

Abstract
Recent basic and clinical studies have shown that the programmed death ligand (PD-L)/PD-1 pathway has a significant role in tumour immunity, and its blockade has a therapeutic potential against several human cancers. We hypothesized that anti-angiogeneic treatment might augment the efficacy of PD-1 blockade. To this end, we evaluated combining the blockade of PD-1 and vascular endothelial growth factor receptor 2 (VEGFR2) in a murine cancer model using Colon-26 adenocarcinoma. Interestingly, simultaneous treatment with anti-PD-1 and anti-VEGFR2 monoclonal antibodies (mAbs) inhibited tumour growth synergistically in vivo without overt toxicity. Blocking VEGFR2 inhibited tumour neovascularization significantly, as demonstrated by the reduced number of microvessels, while PD-1 blockade had no impact on tumour angiogenesis. PD-1 blockade might promote T cell infiltration into tumours and significantly enhanced local immune activation, as shown by the up-regulation of several proinflammatory cytokine expressions. Importantly, VEGFR2 blockade did not interfere with T cell infiltration and immunological activation induced by PD-1 blockade. In conclusion, simultaneous blockade of PD-1 and VEGFR2 induced a synergistic in-vivo anti-tumour effect, possibly through different mechanisms that might not be mutually exclusive. This unique therapeutic strategy may hold significant promise for future clinical application.
Keywords: anti-angiogenesis, anti-tumour immunity, immune checkpoint, PD-1, VEGFR2
Introduction
Blocking immune check-points can potentially activate and sustain T cell response against tumours 1. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) (CD152) is well known to serve as a dominant inhibitory receptor on T cells, and plays a key role in immune tolerance and homeostasis 2,3. CTLA-4 blockade has been long expected as a new cancer immunotherapy. A recent large-scale randomized clinical trial demonstrated usefully that immunotherapy using anti-human CTLA-4 monoclonal antibody had a significant anti-tumour response and improved overall survival in metastatic melanoma 4. This was the first therapy to extend overall survival in humans, and one of the most successful cancer immunotherapies. However, immune-related adverse events have been also reported, and some patients have died due to severe toxicities related to the study drugs. Therefore, another treatment targeting the T cell-negative regulatory pathway with less toxicity, as well as substantial efficacy for anti-cancer treatment, would be desirable.
Programmed death 1 (PD-1, CD279) is another potent immune check-point receptor 5–7. PD-1/programmed death ligand-1 (PD-L1) also functions as a negative regulator of T cell activation and contributes to the prevention of autoimmune diseases. A number of previous studies have shown that the PD-1/PD-L1 pathway has clinical importance in several human malignancies and its blockade has a significant anti-tumour effect in rodent models 8–10. Furthermore, recent Phase I clinical trials have shown that anti-human PD-1 or PD-L1 antibodies were tolerable for clinical use and might hold great promise as a new anti-cancer treatment for several advanced human malignancies 11,12. However, the effect of targeting PD-1/PD-L1 alone may be insufficient, especially for advanced or intractable malignant tumours that are resistant to conventional anti-cancer treatments, including chemotherapy and radiotherapy. Therefore, it is important to investigate the combination treatments for augmenting the potency of PD-1 blockade.
It is known that angiogenesis is a key feature in cancer development and metastasis 13,14. Among various regulators of angiogenesis, vascular endothelial growth factor (VEGF) and its receptor VEGFR receptor 2 (VEGFR2) are thought to be essential 15. Basic findings have shown that blocking of the VEGF/VEGFR pathway disrupts tumour microvessels and inhibits tumour growth. Furthermore, it has been also reported that VEGF/VEGFR blockade could normalize abnormal tumour vessels and increase tumour oxygenation, drug supply and immune cells 16–19. Indeed, anti-VEGF treatment is currently standard therapy for several human malignancies. However, it is also insufficient as a single treatment and usually administered with other cytotoxic anti-cancer drugs.
