

Abstract
Insulin plays important roles in apoptosis and lipid droplet (LD) formation, and it is one of the determinants involved in increasing fat mass. However, the mechanisms underlying insulin-induced enlargement of fat mass remain unclear. Our previous study suggested that insulin-induced increases in LDs are related to c-Jun N-terminal kinase (JNK)2-mediated upregulation of cell death-inducing DNA fragmentation factor-α-like effector (CIDE)C in human adipocytes. However, other genes involved in insulin/JNK2-induced LD formation are unknown. Here, we explored insulin/JNK2-regulated genes to clarify the mechanism of enlargement of LDs. Microarray analysis revealed that an insulin/JNK2 pathway mostly regulates expression of genes involved in lipid metabolism, including sterol regulatory element binding protein (SREBP)-1, a key transcription factor of lipogenesis. The JNK inhibitor SP600125 blocked insulin-induced upregulation of SREBP-1c expression. Small interfering RNA-mediated depletion of JNK2 suppressed insulin-induced nuclear accumulation of the active form of SREBP-1 protein and upregulation of SREBP-1c. Furthermore, depletion of JNK2 attenuated insulin-induced upregulation of SREBP-1c target lipogenic enzymes, leading to reduced de novo fatty acid synthesis. In addition, JNK2 coimmunoprecipitated with SREBP-1, reinforcing the correlation between JNK2 and SREBP-1. These results suggest that SREBP-1c is a novel insulin/JNK2-regulated gene and that the JNK2/SREBP-1c pathway mediates insulin-induced fatty acid synthesis, which may lead to enlargement of LDs in human adipocytes.
Keywords: c-Jun N-terminal kinase 2, cell death-inducing DNA fragmentation factor-α-like effector, microarray, sterol regulatory element-binding protein-1c, de novo lipogenesis, small interfering RNA
White adipose tissue (WAT) is a key organ for energy homeostasis. Excessive lipid accumulation of WAT in obesity contributes to chronic inflammatory diseases, including type 2 diabetes, hypertension, dyslipidemia, cardiovascular disease, nonalcoholic steatohepatitis, nephropathy, and several types of cancer (1–3). WAT mass is determined by the number and size of adipocytes (4, 5) regulated by cell differentiation, apoptosis, and lipid droplet (LD) formation (6–8). Insulin is known to inhibit apoptosis (9, 10) and increase LD formation (11, 12) in adipocytes. Hyperinsulinemia is associated with weight gain in humans (13). Insulin signaling in adipocytes is critical for the development of obesity (14, 15). Therefore, it has been suggested that insulin is one of the determinants involved in increasing the WAT mass.
Our previous study showed that insulin decreases cell death-inducing DNA fragmentation factor-α-like effector (CIDE)A expression and increases CIDEC expression, both of which belong to the CIDE family and play critical roles in apoptosis (16, 17) and LD formation (18–21), and the differential regulation of these genes is related, at least in part, to insulin-induced anti-apoptosis and LD formation in human adipocytes (22). Our recent study also showed that insulin regulates CIDEA and CIDEC expression via phosphatidylinositol 3-kinase (PI3K), and regulates expression of each protein via Akt1/2- and c-Jun N-terminal kinase (JNK)2-dependent pathways downstream of PI3K, respectively (23). These results suggest that insulin regulation of CIDEA and CIDEC contribute separately via different signaling pathways to insulin-induced anti-apoptosis and LD formation, leading to increases in the number and size of adipocytes. Adipocyte size is determined by LD formation regulated by glucose uptake, de novo fatty acid synthesis, triacylglycerol synthesis, and lipolysis (24, 25). Previous studies suggested that regulation of CIDEC expression contributes to LD maintenance by modulating lipolysis. However, regulation of other genes involved in LD formation by an insulin/JNK2 pathway remains unknown. Enlargement of adipocyte size (so-called adipocyte hypertrophy) is thought to contribute markedly to adipose tissue inflammation and its associated metabolic complications (26–28). Therefore, exploring the mechanisms of insulin/PI3K/JNK2-regulated LD formation would provide novel therapeutic targets for the treatment of chronic inflammatory diseases.
The JNK subgroup of mitogen-activated protein kinases is encoded by three genes: JNK1 and JNK2 are expressed ubiquitously, and play key roles in the development of obesity and insulin resistance (29, 30), while JNK3 is expressed mainly in the brain, heart, and testis (31), and is an important component in the pathogenesis of neurotoxicity (32). Small interfering RNA (siRNA) screening revealed that JNK2 is a regulator of LD homeostasis in HeLa cells (33). Furthermore, we recently showed that JNK2 is required for insulin-induced LD formation in human adipocytes (23). These observations implied a role of JNK2 in lipid-related pathologies such as obesity and insulin resistance. Therefore, inhibition of JNK2 activity may be useful in the treatment of obesity and its associated disorders.
Sterol regulatory element binding protein-1c (SREBP)-1c is a key lipogenic transcription factor and is known to mediate lipogenic actions of insulin in the liver (34). SREBP-1c preferentially regulates the lipogenesis by activating expression of genes, such as ATP citrate lyase (ACLY), acetyl-CoA carboxylase 1 (ACC1), and fatty acid synthase (FAS). SREBP-1c is synthesized as a precursor protein and the precursor form of SREBP-1c (pre-SREBP-1c) is translocated into the nucleus by sequential proteolytic processing (35). The active nuclear form of SREBP-1c (n-SREBP-1c) modulates transcription of its own and target genes, thereby promoting the lipogenic process in the liver. The dysregulation of SREBP-1c has been implicated in the pathogenesis of dyslipidemia of obesity, insulin resistance, hepatic steatosis, and diabetic nephropathy (35–38). Despite the established role of SREBP-1c in the liver, the influence of SREBP-1c on genes involved in lipid metabolism in adipocytes has not been fully defined. Moreover, there have been few studies in tissues or cells of human origin. Hyperinsulinemia increases SREBP-1c gene expression in human skeletal muscle and adipose tissue (39). Insulin positively modulates expression and proteolytic processing of SREBP-1 in isolated human adipocytes (40). At present, the mechanism by which insulin induces the expression of SREBP-1c and the contribution of SREBP-1c to the lipogenic action of insulin in adipocytes remain unclear.
