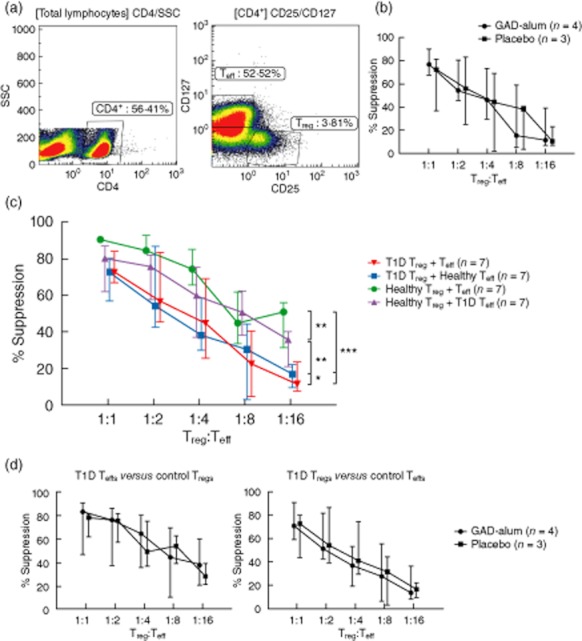
Dose-dependent regulatory T cell (Treg)-mediated suppression of effector T cells (Teff). Proliferation was assessed by [3H]-thymidine incorporation. Percentage of suppression of proliferation in co-cultures of Tregs and Teffs was calculated by dividing the mean counts per minute (cpm) of co-culture wells by the mean cpm obtained from Teffs cultured alone. (a) Left panel illustrates the gate discriminating CD4+ cells and the right panel shows the gates for sorting CD25hiCD127lo (Tregs) and CD25–CD127+/hi cells (Teffs). (b) Suppression exerted by Tregs from patients treated with placebo (n = 3, square) and glutamic acid decarboxylase (GAD)-alum (n = 4, circle) is shown as median for each group. (c) Suppression exerted by Tregs from type 1 diabetes (T1D) patients on autologous Teffs (red downward triangle; n = 7), by healthy Tregs on autologous Teffs (green circle), by T1D Tregs in co-culture with healthy Teffs (blue square) and by healthy Tregs in co-culture with T1D Teffs (purple upward triangle). The suppression is shown as percentage of decrease in Teff proliferation in the presence of Tregs compared to Teffs cultured in the absence of Tregs. Healthy represents repeated measurements (n = 7) of one healthy reference. (d) Left panel illustrates suppression exerted by healthy Tregs in cross-culture with patient Teffs and the right panel shows suppression exerted by patient Tregs in cross-culture with healthy Teffs. Statistical significances are shown as P-values: *P < 0·05; **P < 0·01; ***P < 0·001. Data points indicate medians, error bars designate interquartile range.
To determine whether there was a difference in reduced Treg-mediated suppression due to treatment, we tested if the suppression exerted in cross-over cultures of T1D Tregs versus healthy Teffs and healthy Tregs versus T1D Teffs was different between treatment arms. There was no difference in suppression exerted by Tregs from GAD-alum-treated patients compared to placebo Tregs in cross-over cultures with healthy Teffs, nor in the suppression exerted by healthy Tregs cultured with Teffs from GAD-alum-treated patients and Teffs from placebo subjects (Fig. 3d).
Discussion
Previous findings from the Phase II GAD-alum trial indicated an ability of GAD-alum to up-regulate CD25 and FoxP3 following in-vitro stimulation with GAD65, in addition to increased secretion of several pro- and anti-inflammatory cytokines from PBMC 9,10. Preservation of C-peptide secretion was still present in a 4-year follow-up to the Phase II trial 11, and induction of a T cell subset with memory phenotype was observed upon GAD65 stimulation 12.
Here we demonstrate that a great majority of lymphocytes in this T cell subset with memory phenotype expressed FoxP3 and high levels of CD25. Although some differences in the experimental setup were introduced in the present study, the main difference being that PBMC were cultured for 72 h at 21 and 30 months and for 7 days at the 4-year follow-up, the increased frequencies of CD25hi and FoxP3+ cells detected in this 4-year follow-up of the study are in agreement with our previous findings at 21 and 30 months after treatment 9. In the present study, the CD127 and CD39 markers were included to further define Tregs. Both CD4+CD25hiCD127lo and CD4+CD25+CD127+ cells were expanded by GAD65 stimulation, but a higher proportion of FSChiSSChi CD4+ cells were CD127+ than CD127lo/–, suggesting that the frequency of T cells with both Treg and activated-non-Treg phenotype increased following GAD65 stimulation.