In this study, we hypothesized that anti-angiogenesis treatment may enhance the anti-tumour effect of targeting the PD-1 pathway without enhancing toxicity by efficiently inducing T cell infiltration into tumours. To this end, we employed an anti-VEGF receptor-2 (VEGFR2, CD309) monoclonal antibody (mAb), designated DC101. VEGFR2 is a major receptor for VEGF and plays a central role in tumour angiogenesis 20. Furthermore, this mAb has been proved to have a certain anti-tumour effect in murine models 21–24.
Materials and methods
Animal and cell line
Female BALB/c mice (5–6 weeks old) were obtained from Clea Japan (Tokyo, Japan). All mice were maintained under specific pathogen-free conditions in the animal facility at Nara Medical University. All experiments were conducted under a protocol approved by our institutional review board. A murine Colon-26 adenocarcinoma was obtained from Riken Cell Bank (Tsukuba, Japan). Cells were grown in RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum.
Antibodies
The anti-mouse PD-1 blocking mAb [RB65 mediating protein (RMP)1–14, rat immunoglobulin (Ig)G1] was generated as described previously 25. The anti-mouse VEGFR2 blocking mAb (DC101, rat IgG1) was kindly provided by ImClone Systems (New York, NY, USA). The anti-mouse CD34 mAb (MEC 14·7, rat IgG2a), anti-CD4 antibody (sc-7219, rabbit polyclonal) and anti-CD8 mAb (EP1150Y, rabbit IgG) were purchased from Abcam (Tokyo, Japan), Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), and Novus Biologicals (Littleton, CO, USA), respectively.
Animal experimental protocol
One million Colon-26 cells were inoculated subcutaneously in the flank of syngeneic BALB/c mice with 100 μl of cell suspension with an equal volume of Matrigel (BD Bioscience, San Jose, CA, USA). When the tumour reached 4–5 mm in diameter approximately 3 days after tumour inoculation, treatment was started. In the antibody treatment arm, mice were injected intraperitoneally (i.p.) with 0·25 mg of RMP1–14 and/or 0·8 mg of DC101 every other day for five times. Control mice received control rat IgG. The doses were determined on the basis of our preliminary experiments and previous studies 8,23,24. Tumour size was determined by electric caliper measurements. Some mice were killed and tumours were removed for further analysis at 11 days after tumour establishment.
Cell viability analysis
Cell viability was determined using one solution cell proliferation assay kit (MTS assay), according to the instruction manual (Promega Corporation, Madison, WI, USA). Briefly, aliquots of 3 × 103 cells per well were cultured in 96-well plates with control IgG, RMP1–14, DC101 or both of RMP1–14 and DC101 for 72 h. Antibody was used at a concentration of 1 or 10 μg/ml. Cell-titre 96 aqueous one solution was added to each well and incubated for an additional 1 h. The absorbance at 490 nm was recorded with a 96-well plate reader. Each experiment was performed in triplicate and repeated at least three times.
Immunohistochemistry and tumour vessel density measurement
Formalin-fixed or zinc-fixed, paraffin-embedded tissues of primary tumour were cut into 5-μm sections, deparaffinized and rehydrated in a graded series of ethanol. To block endogenous peroxidase, sections were immersed in 0·3% solution of hydrogen peroxide in absolute methanol for 5 min at room temperature and washed three times in fresh phosphate-buffered saline (PBS), each for 5 min duration. Purified rat anti-mouse CD34 mAb, rabbit anti-CD4 antibody or rabbit anti-CD8 mAb diluted with antibody diluent (Dako, Tokyo, Japan) was added and incubated for 1 h at room temperature or overnight at 4°C. Sections were washed three times in PBS, each for 5 min duration, and then Histofine Simple Stain Max PO (Nichirei, Tokyo, Japan) was added and incubated at room temperature for 30 min. After washing three times, the Histofine diaminobenzidine (DAB) substrate kit (Nichirei) was added and incubated at room temperature for 5 min. Sections were rinsed three times in distilled water, counterstained with haematoxylin, dehydrated in ethanol, cleared in Hemo-De and coverslipped. For tumour vessel density measurement, slides were scanned at low power fields (×40) to identify areas of highest vascularity. Twenty high-powered (×200) fields were then selected randomly within these areas, and tumour vessel densities were calculated based on the number of CD34-positive luminal structures. To rule out the possibility that the staining kit reacted with antibodies which had been used for the treatment in mice, we confirmed that there were no positive signals in the samples stained without primary anti-CD34 mAb.