In this study, we analyzed insulin/JNK2-regulated genes to clarify the mechanism of LD formation in human adipocytes. Our data show that SREBP-1c is a major downstream protein regulated by an insulin/JNK2-dependent pathway and the JNK2/SREBP-1c pathway mediates insulin-induced fatty acid synthesis, which may lead to LD formation in human adipocytes.
MATERIALS AND METHODS
Materials
DMEM/F-12 (1:1, v/v) was purchased from Invitrogen (Carlsbad, CA). Human insulin was purchased from Novo Nordisk (Bagsværd, Denmark), and rosiglitazone was purchased from Alexis Biochemicals (San Diego, CA). 3-Isobutyl-1-methylxanthine, dexamethasone (Dex), pantothenate, and β-actin were purchased from Sigma (St. Louis, MO). Biotin was purchased from Wako Pure Chemical Industries (Osaka, Japan), and FBS was purchased from Biological Industries (Kibbutz Beit Haemek, Israel). SP600125 was purchased from Calbiochem (San Diego, CA). Anti-SREBP-1 antibody (H-160, sc-8984) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-JNK2 antibody was purchased from Cell Signaling Technology (Beverly, MA).
Differentiation of human preadipocytes into adipocytes
Human preadipocytes, derived from subcutaneous adipose tissue of six male subjects (used in Fig. 2) or six female subjects (used in Figs. 1, 3–6), were obtained from Zen-Bio (Research Triangle Park, NC). Institutional approval was obtained for the study and all participants gave their informed consent. The six male subjects were nonsmokers with a mean body mass index of 27.2 (range 26.4–28.4) and an average age of 41 years (range 29–57 years). The six female subjects were nonsmokers with a mean body mass index of 27.9 (range 25.7–29.9) and an average age of 40 years (range 29–52 years). Human preadipocytes were differentiated into adipocytes as described previously (22). Human preadipocytes were seeded on 24-well plates and cultured in DMEM/F-12 medium with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C under an atmosphere of 5% CO2. Cells were grown to confluence and treated with differentiation medium consisting of DMEM/F-12 medium containing 3% FBS, 500 μM 3-isobutyl-1-methylxanthine, 1 μM rosiglitazone, 100 nM insulin, 1 μM Dex, 33 μM biotin, 17 μM pantothenate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B for 6 days. Cells were then cultured in maintenance medium consisting of DMEM/F-12 medium containing 3% FBS, 100 nM insulin, 1 μM Dex, 33 μM biotin, 17 μM pantothenate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B for 5 days. Cells were treated again with differentiation medium for 6 days, and then cultured in maintenance medium for 2 days. These cells were used as differentiated adipocytes in all experiments. The medium was changed for fresh medium every 3 days in all cases.
Fig. 2.

Insulin increases SREBP-1c mRNA expression in a time- and concentration-dependent manner. A: Time course of insulin-regulated SREBP-1c mRNA expression. Differentiated adipocytes were starved in serum/Dex/insulin-free maintenance medium for 16 h and then incubated in serum/Dex-free maintenance medium in the presence or absence of 100 nM insulin for the indicated times. B: Concentration-response effect of insulin on SREBP-1c mRNA expression. Differentiated adipocytes were starved in serum/Dex/insulin-free maintenance medium for 16 h and then incubated in serum/Dex-free maintenance medium in the presence or absence of insulin at the indicated concentrations for 24 h. The mRNA expression levels of SREBP-1c were measured by quantitative real-time PCR. Data are presented as means ± SEM of three independent experiments. *P < 0.05, **P < 0.01.
Fig. 1.

Global analysis of gene expression regulated by insulin/JNK1- or insulin/JNK2-dependent pathways. A: Hierarchical cluster analysis of upregulated or downregulated genes. Differentiated adipocytes were treated with control siRNA (siControl), JNK1 siRNA (siJNK1), or JNK2 siRNA (siJNK2) in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. Fold changes were calculated using changes in gene expression in siJNK1 or siJNK2 with insulin compared with siControl with insulin (siJNK1/insulin or siJNK2/insulin), and insulin compared with control (Insulin). Expression values are presented as Log2 scale using different gradations of red for genes upregulated by ≥1.2-fold, and blue for genes downregulated by ≥1.2-fold. B, C: Venn diagrams illustrating overlapping upregulated (↑) and/or downregulated (↓) genes in siJNK1/insulin (blue), siJNK2/insulin (green), and insulin (red). Lists of siJNK2-specific insulin-response genes (yellow regions) were integrated into the IPA (Tables 1–3).
Fig. 3.

Depletion of JNK2, but not JNK1, attenuates insulin-induced SREBP-1c expression. A: Concentration-response effect of the JNK inhibitor on SREBP-1c mRNA expression. Differentiated adipocytes were starved in serum/Dex/insulin-free maintenance medium for 16 h. Cells were then treated with SP600125 at the indicated concentrations for 30 min followed by stimulation with or without 100 nM insulin for 24 h. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus control without insulin; ##P < 0.01 versus control with insulin. B, C: Effects of JNK1 or JNK2 depletion on mRNA expression of SREBP-1c (B), JNK1, and JNK2 (C). Differentiated adipocytes were treated with control siRNA (siControl), JNK1 siRNA (siJNK1), or JNK2 siRNA (siJNK2) in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; ##P < 0.01 versus siControl with insulin. D: Western blot analysis of pre-SREBP-1 (∼125 kDa) and n-SREBP-1 (∼68 kDa) protein expression in cytoplasmic and nuclear extracts, respectively. β-actin served as a loading control. An arrowhead indicates the 54 kDa isoform of JNK2. These experiments were performed three times and the results of one representative experiment are shown. The expression levels of each protein were normalized relative to β-actin protein expression and are shown as relative protein levels. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; #P < 0.05, ##P < 0.01 versus siControl with insulin.
Fig. 6.

