

Abstract
The LDL receptor (LDLR) relies upon endocytic adaptor proteins for internalization of lipoproteins. The results of this study show that the LDLR adaptor autosomal recessive hypercholesterolemia protein (ARH) requires nitric oxide to support LDL uptake. Nitric oxide nitrosylates ARH at C199 and C286, and these posttranslational modifications are necessary for association of ARH with the adaptor protein 2 (AP-2) component of clathrin-coated pits. In the absence of nitrosylation, ARH is unable to target LDL-LDLR complexes to coated pits, resulting in poor LDL uptake. The role of nitric oxide on LDLR function is specific for ARH because inhibition of nitric oxide synthase activity impairs ARH-supported LDL uptake but has no effect on other LDLR-dependent lipoprotein uptake processes, including VLDL remnant uptake and dab2-supported LDL uptake. These findings suggest that cells that depend upon ARH for LDL uptake can control which lipoproteins are internalized by their LDLRs through changes in nitric oxide.
Keywords: nitric oxide, low density lipoprotein receptor, autosomal recessive hypercholesterolemia protein
The LDL receptor (LDLR) internalizes a broad spectrum of lipoproteins, including chylomicrons, chylomicron remnants, VLDL, VLDL remnants, LDL, and some forms of HDL. Of these lipoproteins, the LDLR is the primary endocytic receptor responsible for the uptake of LDL and VLDL remnants (1, 2). LDLR-dependent endocytosis of both lipoproteins occurs through clathrin-coated pits; however, the mechanisms used by the LDLR to concentrate LDL and VLDL remnants in coated pits are distinct. Clustering of LDL-LDLR complexes in coated pits requires the FDNPVY807 sequence on the LDLR cytoplasmic domain (3, 4). This sequence drives constitutive targeting of the LDLR and lipoprotein-LDLR complexes to coated pits via interaction with either of two endocytic adaptor proteins, the autosomal recessive hypercholesterolemia protein (ARH) and the disabled-like protein 2 (dab2). Both ARH and dab2 target LDLRs to coated pits through binding sites for the LDLR, clathrin, and the adaptor protein 2 (AP-2) complex. ARH binds to the FDNPVY807 sequence of the LDLR through a phosphotyrosine-binding (PTB) domain, to the heavy chain of clathrin through a clathrin box sequence, and to the β2-subunit of AP-2 through a sequence with strong homology to the AP-2-binding sequences of β-arrestins (5–8) (Fig. 1A). Y807 of the FDNPVY807 plays a critical role in the interaction of ARH and dab2 with the LDLR, and mutation of Y807 to either cysteine or alanine cripples LDLR-dependent LDL uptake (3, 5, 8, 9). By contrast, uptake of VLDL remnants does not require ARH, dab2, or a functional FDNPVY sequence because binding of VLDL remnants to the LDLR induces a separate endocytic process that involves a second internalization motif on the LDLR cytoplasmic domain (10). It is not clear why the LDLR needs an induced process for VLDL remnant uptake when the FDNPVY-dependent process can internalize both LDL and VLDL remnants, nor why the FDNPVY process utilizes both the ARH and dab2 adaptors when either adaptor is sufficient to support lipoprotein uptake by the LDLR.
Fig. 1.