Extraction of total RNAs and real-time reverse transcriptase polymerase chain reaction (PCR) analysis
Total RNA was isolated using RNAspin Mini (GE Healthcare, Tokyo, Japan) and the first-strand cDNA was synthesized from 1 μg RNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's protocol. Real-time quantitative PCR analysis was carried out using an ABI Prism 7700 sequence detector system (Applied Biosystems). All primer/probe sets were purchased from Applied Biosystems. PCR was carried out using the TaqMan Universal PCR Master Mix (Applied Biosystems) using 1 μl of cDNA in a 20 μl final reaction volume. The PCR thermal cycle conditions were as follows: initial step at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The expression level of the housekeeping gene β2-microglobulin was measured as an internal reference with a standard curve to determine the integrity of template RNA for all specimens. The ratio of the mRNA level of each gene was calculated as follows: (absolute copy number of each gene)/(absolute copy number of β2-microglobulin).
Statistical analysis
Results were expressed as mean values ± standard error, and Student's t-test or Welch's t-test were used for evaluating statistical significance. A value less than 0·05 was considered statistically significant.
Results
Simultaneous blockade of PD-1 and VEGFR2 induced synergistic anti-tumour effect
First, we investigated the efficacy of simultaneous blockade with both PD-1 and VEGFR2 in vivo using the murine colon cancer model. Tumour cells were inoculated subcutaneously with 1 × 106 in the right flank of BALB/c mice and treated with anti-PD-1 mAb (RMP1–14) and/or anti-murine VEGFR2 mAb (DC101). Control rat IgG was used as a control. In-vivo treatment either with anti-PD-1 mAb or anti-VEGFR2 mAb induced a substantial anti-tumour effect and inhibited tumour growth significantly compared to control (Fig. 1). There was no significant difference in tumour growth between PD-1 and VEGFR2 blockade. Furthermore, dual blockade of both PD-1 and VEGFR2 inhibited tumour growth significantly compared to each mAb treatment (Fig. 1). Thus, the combination therapy of anti-PD-1 and anti-VEGFR2 mAb showed a synergistic anti-tumour effect in tumour growth. There were no overt toxicities in treated mice.
Fig. 1.
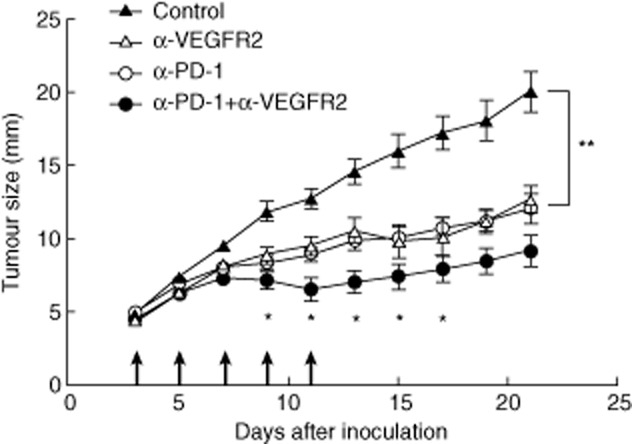
Simultaneous blockade of programmed death (PD)-1 and vascular endothelial growth factor receptor 2 (VEGFR2) induced synergistic anti-tumour effect in vivo. BALB/c mice were inoculated subcutaneously with Colon-26 cells and were given with control rat immunoglobulin (Ig)G, anti-PD-1 monoclonal antibody (mAb), anti-VEGFR2 mAb or both mAbs five times (arrow). Data are presented as mean ±standard error of seven to 10 mice of each group. *P < 0·05; **P < 0·01.