Depletion of JNK2 inhibits insulin-induced SREBP-1 cleavage processing and nuclear translocation. A: Western blot analysis of pre-SREBP-1 and n-SREBP-1 protein expression in cytoplasmic and nuclear extracts, respectively. Differentiated adipocytes were treated with control siRNA (siControl) or JNK2 siRNA (siJNK2) in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 1 h. β-actin served as a loading control. An arrowhead indicates the 54 kDa isoform of JNK2. These experiments were performed three times and the results of one representative experiment are shown. The protein expression levels of n-SREBP-1 were normalized relative to β-actin protein expression and are shown as relative protein levels. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; ##P < 0.01 versus siControl with insulin. B: Confocal immunofluorescence microscopy of adipocytes stained with SREBP-1 (green) and DAPI (blue). DIC, differential interference contrast. Scale bar, 30 μm.
siRNA study
Differentiated adipocytes were transfected with 10 nM control siRNA (12935-114; Invitrogen), JNK1 siRNA (12936-42 Duplex1; Invitrogen), JNK2 siRNA (12936-44 Duplex1; Invitrogen), or SREBP-1 siRNA (HSS110187) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Transfection was performed once 5 days prior to the assays.
Microarray analysis
Total RNA was isolated and treated with DNase using an RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Equal amounts of RNA prepared from three independent experiments were pooled and utilized for two-color microarray analysis with the Agilent whole-human genome 4×44K array platform according to the manufacturer's instructions (Agilent Technologies, Santa Clara, CA). Comparative analysis between expression profiles for Agilent experiments was performed with the use of GeneSpring GX 11.5.1 (Agilent Technologies). A gene list was generated containing genes flagged as detected. The “detected” list was then filtered with use of “filter by expression.” Genes were selected with a cutoff of signal intensity >80 and fold change >1.2. Gene sets were divided into groups showing similar gene expression profiles by hierarchical clustering using the uncentered Pearson correlation and the average linkage method.
Ingenuity pathway analysis
The selected genes were analyzed through the use of Ingenuity Pathway Analysis (IPA) (Ingenuity® Systems, www.ingenuity.com). Molecular and cellular functions, which were predicted to be influenced by the differentially expressed genes, were ranked in order of significance.
Quantitative real-time PCR
Total RNA was isolated and treated with DNase using an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Total RNA (100–200 ng) was reverse transcribed to cDNA in 20 μl reactions using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Quantitative gene expression analysis was performed on an ABI 7500 Fast instrument (Applied Biosystems) by TaqMan gene expression assay. Gene expression levels were normalized relative to 18S rRNA, and are shown as the mRNA levels relative to control. PCR was performed using Hs00982738_m1 for ACLY, Hs01046047_m1 for ACC1, Hs00188012_m1 for FAS, Hs00535723_m1 for CIDEC mRNA, and Hs99999901_s1 for 18S rRNA (Applied Biosystems). For SREBP-1c mRNA, the primer and probe sequences were as follows: forward primer, 5′ CCATGGATTGCACTTTCGAA 3′ reverse primer, 5′ CCAGCATAGGGTGGGTCAAA 3′ and probe, 5′ TATCAACAACCAAGACAGTGACTTCCCTGGC 3′.
Western blot analysis
Western blot analysis was performed as described previously (22). Nuclear and cytoplasmic extracts were prepared using an NE-PER nuclear and cytoplasmic extraction reagent kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Aliquots of cytoplasmic extracts (15 μg) and nuclear extracts (5 μg) were separated on 7.5–10% SDS-PAGE and transferred onto Immobilon-P membranes (Millipore Corporation, Bedford, MA). Membranes were blocked for 1 h with 5% BSA in TBS with 0.05% Tween-20 and incubated overnight at 4°C with antibodies specific to SREBP-1, JNK2, and β-actin. The blots were then treated with horseradish peroxidase-conjugated anti-rabbit IgG antibody (GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 1 h. Proteins were visualized using ECL detection reagents (GE Healthcare).
Confocal immunofluorescence microscopy
Preadipocytes were grown and differentiated into adipocytes on glass coverslips as described above. Differentiated adipocytes were treated with control siRNA or JNK2 siRNA in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. After treatment, the cells were fixed with 2% paraformaldehyde in PBS for 20 min at room temperature and washed three times with PBS, followed by permeabilization with 0.2% Triton X-100 in 0.1% sodium citrate for 10 min on ice. After fixation, cells were blocked with Image-iT FX Signal Enhancer (Invitrogen) for 30 min at room temperature, incubated with polyclonal SREBP-1 antibody (H-160, 2 μg/ml) overnight at 4°C, and then incubated with Alexa-488-conjugated goat anti-rabbit IgG for 1 h at room temperature. Cells were then washed, incubated with 0.2 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma) in PBS for 5 min at room temperature, and washed three times. After the final washes, cells were mounted on slides with ProLong Gold Antifade Reagent (Invitrogen) and visualized by confocal laser microscopy (LSM 700; Carl Zeiss, Jena, Germany). Photomicrographs were captured under green (SREBP-1) and blue (DAPI) channels at ×63 magnification, and merged using ZEN software (Carl Zeiss).
Analysis of de novo fatty acid synthesis
Differentiated adipocytes were starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. Cells were incubated in the same medium containing 1.5 μCi/ml [1-14C]acetate (New England Nuclear, Boston, MA) with cold acetic acid (500 μM) for an additional 2 h at 37°C. After incubation, cells were dissolved with 15% KOH in ethanol, and cell suspensions were incubated for 2 h at 85°C for alkaline saponification. Then, the nonpolar lipids were extracted in petroleum ether and removed. After addition of concentrated HCl, polar lipids (fatty acids) were extracted in petroleum ether and evaporated to dryness. The lipids were resolved in methanol followed by addition of ULTIMA Gold MV scintillation cocktail (PerkinElmer, Waltham, MA). The radioactivity of the cellular lipid was counted with a liquid scintillation counter (Packard Instrument Co., Meriden, CT). The results are expressed as nanomoles of fatty acid per hour per milligram of protein.
Coimmunoprecipitation experiments
Coimmunoprecipitation experiments were performed using Catch and Release Reversible v2.0 Immunoprecipitation System (Millipore Corporation) according to the manufacturer's instructions. Cells were lysed in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 50 μM MG-132, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Equal amounts of protein (500 μg) in the soluble fraction were incubated with antibodies specific to JNK2 at 4°C overnight. The immunoprecipitates were then analyzed by Western blot analysis.
Statistical analyses
The significance of differences was assessed by unpaired t-test. In all analyses, P < 0.05 was taken to indicate statistical significance. Data are expressed as the means ± SEM of three independent experiments.