Expression of CD39, an ectonucleotidase expressed on a subset of Tregs which hydrolyzes ATP into adenosine monophosphate (AMP) 23,29, was also increased upon antigen recall in GAD-alum-treated patients. It has been postulated that removal of proinflammatory ATP could be a suppressive mechanism mediated by CD39 on Tregs. In a recent study, CD39+ but not CD39–CD4+CD25hi cells were able to suppress IL-17 production 30. As the levels of IL-17 were undetectable in the supernatants of both expanded Teffs and Teff/Treg cultures, we cannot draw any conclusion on the ability of Tregs to suppress production of this cytokine in our settings. However, we have shown previously that secretion of IL-17, along with that of several other cytokines, was increased by GAD65 stimulation in PBMC supernatants 12. Although the current study does not include healthy subjects, the expression of CD39 on resting CD4+CD25hiCD127lo cells detected by us in these T1D patients seems to be lower than what has been reported in healthy individuals by others using the same anti-CD39 clone and fluorochrome 30.
In line with previous findings 31, expanded CD25+CD127lo Tregs were suppressive and retained their phenotype after expansion and cryopreservation. Although we were able to sort, expand and assess suppression in a limited number of individuals, there was no readily evident difference in the suppressive capacity of Tregs between placebo and GAD-alum-treated patients 4 years after administration of the treatment. Cross-over culture experiments revealed that Tregs isolated from patients with T1D participating in the GAD-alum trial had an impaired suppressive effect on autologous Teffs and also on Teffs from a healthy individual. In addition, Tregs from T1D patients were not capable of suppressing the proliferation of Teffs more readily from the healthy control than they did with their own Teffs. Further, Teffs from T1D patients were suppressed to a greater extent by Tregs from the healthy control than by their own Tregs. Taken together, these findings suggest that the reduced regulation observed in autologous co-cultures of cells isolated from T1D patients was due to reduced Treg-mediated suppression intrinsic to the Treg population. Our results are in contrast with previous findings, showing that responder T cells from T1D were more resistant to suppression 25,26. This could be explained by differences in the definition of cellular phenotypes and expansion conditions. While Schneider et al. used adaptive Tregs generated in vitro from CD4+CD25– cells 25, the Tregs used by us in this study were expanded from the CD4+CD25hiCD127lo population. In the study by Lawson et al., sorted CD4+CD25hi cells without in-vitro expansion from patients with long-standing T1D were used, and CD127 was not included to discriminate Tregs 26. Although we have identified a deficient Treg-mediated suppression of polyclonal T cell stimulation in T1D patients who participated in the GAD-alum Phase II trial, treatment with GAD-alum did not affect the suppressive activity of Tregs. It should be kept in mind that samples included in the current study were drawn 4 years after treatment, and that an effect on suppression shortly after treatment cannot be excluded. Furthermore, due to the random selection of patients based on the availability of samples, none of the GAD-alum-treated patients classified as responders to treatment were included in suppression assays 10, and we were thus unable to relate suppression to clinical outcome. Because our assay measures suppression of polyclonal activation, an effect on the specific suppression in response to GAD65 stimulation cannot be excluded. In fact, changes in the frequency of T cells with a Treg phenotype during the trial have been observed only upon GAD65 stimulation 9, while the frequency of Tregs after culture in medium alone has been similar in GAD-alum and placebo-treated patients throughout the study. Proliferative responses of PBMC from GAD-alum-treated patients in response to GAD65 stimulation were significantly stronger compared to placebo in a thymidine incorporation assay, as we have reported previously 12, suggesting that the GAD65-specific responses initiated by in-vitro antigen recall are not anergic.
In conclusion, we demonstrate GAD65 recall-induced populations of CD4+CD25hiCD127lo Tregs as well as FSChiSSChiCD4+CD25+CD127+ activated T cells, detectable 4 years after treatment. A deficiency in Treg-mediated suppression detected in T1D patients was intrinsic to the Treg population, but was not affected by GAD-alum treatment. Overall, these results suggest that higher numbers of Treg cells readily expandable upon GAD65 stimulation, rather than increased Treg function, may be responsible for the persistent clinical benefit of GAD-alum treatment after 4 years 11.