Effect of PD-1 and VEGFR2 blockade on cancer cells in vitro
To analyse the underlying mechanisms in tumour growth inhibition induced by PD-1 and VEGFR2 blockade, we evaluated the in-vitro effect of anti-PD-1 and anti-VEGFR2 mAb on Colon-26. A total of 3000 Colon-26 were co-cultured with anti-PD-1 mAb, anti-VEGFR2 mAb or both mAbs. Control rat IgG was used as a control. The survival rate of Colon-26 was determined by MTS assay. As a result, anti-PD-1 mAb and anti-VEGFR2 mAb did not affect cell survival. Thus, blockade of PD-1 and VEGFR2 does not have any direct effect on cancer cell growth (Fig. 2).
Fig. 2.

Programmed death (PD)-1and vascular endothelial growth factor receptor 2 (VEGFR2) blockade did not have any direct effect on cancer cell growth in vitro. A total of 3000 Colon-26 cells were co-cultured with anti-PD-1 monoclonal antibody (mAb), anti-VEGFR2 mAb or control rat immunoglobulin (Ig)G for 72 h, and cell proliferation was determined by MTS assay.
VEGFR2 blockade inhibited tumour neovascularization
We then analysed tumour neovascularization by immunohistochemistry with antibody against CD34 (Fig. 3a). Treatment with anti-VEGFR2 mAb or combination therapy inhibited the development of tumour microvessels significantly compared with control (Fig. 3b). Furthermore, anti-PD-1 mAb had no effect on tumour neovascularization (Fig. 3b). Thus, PD-1 blockade did not interfere with the anti-cancer treatment targeting tumour angiogenesis.
Fig. 3.

Treatment with anti-vascular endothelial growth factor receptor 2 (VEGFR2) monoclonal antibody (mAb) inhibited tumour neovascularization. (a) Immunohistochemistry analysis by staining with CD34. Representative tumours from mice treated with control rat IgG, anti-programmed death (PD)-1 mAb, anti-VEGFR2 mAb or both mAbs. (b) Tumour microvessels were counted at ×200 magnification. Data are collected from four to seven mice of each group. *P < 0·01; **P < 0·001.
PD-1 blockade enhanced T cell recruitment into tumours
We next evaluated tumour T cell infiltrations by immunohistochemistry and quantitative real-time PCR analysis. Although there were no significant statistical differences, there was a constant tendency of increase in CD4+ and CD8+ T cell infiltration in tumour tissues treated with anti-PD-1 mAb alone or combination of anti-PD-1 mAb and anti-VEGFR2 mAb compared to control or anti-VEGFR2 mAb alone (Fig. 4). Even though anti-VEGFR2 mAb disrupted tumour vessels, as shown above, T cell infiltration in tumours treated with anti-VEGFR2 mAb or combination did not decrease. Thus, VEGFR2 blockade did not abrogate recruitment of T lymphocytes into tumours induced by PD-1 blockade. In addition, we examined forkhead box protein 3 (FoxP3) expression in tumours as a marker for regulatory T cells. We found that FoxP3 expression was not reduced by anti-VEGFR2 treatment and elevated by anti-PD-1 treatment (data not shown).
Fig. 4.

(a) Immunohistochemical staining of CD4+ and (b) CD8+ T cells in tumour tissue at day 11. Representative pictures of mice for each treatment are shown. Programmed death (PD)-1 blockade and combination treatment seemed to induce more CD4+ and CD8+ T cell infiltration compared to control and vascular endothelial growth factor receptor 2 (VEGFR2) blockade. (c) Quantification of tumour-infiltrating CD4+ and (d) CD8+ T cells by real-time polymerase chain reaction (PCR). There was a tendency towards increased T cell infiltration by the treatment of anti-PD-1 monoclonal antibody (mAb) and combination treatment. Anti-VEGFR2 mAb treatment did not interfere with T cell infiltration. Data are collected from four to seven mice of each group.