RESULTS
Insulin/JNK2 pathway mainly regulates expression of genes involved in lipid metabolism in human adipocytes
Whole genome microarray analysis was performed to clarify the mechanism by which insulin/JNK2 regulates LD size in human adipocytes. We performed hierarchical cluster analysis of genes showing a greater than 1.2-fold change in expression in insulin alone, JNK1 siRNA (siJNK1)/insulin, and JNK2 siRNA (siJNK2)/insulin treatment groups (Fig. 1A). Interestingly, each group showed differential gene expression profiles. Among the 3,152 genes that were upregulated in the insulin alone group, 455 genes were specifically downregulated in the siJNK2/insulin group, suggesting that these 455 genes are downstream targets of the insulin/JNK2 pathway (Fig. 1B). In addition, 361 of the 3,032 genes that were downregulated in the insulin alone group were specifically upregulated in the siJNK2/insulin group (Fig. 1C).
IPA showed that the top molecular and cellular functions of 816 of these genes were involved in lipid metabolism (Table 1), and 8 genes belonged to the synthesis of triacylglycerol in lipid metabolism category with z-score <−2 (Table 2). Furthermore, based on IPA, SREBP-1 (SREBF1) was a predicted transcription regulator of these genes with the highest absolute value of the z-score (−4.119; Table 3). In contrast, the 868 siJNK1-specific insulin-response genes did not show any of these functions. These results suggest that the insulin/JNK2 pathway mainly regulates expression of genes involved in lipid metabolism in human adipocytes.
TABLE 1.
Top five bio-functions of genes regulated by an insulin/JNK2-dependent pathway analyzed by Ingenuity Pathway Analysis
| Molecular and Cellular Functions | |
| Category | P |
| 1 Lipid metabolism | 3.84E-09–1.64E-02 |
| 2 Small molecule biochemistry | 3.84E-09–1.64E-02 |
| 3 Cellular growth and proliferation | 6.52E-08–1.37E-02 |
| 4 Vitamin and mineral metabolism | 1.68E-07–1.37E-02 |
| 5 Cellular development | 1.55E-06–1.56E-02 |
TABLE 2.
Functional annotation of genes regulated by an insulin/JNK2-dependent pathway
| Category | Functions Annotation | P | Predicted Activation State | Regulation z-Score | Molecules |
| Lipid metabolism | Synthesis of triacylglycerol | 8.38E-05 | Decreased | −2.005 | AGPAT6, FASN, LPL, NR1H3, PLIN2, PPARG, SCD, SREBF1 |
TABLE 3.
Predicted transcription regulator of genes regulated by an insulin/JNK2-dependent pathway
| Transcription Regulator | Fold Change | Predicted Activation State | Regulation z-Score | P Value of Overlap | Target Molecules in Dataset |
| SREBF1 | −1.499 | Inhibited | −4.119 | 1.22E-12 | AACS, AARS, ACACA, ACACB, ACLY, ACSS2, ALDOC, DBI, DHCR7, ELOVL6, FASN, FDPS, G6PD, IDI1, IMMT, INSIG1, IRS2, LDLR, LPL, LSS, MVD, NR1H3, NSDHL, PPARG, SCD, SREBF1, TMEM97 |
JNK2 mediates insulin-induced upregulation of SREBP-1c
As microarray data showed that SREBP-1 would be one of the major regulators of genes regulated by the insulin/JNK2 pathway, we next analyzed the expression levels of SREBP-1c, which is the abundant isoform of SREBP-1 in adipose tissue (41), by quantitative real-time PCR. Time course and concentration-response analyses showed that insulin increased the levels of SREBP-1c mRNA in a time-dependent manner with its maximal effect observed at 24 h (Fig. 2A) and in a concentration-dependent manner with its maximal effect observed at concentrations >100 nM insulin, respectively (Fig. 2B).
We next examined the contribution of JNK to the regulation of SREBP-1c expression by insulin. The JNK inhibitor SP600125 blocked insulin-induced upregulation of SREBP-1c in a concentration-dependent manner (Fig. 3A). siJNK1 had no effect on SREBP-1c expression, whereas siJNK2 attenuated SREBP-1c expression induced by insulin (Fig. 3B). Each siRNA-mediated knockdown resulted in specific reductions in the levels of JNK1 and JNK2 mRNAs, respectively (Fig. 3C). To further verify the regulation of SREBP-1 expression, we examined protein expression of pre-SREBP-1 (∼125 kDa) and n-SREBP-1 (∼68 kDa) by Western blot analysis. Insulin markedly enhanced expression of both pre-SREBP-1 and n-SREBP-1 proteins (Fig. 3D). Insulin-enhanced expression of both forms of SREBP-1 protein showed marked attenuation by siJNK2 (Fig. 3D). siRNA-mediated knockdown resulted in specific reductions in the levels of JNK2 protein (Fig. 3D). These results indicate that insulin-induced upregulation of SREBP-1c is mediated by JNK2 but not JNK1 in human adipocytes.
JNK2 mediates insulin-induced upregulation of lipogenic enzymes and de novo fatty acid synthesis
Quantitative real-time PCR was performed to evaluate the specific involvement of JNK2 in regulation of lipogenic enzyme gene expression by insulin. Insulin markedly induced ACLY, ACC1, and FAS mRNA expression (Fig. 4A). siJNK1 had no effect on ACLY, ACC1, or FAS mRNA expression, whereas siJNK2 attenuated expression of these lipogenic enzymes induced by insulin. We next evaluated the role of JNK2 in de novo fatty acid synthesis. Insulin markedly enhanced de novo fatty acid synthesis (Fig. 4B). siJNK2 attenuated insulin-induced de novo fatty acid synthesis, whereas siJNK1 did not. These results suggest that insulin-induced upregulation of lipogenic enzymes and de novo fatty acid synthesis are mediated by JNK2 but not by JNK1 in human adipocytes.
Fig. 4.

Depletion of JNK2, but not JNK1, attenuates insulin-induced upregulation of SREBP-1c target genes and increase of fatty acid synthesis. A: Effects of JNK1 or JNK2 depletion on SREBP-1c target gene expression. Differentiated adipocytes were treated with control siRNA (siControl), JNK1 siRNA (siJNK1), or JNK2 siRNA (siJNK2) in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. The mRNA expression levels of each gene were measured by quantitative real-time PCR. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; ##P < 0.01 versus siControl with insulin. B: Analysis of fatty acid synthesis. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; ##P < 0.01 versus siControl with insulin.