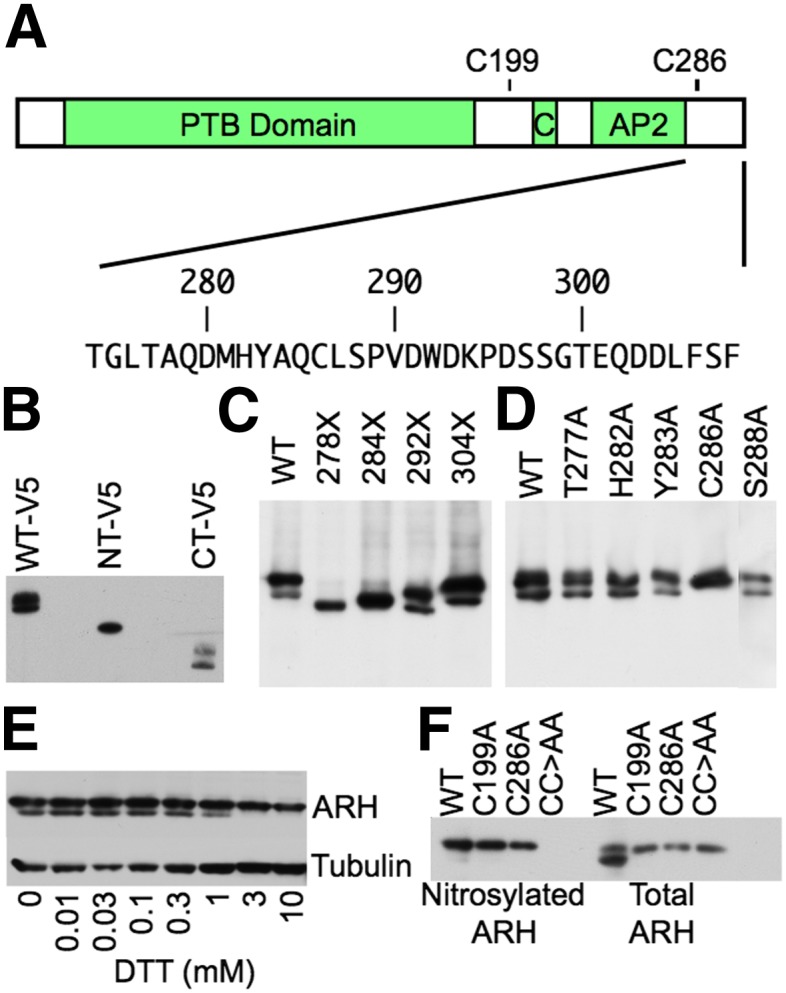
ARH is nitrosylated at C199 and C286. (A) Domain structure of ARH. ARH has a PTB domain (PTB), Clathrin Box module (C), and an AP-2-binding module (AP2). Below the domain structure is the amino acid sequence of the C-terminal 44 amino acids. (B) Identification of a posttranslational modification in the C-terminal half of ARH. HEK293 cells were transfected with C-terminal V5-tagged human ARH constructs encompassing the entire ARH protein (WT-V5), residues 1–187 (NT-V5), or residues 188–308 (CT-V5), and then lysed. Lysates were run on SDS-PAGE and immunoblotted for V5. (C) Posttranslational modification resides between residues 284 and 292. HEK293 cells were transfected with untagged human ARH variants bearing premature stop codons at the indicated residue positions and lysed. Lysates were run on SDS-PAGE and immunoblotted for ARH. (D) C286 is posttranslationally modified. HEK293 cells were transfected with human ARH variants bearing alanine mutations at the indicated positions and lysed. Lysates were run on SDS-PAGE and immunoblotted for ARH. (E) C286 participates in an intramolecular disulfide bond. Lysates of normal human fibroblasts were treated with the indicated concentrations of DTT for 30 min at room temperature, run on SDS-PAGE, and immunoblotted for ARH. (F) C286 forms a disulfide bridge with C199, and both C199 and C286 are nitrosylated. The C-terminal 121 residues of human ARH have two cysteines, C199 and C286. HEK293 cells were transfected with the indicated ARH variants and lysed. Lysates were divided into two portions. The first portion was processed by biotin switch to label nitrosylated cysteines with biotin. Biotinylated proteins were precipitated with neutravidin agarose and run with the second, untreated portion of the lysate on SDS-PAGE and immunoblotted for ARH. CC>AA indicates the ARH variant with both the C199A and C286A mutations.
A key question is why an ARH is necessary. ARH has highest expression in kidney, liver, and placenta (11); however, dab2 is also highly expressed in both kidney and placenta, and it is dab2, not ARH, that is required for normal placental and kidney function (12). Hepatocytes and peripheral blood leukocytes lack dab2 (13, 14), and ARH deficiency in both humans and mice sharply reduces LDL clearance rates, resulting in hypercholesterolemia (15–18). Why hepatocytes and leukocytes normally rely upon ARH is unclear because both cell types lose their dependence on ARH for LDL uptake when dab2 expression is induced (19, 20), indicating that dab2 could support LDL uptake in these cells were it expressed.
Here we show that the activity of ARH is regulated by nitric oxide. Nitric oxide nitrosylates ARH at two cysteines, and nitrosylation is necessary for ARH to support LDL uptake by the LDLR. Nitric oxide is not required for either VLDL remnant uptake or dab2-supported LDL uptake. We suggest that the ability of ARH to be regulated is the reason why hepatocytes and leukocytes normally use ARH and not dab2 for LDL uptake.