PD-1 blockade activated local immunity
To analyse local immune status in tumours, we evaluated the several potent proinflammatory cytokines and mediators. Treatment with anti-PD-1 mAb or combination therapy induced a significant increase in the expression of interferon (IFN)-γ, tumour necrosis factor (TNF)-α and granzyme B in comparison with control (Fig. 5). Thus, PD-1 blockade enhanced T cell recruitment and activated local immune status, thereby resulting in tumour reduction. However, VEGFR2 blockade alone did not induce local immune activation in this model.
Fig. 5.
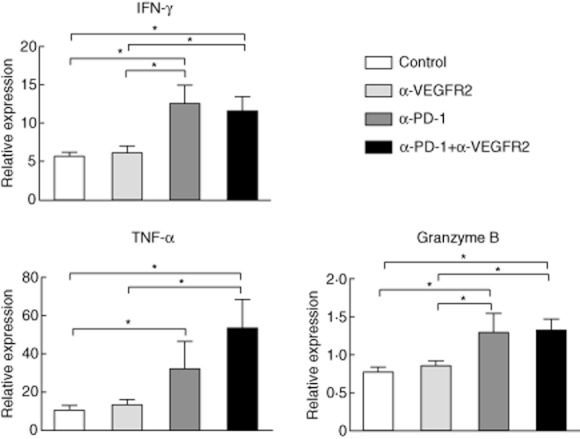
Expression of interferon (IFN)-γ, tumour necrosis factor (TNF)-α and granzyme B was up-regulated significantly by anti-programmed death (PD)-1 monoclonal antibody (mAb) or combination mAb treatment compared with control. Treatment of anti-VEGFR2 mAb alone did not increase each cytokine expression. Data are collected from four to seven mice of each group. *P < 0·05.
Discussion
This is the first study to investigate the synergistic anti-tumour effect induced by dual blockade of PD-1 and VEGFR2. Recent multi-centre Phase I clinical trials have shown that PD-1 or PD-L1 mAbs were safe in patients with various types of cancer, and hold promise as new anti-cancer agents. However, in order to enhance anti-tumour efficacy of strategies targeting the PD-1/PD-L pathway, combination therapy may be desirable, especially for refractory tumours such as pancreatic cancer. Because practical reagents targeting PD-1 and VEGF pathways are currently available, our proposed strategy may have actual clinical relevance. There are several advantages in dual blockade of completely different pathways. First, the combination therapy may reduce the harmful effect if they have different profiles of toxicity, as they can be used at reduced doses while preserving efficacy. This is important, because both PD-1 and VEGFR blockades are thought to cause unique adverse events. For instance, it is known that PD-1 blockade has a risk of inducing autoimmune reactions and diseases 10,11,26. The therapeutic dose of anti-PD-1 antibody may cause significant harmful effects. Its reduced, but optimal, dose can be achieved by combination therapy. Although we have observed no overt toxicity in mice during and after treatment, careful observations will be required in clinical applications. Secondly, when combining two reagents that have different properties, enhanced efficacy may be anticipated because of synergistic interactions. Our data indicate clearly that a synergistic in-vivo anti-tumour effect can be induced successfully by combining PD-1 and VEGFR2 blockade.