SREBP-1 mediates insulin-induced upregulation of lipogenic enzymes and de novo fatty acid synthesis
Although the lipogenic actions of SREBP-1c are well known in hepatocytes, the actions in adipocytes are unclear. Therefore, we examined whether SREBP-1c contributes to regulation of lipogenic gene expression and fatty acid synthesis by insulin in human adipocytes. SREBP-1 siRNA (siSREBP-1) attenuated insulin-induced mRNA expression of ACLY, ACC1, and FAS (Fig. 5A). siRNA-mediated knockdown resulted in specific reductions in the levels of SREBP-1c mRNA (Fig. 5B). Furthermore, siSREBP-1 attenuated de novo fatty acid synthesis induced by insulin (Fig. 5C). These results suggest that insulin-induced upregulation of lipogenic enzymes and de novo fatty acid synthesis are mediated by SREBP-1 in human adipocytes.
Fig. 5.

Depletion of SREBP-1 attenuates insulin-induced upregulation of lipogenic enzymes and fatty acid synthesis. A, B: Effects of SREBP-1c depletion on expression of lipogenic enzymes. Differentiated adipocytes were treated with control siRNA (siControl), or SREBP-1 siRNA (siSREBP-1) in maintenance medium for 7 days. Cells were then starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 24 h. The mRNA expression levels of lipogenic enzymes (A) and SREBP-1c (B) were measured by quantitative real-time PCR. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; #P < 0.05, versus siControl with insulin. C: Analysis of fatty acid synthesis. Data are presented as means ± SEM of three independent experiments. **P < 0.01 versus siControl without insulin; ##P < 0.01 versus siControl with insulin.
JNK2 mediates insulin-induced SREBP-1 cleavage processing and nuclear translocation
As SREBP-1 activity is thought to be dependent on its subcellular localization (42), the effects of JNK2 on SREBP-1 subcellular distribution were examined by Western blot analysis of cytosolic and nuclear extracts and by confocal immunofluorescence microscopy. Treatment with insulin for 1 h decreased cytoplasmic pre-SREBP-1 protein levels and increased those of n-SREBP-1 protein (Fig. 6A). siJNK2 attenuated accumulation of n-SREBP-1 induced by insulin (Fig. 6A). Strong staining for SREBP-1 was primarily located in the nuclei of differentiated adipocytes treated with insulin (Fig. 6B). In contrast, siJNK2 reduced nuclear translocation of SREBP-1 (Fig. 6B). Incubation with nonimmune IgG showed no detectable fluorescence under similar conditions, suggesting that the signal for SREBP-1 was specific (data not shown). These results suggest that insulin-induced SREBP-1 cleavage processing and nuclear translocation are mediated via a JNK2-dependent pathway in human adipocytes.
JNK2 associates with the precursor forms of SREBP-1 in response to insulin
To know how JNK2 affects the processing and expression of SREBP-1c, we examined whether JNK2 forms a protein complex with SREBP-1 by coimmunoprecipitation and Western blot analysis. Endogenous pre-SREBP-1 protein was effectively coimmunoprecipitated with JNK2 in lysates from cells treated with insulin for 30 min (Fig. 7). This result was supported by our previous observation that JNK phosphorylation was stimulated by insulin treatment for 30 min (23). The control experiments using normal IgG did not precipitate any of the proteins. These results demonstrate that JNK2 associates with pre-SREBP-1 in response to insulin in human adipocytes.
Fig. 7.
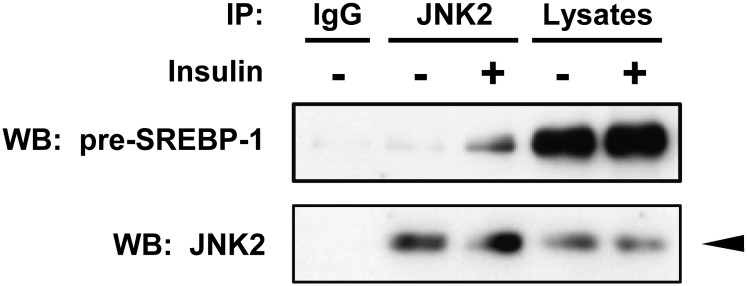
JNK2 associates with the precursor forms of SREBP-1 in response to insulin. Differentiated adipocytes were starved in serum/Dex/insulin-free maintenance medium for 16 h followed by stimulation with or without 100 nM insulin for 30 min. Endogenous JNK2 protein from cell lysates was immunoprecipitated with JNK2 antibody, followed by immunoblots with SREBP-1 and JNK2 antibodies as indicated. An arrowhead indicates the 54 kDa isoform of JNK2. These experiments were performed three times and the results of one representative experiment are shown.
DISCUSSION
It has been reported that JNK1 and/or JNK2 depletion affect basal levels of gene expression involved in lipid metabolism in mouse adipocytes (43). Our recent study showed that JNK2, but not JNK1, mediates regulation of CIDEC expression by insulin in human adipocytes (23). The results of the present study represent the first evidence regarding the specific regulation of global gene expression by an insulin/JNK2 pathway in human adipocytes. Microarray analysis and IPA indicated that genes specifically regulated by an insulin/JNK2 pathway are related mainly to lipid metabolism (Fig. 1; Tables 1, 2). Furthermore, IPA predicted that SREBP-1, a key regulator of lipogenic enzymes in hepatocytes (34), is a transcription regulator of genes regulated by an insulin/JNK2 pathway (Table 3). These results suggest that the insulin/JNK2 pathway mostly regulates expression of genes involved in lipid metabolism in addition to CIDEC in human adipocytes.