Acknowledgments
We thank Ingela Johansson, Gosia Smolinska-Konefal and Lena Berglert for skilful laboratory work. This project was supported by grants from the Swedish Research Council (K2008-55x-20652-01-3), the Swedish Child Diabetes Foundation (Barndiabetesfonden) and the Medical Research Council of Southeast Sweden. R.M. received support from JDRF (grant 1-2008-106), the Ile-de-France CODDIM and the Inserm Avenir Program. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Disclosure
The authors declare that they have no conflicts of interest.
References
- 1.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 2.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ludvigsson J, Faresjo M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–1920. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 4.Skyler JS. Update on worldwide efforts to prevent type 1 diabetes. Ann NY Acad Sci. 2008;1150:190–196. doi: 10.1196/annals.1447.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison LC. The prospect of vaccination to prevent type 1 diabetes. Hum Vaccin. 2005;1:143–150. doi: 10.4161/hv.1.4.1923. [DOI] [PubMed] [Google Scholar]
- 6.Atkinson MA, Kaufman DL, Campbell L, et al. Response of peripheral-blood mononuclear cells to glutamate decarboxylase in insulin-dependent diabetes. Lancet. 1992;339:458–459. doi: 10.1016/0140-6736(92)91061-c. [DOI] [PubMed] [Google Scholar]
- 7.Panina-Bordignon P, Lang R, van Endert PM, et al. Cytotoxic T cells specific for glutamic acid decarboxylase in autoimmune diabetes. J Exp Med. 1995;181:1923–1927. doi: 10.1084/jem.181.5.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallone R, Martinuzzi E, Blancou P, et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes. 2007;56:613–621. doi: 10.2337/db06-1419. [DOI] [PubMed] [Google Scholar]
- 9.Hjorth M, Axelsson S, Ryden A, et al. GAD-alum treatment induces GAD65-specific CD4+CD25highFOXP3+ cells in type 1 diabetic patients. Clin Immunol. 2011;138:117–126. doi: 10.1016/j.clim.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Axelsson S, Hjorth M, Akerman L, et al. Early induction of GAD(65)-reactive Th2 response in type 1 diabetic children treated with alum-formulated GAD(65) Diabetes Metab Res Rev. 2010;26:559–568. doi: 10.1002/dmrr.1126. [DOI] [PubMed] [Google Scholar]
- 11.Ludvigsson J, Hjorth M, Cheramy M, et al. Extended evaluation of the safety and efficacy of GAD treatment of children and adolescents with recent-onset type 1 diabetes: a randomised controlled trial. Diabetologia. 2011;54:634–640. doi: 10.1007/s00125-010-1988-1. [DOI] [PubMed] [Google Scholar]
- 12.Axelsson S, Cheramy M, Hjorth M, et al. Long-lasting immune responses 4 years after GAD-alum treatment in children with type 1 diabetes. PLoS ONE. 2011;6:e29008. doi: 10.1371/journal.pone.0029008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludvigsson J, Krisky D, Casas R, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. 2012;366:433–442. doi: 10.1056/NEJMoa1107096. [DOI] [PubMed] [Google Scholar]
- 14.Wherrett DK, Bundy B, Becker DJ, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet. 2011;378:319–327. doi: 10.1016/S0140-6736(11)60895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakaguchi S, Miyara M, Costantino CM, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 16.Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 17.Tian J, Kaufman DL. Antigen-based therapy for the treatment of type 1 diabetes. Diabetes. 2009;58:1939–1946. doi: 10.2337/db09-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baecher-Allan C, Brown JA, Freeman GJ, et al. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 19.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 20.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ Treg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 24.Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Schneider A, Rieck M, Sanda S, et al. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–7355. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawson JM, Tremble J, Dayan C, et al. Increased resistance to CD4+CD25hi regulatory T cell-mediated suppression in patients with type 1 diabetes. Clin Exp Immunol. 2008;154:353–359. doi: 10.1111/j.1365-2249.2008.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindley S, Dayan CM, Bishop A, et al. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–99. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- 28.Brusko TM, Wasserfall CH, Clare-Salzler MJ, et al. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes. 2005;54:1407–1414. doi: 10.2337/diabetes.54.5.1407. [DOI] [PubMed] [Google Scholar]
- 29.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fletcher JM, Lonergan R, Costelloe L, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183:7602–7610. doi: 10.4049/jimmunol.0901881. [DOI] [PubMed] [Google Scholar]
- 31.Putnam AL, Brusko TM, Lee MR, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes. 2009;58:652–662. doi: 10.2337/db08-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]