Although the underlying mechanisms are not elucidated fully, several interpretations may be drawn from our data. First, as expected, anti-VEGFR2 mAb treatment resulted in a significant decrease of tumour microvessels. Reducing tumour vasculature deprives the tumours of blood supply, thereby leading to the necrosis or apoptosis of tumour cells 19,21. This was not observed in tumours treated with anti-PD-1 mAb. Secondly, anti-PD-1 mAb treatment enhanced the infiltration of T cells into tumours. Furthermore, significant increases of several proinflammatory cytokines were also confirmed in tumours treated with anti-PD-1 mAb. Thus, PD-1 blockade induced T cell infiltrations, thereby resulting in local immune activation against tumours. Interestingly, although tumour vessels were reduced significantly by VEGFR2 blockade, tumour T cell infiltration was not interfered with the treatment. This paradoxical phenomenon may be explained by the normalization of tumour vessels induced by anti-angiogenesis treatment 16. The normalized tumour vessels restore blood flow and improve the ability to transport oxygen and anti-cancer drugs as well as immune cells to the tumour 27–30. Consistent with our data, previous studies investigating the combination of tumour immunotherapy with anti-angiogenic therapy have also shown that anti-angiogenesis treatment does not impede the infiltration of immune-competent cells into tumours 19,21,31. In addition, as regulatory T cells also express PD-1 selectively, it is possible that PD-1 blockade suppressed regulatory T cells and inhibited tumour growth 32. However, our data analysing FoxP3 expression suggested that regulatory T cells did not play a significant role in this model. Interestingly, there were no direct effects of PD-1 or VEGFR2 blockade on cancer cell growth, as demonstrated by in-vitro studies. Therefore, combining PD-1 and VEGFR2 blockades may exert their anti-tumour efficacy through controlling tumour microenvironments by activating tumour-infiltrating lymphocytes and inhibiting tumour neovascularization. Taken together, anti-angiogenesis strategy may be a good candidate for combination with immune check-point blockade in cancer therapy.
Immunotherapy has long been expected to become a powerful anti-cancer treatment that can be tumour-specific and less toxic 33. It includes cancer vaccine and adoptive cell therapy. To date, however, there are few definitive evidences for their efficacy in clinical cancers. Besides these conventional immunotherapies, monoclonal antibody-based treatments of targeting T cell negative regulatory pathways, CTLA-4 and PD-1, have been recently introduced and evaluated. A recent large-scale randomized clinical trial demonstrated that immunotherapy using anti-human CTLA-4 monoclonal antibody improved overall survival in metastatic melanoma 4. To our knowledge, this is the first strong evidence that immunotherapy has worked in actual human cancer. In general, there are many pathways and mechanisms involved in tumour development and progression. Thus, it may be difficult to induce a complete cure by monotherapy or a single anti-cancer method, especially for intractable tumours. Regarding future clinical applications, other combination therapies with blockade of immune check-points should be evaluated in order to achieve a synergistic anti-tumour effect and less systematic toxicity. In fact, several previous preclinical in-vivo studies have shown that the combination of blockade of PD-L1/PD-1 pathway with the simultaneous use of gemcitabine 8, anti-LAG-3 34 or anti-TIM3 mAb 35 exerted a significant anti-tumour efficacy without overt toxicity. Furthermore, other immune check-points, including B7-H3 36, LAG3 34 or TIM3 35, should also be evaluated in the combination of anti-angiogenesis treatment. In addition, VEGFR1 has become recognized to have unique and diverse activities, including cancer cell survival and migration 37. Therefore, a combination of PD-1 and VEGFR1 blockades warrants further investigation.
Clearly, further studies will be required to achieve definitive conclusions. First, long-term treatment of combination of PD-1 and VEGFR2 blockade needs to be assessed. In this study, tumour growth became noticeable after withdrawal of antibody treatment. It may be desirable that immunotherapy can induce tumour-specific memory cells that prevent tumour recurrence. Therefore, the sustained beneficial and adverse effects by long-term administration of both mAbs need to be evaluated. Secondly, more fundamental mechanistic studies should be also performed, as some of our data failed to demonstrate statistical significance.
In conclusion, we have shown for the first time that the combination of PD-1 and VEGFR2 had induced a synergistic in-vivo anti-tumour effect without overt toxicity. This unique strategy may have clinical relevance and should have the potential to be evaluated in future clinical trials.
Acknowledgments
This work was supported by the following grants: Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, no. 19591491, 21591648; Research Grant from the Pancreas Research Foundation of Japan; Research Grant from the Foundation for Promotion of Cancer Research in Japan; Research Grant from Daiwa Securities Health Foundation; Research Grant from The Japanese Society of Gastroenterology; and Research Grant from Nakayama Cancer Research Institute (M. Sho).