As microarray data showed that SREBP-1 would be one of the major regulators of genes regulated by the insulin/JNK2 pathway, we next examined the expression levels of SREBP-1c by quantitative real-time PCR and Western blot analysis (Figs. 2, 3). The specific JNK inhibitor SP600125, which inhibits both JNK1 and JNK2 with similar potency (44), blocked insulin-induced upregulation of SREBP-1c mRNA expression in a concentration-dependent manner. Depletion of JNK2, but not JNK1, attenuated insulin-induced upregulation of SREBP-1c mRNA expression. To further verify insulin regulation of SREBP-1 expression, we examined SREBP-1 protein expression by Western blot analysis. Consistent with previous reports (40), insulin enhanced expression of both the precursor and the nuclear form of the SREBP-1 protein. In addition, our data showed that depletion of JNK2 reduced the levels of expression of both forms of SREBP-1 protein induced by insulin. These results indicated that JNK2 mediates insulin-induced SREBP-1c expression in human adipocytes. Regulation of SREBP-1c expression and/or activity by insulin is known to also be mediated by other pathways, such as Akt, protein kinase A, protein kinase C, and insulin-induced gene-2 (Insig-2) in hepatocytes (45–49). These observations suggest that insulin regulates SREBP-1c expression via differential signaling pathways depending on various cellular conditions.
We next investigated the contributions of JNK2 to regulation of lipogenic gene expression and de novo fatty acid synthesis by insulin (Fig. 4). Depletion of JNK2, but not JNK1, attenuated insulin-induced expression of lipogenic enzymes and de novo fatty acid synthesis. This is the first evidence indicating that JNK2, but not JNK1, mediates insulin-induced lipogenic gene expression and de novo fatty acid synthesis reported to date. Although lipogenic actions of SREBP-1c are well known in hepatocytes, the actions in adipocytes are unclear. Therefore, we examined the contributions of SREBP-1c to regulation of lipogenic gene expression and de novo fatty acid synthesis by insulin in human adipocytes (Fig. 5). Depletion of SREBP-1 attenuated insulin-induced expression of lipogenic enzymes and de novo fatty acid synthesis, suggesting that this transcription factor mediates insulin-induced lipogenic gene expression and de novo fatty acid synthesis. Taken together, these results suggest that JNK2, but not JNK1, contributes to insulin-induced de novo fatty acid synthesis by stimulating the expression of SREBP-1c and its target lipogenic enzymes in human adipocytes. Similar to these effects of JNK2 on the actions of insulin in human adipocytes, the JNK/SREBP-1 pathway has been shown to mediate keratinocyte growth factor-induced lipogenic gene expression in H292 cells (50).
As SREBP-1 activity is thought to depend on its subcellular localization (42), we assessed the effects of JNK2 on SREBP-1 subcellular distribution. Insulin acutely promoted SREBP-1 cleavage and nuclear accumulation (Fig. 6), consistent with previous reports in hepatocytes (51). Depletion of JNK2 inhibited the regulation of SREBP-1 localization by insulin. These results suggest that insulin-induced SREBP-1 cleavage processing and nuclear translocation are mediated via a JNK2-dependent pathway in human adipocytes. To gain further insights into the mechanism underlying the JNK2 action on the processing and expression of SREBP-1, we examined the association of JNK2 with SREBP-1 (Fig. 7). Results of coimmunoprecipitation experiments demonstrate that JNK2 associates with pre-SREBP-1 in response to insulin in human adipocytes. In this study, we also demonstrate that JNK2 mediates both insulin-induced processing and upregulation of SREBP-1. There is evidence that n-SREBP-1 can control the expression of SREBP-1c in an autoregulation loop in response to insulin in human cells (52), suggesting that increased processing induces nuclear accumulation of n-SREBP-1 and may contribute to regulation of its own gene expression. Taken together, these results suggest that the association of JNK2 with SREBP-1 might be responsible for JNK2-mediated processing and expression of SREBP-1c. It has been reported that insulin enhances the processing of pre-SREBP-1c in rat hepatocytes by promoting its phosphorylation and association with coatomer protein II vesicles where two sequential cleavages generate the n-SREBP-1c (53). A recent study showed that SREBP-1a is phosphorylated by extracellular signal-regulated kinase, JNK, and p38 mitogen-activated protein kinases, and preventing phosphorylation of SREBP-1a protects mice from fatty liver and visceral obesity (54). The insulin/JNK2 pathway may directly stimulate SREBP-1a or SREBP-1c activity, induce SREBP-1c expression, and may be involved in obesity-related disorders. Future studies will be required to clarify the precise mechanism by which JNK2 mediates insulin-induced SREBP-1c expression.
Our previous study showed that JNK2 is required for insulin-induced upregulation of CIDEC and enlargement of LDs (23). Depletion of SREBP-1 had no effect on CIDEC mRNA expression in the presence or absence of insulin (data not shown), suggesting that SREBP-1 is not involved in the regulation of CIDEC expression. It has been shown that CIDEC stabilization is associated with triacylglycerol synthesis and LD formation (55). The present study suggested that SREBP-1c mediates insulin/JNK2-induced de novo fatty acid synthesis. Thus, the regulation of LD size by JNK2 can be accounted for, at least in part, by modulation of SREBP-1c-mediated de novo fatty acid synthesis and accompanying regulation of CIDEC expression. We found that the JNK2/SREBP-1c pathway partially contributes to de novo fatty acid synthesis, suggesting that the regulation of LD size by JNK2 may also be mediated by other lipogenic pathways. Further studies are required to clarify the precise mechanism of LD formation.
In conclusion, the present study provides the first evidence that an insulin/JNK2 pathway mainly regulates expression of genes involved in lipid metabolism, most notably SREBP-1c. The JNK2/SREBP-1c pathway involved in insulin-induced fatty acid synthesis would be a part of the mechanisms accounting for regulation of LD size in human adipocytes. The identification of a novel pathway, such as JNK2/SREBP-1c, will provide new therapeutic strategies to selectively regulate the size of adipocytes, leading to improvement of quality of adipocytes, which contributes to adipose tissue inflammation and its associated disorders.
Acknowledgments
The authors thank Prof. Kiyoto Motojima of Meiji Pharmaceutical University for helpful advice and comments on the manuscript.
Footnotes
Abbreviations:
- ACC1
- acetyl-CoA carboxylase 1
- ACLY
- ATP citrate lyase
- CIDE
- cell death-inducing DNA fragmentation factor-α-like effector
- DAPI
- 4′,6′-diamidino-2-phenylindole
- Dex
- dexamethasone
- FAS
- fatty acid synthase
- IPA
- Ingenuity Pathway Analysis
- JNK
- c-Jun N-terminal kinase
- LD
- lipid droplet
- n-SREBP-1c
- nuclear form of sterol regulatory element binding protein-1c
- PI3K
- phosphatidylinositol 3-kinase
- pre-SREBP-1c
- precursor form of sterol regulatory element binding protein-1c
- siJNK
- JNK siRNA
- siRNA
- small interfering RNA
- siSREBP-1
- SREBP-1 siRNA
- SREBP
- sterol regulatory element binding protein
- WAT
- white adipose tissue
REFERENCES
- 1.Haslam D. W., James W. P. 2005. Obesity. Lancet. 366: 1197–1209 [DOI] [PubMed] [Google Scholar]
- 2.Tilg H., Moschen A. R. 2010. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 52: 1836–1846 [DOI] [PubMed] [Google Scholar]
- 3.Praga M., Morales E. 2006. Obesity, proteinuria and progression of renal failure. Curr. Opin. Nephrol. Hypertens. 15: 481–486 [DOI] [PubMed] [Google Scholar]
- 4.Hirsch J., Batchelor B. 1976. Adipose tissue cellularity in human obesity. Clin. Endocrinol. Metab. 5: 299–311 [DOI] [PubMed] [Google Scholar]
- 5.Sakai T., Sakaue H., Nakamura T., Okada M., Matsuki Y., Watanabe E., Hiramatsu R., Nakayama K., Nakayama K. I., Kasuga M. 2007. Skp2 controls adipocyte proliferation during the development of obesity. J. Biol. Chem. 282: 2038–2046 [DOI] [PubMed] [Google Scholar]
- 6.Després J. P., Lemieux I. 2006. Abdominal obesity and metabolic syndrome. Nature. 444: 881–887 [DOI] [PubMed] [Google Scholar]
- 7.Rosen E. D., Spiegelman B. M. 2006. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 444: 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prins J. B., O'Rahilly S. 1997. Regulation of adipose cell number in man. Clin. Sci. (Lond.). 92: 3–11 [DOI] [PubMed] [Google Scholar]
- 9.Ursø B., Niesler C. U., O'Rahilly S., Siddle K. 2001. Comparison of anti-apoptotic signalling by the insulin receptor and IGF-I receptor in preadipocytes and adipocytes. Cell. Signal. 13: 279–285 [DOI] [PubMed] [Google Scholar]
- 10.Qian H., Hausman D. B., Compton M. M., Martin R. J., Della-Fera M. A., Hartzell D. L., Baile C. A. 2001. TNFalpha induces and insulin inhibits caspase 3-dependent adipocyte apoptosis. Biochem. Biophys. Res. Commun. 284: 1176–1183 [DOI] [PubMed] [Google Scholar]
- 11.Kahn B. B., Flier J. S. 2000. Obesity and insulin resistance. J. Clin. Invest. 106: 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kersten S. 2001. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2: 282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thörne A., Lönnqvist F., Apelman J., Hellers G., Arner P. 2002. A pilot study of long-term effects of a novel obesity treatment: omentectomy in connection with adjustable gastric banding. Int. J. Obes. Relat. Metab. Disord. 26: 193–199 [DOI] [PubMed] [Google Scholar]
- 14.Loftus T. M., Kuhajda F. P., Lane M. D. 1998. Insulin depletion leads to adipose-specific cell death in obese but not lean mice. Proc. Natl. Acad. Sci. USA. 95: 14168–14172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blüher M., Michael M. D., Peroni O. D., Ueki K., Carter N., Kahn B. B., Kahn C. R. 2002. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell. 3: 25–38 [DOI] [PubMed] [Google Scholar]
- 16.Inohara N., Koseki T., Chen S., Wu X., Nunez G. 1998. CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J. 17: 2526–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Omae N., Ito M., Hase S., Nagasawa M., Ishiyama J., Ide T., Murakami K. 2012. Suppression of FoxO1/cell death-inducing DNA fragmentation factor alpha-like effector A (Cidea) axis protects mouse beta-cells against palmitic acid-induced apoptosis. Mol. Cell. Endocrinol. 348: 297–304 [DOI] [PubMed] [Google Scholar]
- 18.Nishino N., Tamori Y., Tateya S., Kawaguchi T., Shibakusa T., Mizunoya W., Inoue K., Kitazawa R., Kitazawa S., Matsuki Y., et al. 2008. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Invest. 118: 2808–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toh S. Y., Gong J., Du G., Li J. Z., Yang S., Ye J., Yao H., Zhang Y., Xue B., Li Q., et al. 2008. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS ONE. 3: e2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puri V., Konda S., Ranjit S., Aouadi M., Chawla A., Chouinard M., Chakladar A., Czech M. P. 2007. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 282: 34213–34218 [DOI] [PubMed] [Google Scholar]
- 21.Keller P., Petrie J. T., De Rose P., Gerin I., Wright W. S., Chiang S. H., Nielsen A. R., Fischer C. P., Pedersen B. K., MacDougald O. A. 2008. Fat-specific protein 27 regulates storage of triacylglycerol. J. Biol. Chem. 283: 14355–14365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito M., Nagasawa M., Hara T., Ide T., Murakami K. 2010. Differential roles of CIDEA and CIDEC in insulin-induced anti-apoptosis and lipid droplet formation in human adipocytes. J. Lipid Res. 51: 1676–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito M., Nagasawa M., Omae N., Ide T., Akasaka Y., Murakami K. 2011. Differential regulation of CIDEA and CIDEC expression by insulin via Akt1/2- and JNK2-dependent pathways in human adipocytes. J. Lipid Res. 52: 1450–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevenson R. W., Kreutter D. K., Andrews K. M., Genereux P. E., Gibbs E. M. 1998. Possibility of distinct insulin-signaling pathways beyond phosphatidylinositol 3-kinase-mediating glucose transport and lipogenesis. Diabetes. 47: 179–185 [DOI] [PubMed] [Google Scholar]
- 25.Greenberg A. S., Coleman R. A., Kraemer F. B., McManaman J. L., Obin M. S., Puri V., Yan Q. W., Miyoshi H., Mashek D. G. 2011. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest. 121: 2102–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anand S. S., Tarnopolsky M. A., Rashid S., Schulze K. M., Desai D., Mente A., Rao S., Yusuf S., Gerstein H. C., Sharma A. M. 2011. Adipocyte hypertrophy, fatty liver and metabolic risk factors in South Asians: the Molecular Study of Health and Risk in Ethnic Groups (mol-SHARE). PLoS ONE. 6: e22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attie A. D., Scherer P. E. 2009. Adipocyte metabolism and obesity. J. Lipid Res. 50(Suppl.):– S395–S399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guilherme A., Virbasius J. V., Puri V., Czech M. P. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9: 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirosumi J., Tuncman G., Chang L., Gorgun C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. 2002. A central role for JNK in obesity and insulin resistance. Nature. 420: 333–336 [DOI] [PubMed] [Google Scholar]