Disclosure
None.
References
- 1.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 3.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura H, Honjo T. PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001;22:265–268. doi: 10.1016/s1471-4906(01)01888-9. [DOI] [PubMed] [Google Scholar]
- 7.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nomi T, Sho M, Akahori T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 9.Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947–2953. doi: 10.1158/1078-0432.CCR-04-1469. [DOI] [PubMed] [Google Scholar]
- 10.Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 11.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 14.Folkman J. Angiogenesis inhibitors: a new class of drugs. Cancer Biol Ther. 2003;2:S127–133. [PubMed] [Google Scholar]
- 15.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 17.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 18.Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101:937–949. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- 19.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibuya M. Vascular endothelial growth factor receptor-2: its unique signaling and specific ligand, VEGF-E. Cancer Sci. 2003;94:751–756. doi: 10.1111/j.1349-7006.2003.tb01514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manning EA, Ullman JG, Leatherman JM, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–3959. doi: 10.1158/1078-0432.CCR-07-0374. [DOI] [PubMed] [Google Scholar]
- 22.Prewett M, Huber J, Li Y, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59:5209–5218. [PubMed] [Google Scholar]
- 23.Yoshiji H, Kuriyama S, Yoshii J, et al. Halting the interaction between vascular endothelial growth factor and its receptors attenuates liver carcinogenesis in mice. Hepatology. 2004;39:1517–1524. doi: 10.1002/hep.20218. [DOI] [PubMed] [Google Scholar]
- 24.Yoshiji H, Kuriyama S, Hicklin DJ, et al. The vascular endothelial growth factor receptor KDR/Flk-1 is a major regulator of malignant ascites formation in the mouse hepatocellular carcinoma model. Hepatology. 2001;33:841–847. doi: 10.1053/jhep.2001.23312. [DOI] [PubMed] [Google Scholar]
- 25.Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun. 2003;307:672–677. doi: 10.1016/s0006-291x(03)01257-9. [DOI] [PubMed] [Google Scholar]
- 26.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 27.Wei YQ, Huang MJ, Yang L, et al. Immunogene therapy of tumors with vaccine based on Xenopus homologous vascular endothelial growth factor as a model antigen. Proc Natl Acad Sci USA. 2001;98:11545–11550. doi: 10.1073/pnas.191112198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niethammer AG, Xiang R, Becker JC, et al. A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med. 2002;8:1369–1375. doi: 10.1038/nm1202-794. [DOI] [PubMed] [Google Scholar]
- 29.Luo Y, Zhou H, Krueger J, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dirkx AE, oude Egbrink MG, Castermans K, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte–endothelium interactions and infiltration in tumors. FASEB J. 2006;20:621–630. doi: 10.1096/fj.05-4493com. [DOI] [PubMed] [Google Scholar]
- 31.Huang X, Wong MK, Yi H, et al. Combined therapy of local and metastatic 4T1 breast tumor in mice using SU6668, an inhibitor of angiogenic receptor tyrosine kinases, and the immunostimulator B7.2-IgG fusion protein. Cancer Res. 2002;62:5727–5735. [PubMed] [Google Scholar]
- 32.Suzuki H, Onishi H, Wada J, et al. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol. 2010;40:197–203. doi: 10.1002/eji.200939887. [DOI] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–3551. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 36.Yamato I, Sho M, Nomi T, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–1716. doi: 10.1038/sj.bjc.6605375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz JD, Rowinsky EK, Youssoufian H, Pytowski B, Wu Y. Vascular endothelial growth factor receptor-1 in human cancer: concise review and rationale for development of IMC-18F1 (human antibody targeting vascular endothelial growth factor receptor-1) Cancer. 2010;116:1027–1032. doi: 10.1002/cncr.24789. [DOI] [PubMed] [Google Scholar]