- 30.Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G. S. 2006. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA. 103: 10741–10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis R. J. 2000. Signal transduction by the JNK group of MAP kinases. Cell. 103: 239–252 [DOI] [PubMed] [Google Scholar]
- 32.Yang D. D., Kuan C. Y., Whitmarsh A. J., Rincon M., Zheng T. S., Davis R. J., Rakic P., Flavell R. A. 1997. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 389: 865–870 [DOI] [PubMed] [Google Scholar]
- 33.Grimard V., Massier J., Richter D., Schwudke D., Kalaidzidis Y., Fava E., Hermetter A., Thiele C. 2008. siRNA screening reveals JNK2 as an evolutionary conserved regulator of triglyceride homeostasis. J. Lipid Res. 49: 2427–2440 [DOI] [PubMed] [Google Scholar]
- 34.Foufelle F., Ferre P. 2002. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem. J. 366: 377–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raghow R., Yellaturu C., Deng X., Park E. A., Elam M. B. 2008. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 19: 65–73 [DOI] [PubMed] [Google Scholar]
- 36.Ide T., Shimano H., Yahagi N., Matsuzaka T., Nakakuki M., Yamamoto T., Nakagawa Y., Takahashi A., Suzuki H., Sone H., et al. 2004. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 6: 351–357 [DOI] [PubMed] [Google Scholar]
- 37.Browning J. D., Horton J. D. 2004. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114: 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimano H. 2009. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J. 276: 616–621 [DOI] [PubMed] [Google Scholar]
- 39.Ducluzeau P. H., Perretti N., Laville M., Andreelli F., Vega N., Riou J. P., Vidal H. 2001. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes. 50: 1134–1142 [DOI] [PubMed] [Google Scholar]
- 40.Sewter C., Berger D., Considine R. V., Medina G., Rochford J., Ciaraldi T., Henry R., Dohm L., Flier J. S., O'Rahilly S., et al. 2002. Human obesity and type 2 diabetes are associated with alterations in SREBP1 isoform expression that are reproduced ex vivo by tumor necrosis factor-alpha. Diabetes. 51: 1035–1041 [DOI] [PubMed] [Google Scholar]
- 41.Brown M. S., Goldstein J. L. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 89: 331–340 [DOI] [PubMed] [Google Scholar]
- 42.Taghibiglou C., Martin H. G., Lai T. W., Cho T., Prasad S., Kojic L., Lu J., Liu Y., Lo E., Zhang S., et al. 2009. Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nat. Med. 15: 1399–1406 [DOI] [PubMed] [Google Scholar]
- 43.Rozo A. V., Vijayvargia R., Weiss H. R., Ruan H. 2008. Silencing Jnk1 and Jnk2 accelerates basal lipolysis and promotes fatty acid re-esterification in mouse adipocytes. Diabetologia. 51: 1493–1504 [DOI] [PubMed] [Google Scholar]
- 44.Bennett B. L., Sasaki D. T., Murray B. W., O'Leary E. C., Sakata S. T., Xu W., Leisten J. C., Motiwala A., Pierce S., Satoh Y., et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 98: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong R. H., Sul H. S. 2010. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr. Opin. Pharmacol. 10: 684–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fleischmann M., Iynedjian P. B. 2000. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: role of insulin and protein kinase B/cAkt. Biochem. J. 349: 13–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto T., Shimano H., Inoue N., Nakagawa Y., Matsuzaka T., Takahashi A., Yahagi N., Sone H., Suzuki H., Toyoshima H., et al. 2007. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. J. Biol. Chem. 282: 11687–11695 [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto T., Watanabe K., Inoue N., Nakagawa Y., Ishigaki N., Matsuzaka T., Takeuchi Y., Kobayashi K., Yatoh S., Takahashi A., et al. 2010. Protein kinase Cbeta mediates hepatic induction of sterol-regulatory element binding protein-1c by insulin. J. Lipid Res. 51: 1859–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yellaturu C. R., Deng X., Park E. A., Raghow R., Elam M. B. 2009. Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional down-regulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP).SREBP-1c complex. J. Biol. Chem. 284: 31726–31734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang Y., Wang J., Lu X., Thewke D. P., Mason R. J. 2005. KGF induces lipogenic genes through a PI3K and JNK/SREBP-1 pathway in H292 cells. J. Lipid Res. 46: 2624–2635 [DOI] [PubMed] [Google Scholar]
- 51.Hegarty B. D., Bobard A., Hainault I., Ferre P., Bossard P., Foufelle F. 2005. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc. Natl. Acad. Sci. USA. 102: 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dif N., Euthine V., Gonnet E., Laville M., Vidal H., Lefai E. 2006. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem. J. 400: 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yellaturu C. R., Deng X., Cagen L. M., Wilcox H. G., Mansbach C. M., 2nd, Siddiqi S. A., Park E. A., Raghow R., Elam M. B. 2009. Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J. Biol. Chem. 284: 7518–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotzka J., Knebel B., Haas J., Kremer L., Jacob S., Hartwig S., Nitzgen U., Muller-Wieland D. 2012. Preventing phosphorylation of sterol regulatory element-binding protein 1a by MAP-kinases protects mice from fatty liver and visceral obesity. PLoS ONE. 7: e32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nian Z., Sun Z., Yu L., Toh S. Y., Sang J., Li P. 2010. Fat-specific protein 27 undergoes ubiquitin-dependent degradation regulated by triacylglycerol synthesis and lipid droplet formation. J. Biol. Chem. 285: 9604–9615 [DOI] [PMC free article] [PubMed] [Google Scholar]