MATERIALS AND METHODS
Materials
Human LDL and rabbit β-VLDL were provided by Drs. Michael Brown and Joseph Goldstein (UT Southwestern). Rabbit polyclonal antibodies against human ARH (4034) and LDLR (4548) were provided by Dr. Helen Hobbs (UT Southwestern) and Dr. Joachim Herz (UT Southwestern), respectively. Cre-recombinase adenovirsus was provided by Dr. Joachim Herz with the consent of Frank Graham (McMaster University). Monoclonal antibodies were purchased for human ARH (Santa Cruz Biotechnology), V5 (Invitrogen), adaptor protein 2 (AP-2) (BD Biosciences), disabled protein 2 (dab2) (BD Biosciences), and clathrin heavy chain (BD Biosciences). Rabbit polyclonal antibodies against clathrin heavy chain and AP-2 were purchased from ABCAM and ABR Affinity Bioreagents, respectively.
DTT reduction assay
Cell lysate (20 µg) was incubated with DTT (0 to 10 mM) at room temperature for 30 min. Lysates were then mixed with protein loading buffer, separated on SDS-PAGE, and immunoblotted for ARH using the 4034 antibody.
Generation of mouse embryonic fibroblasts lacking both ARH and dab2 expression
Fibroblasts from Arh−/−;Dab2flox/flox;hLDLR+/+ mice were kindly provided by Dr. Helen Hobbs. These cells were infected with adenovirus encoding Cre-recombinase at an MOI of 500. Cells were then clonally selected in 96-well plates for cells that lack expression of dab2 (Arh−/−;Dab2−/−;hLDLR+/+ fibroblasts). An outline of the procedure is shown in Fig. 2.
Fig. 2.
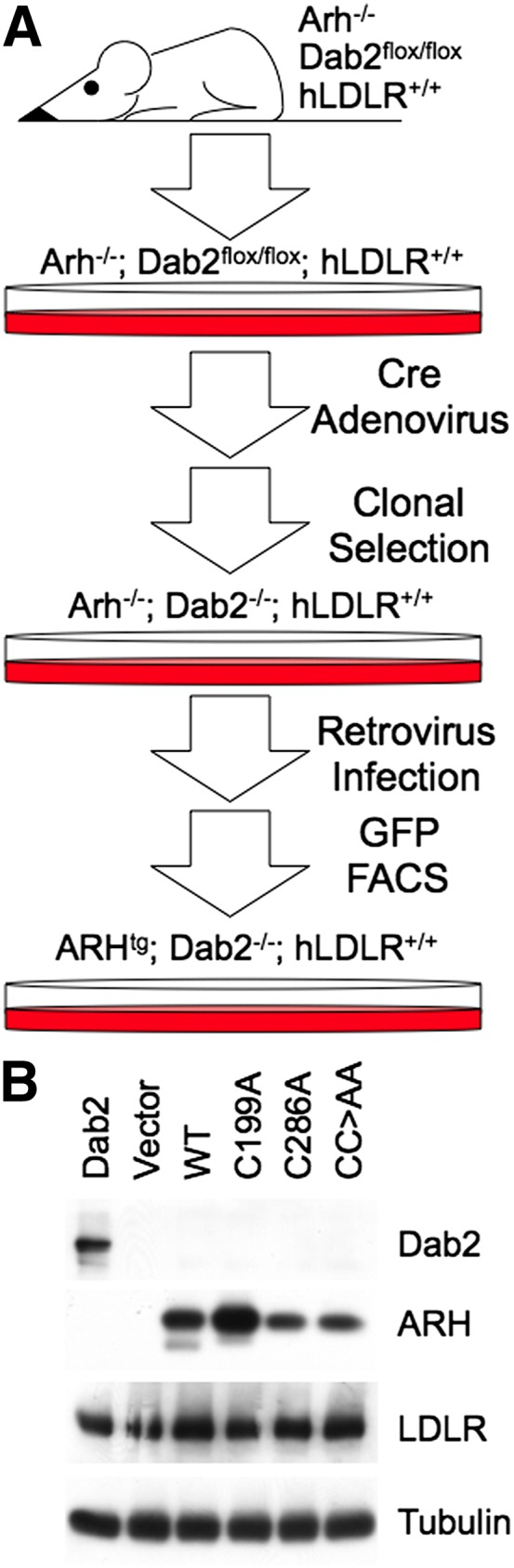
Generation of Arh−/−;Dab2−/−;hLDLR+/+ mouse embryonic fibroblasts and stable cell lines. (A) Isolated embryonic fibroblasts from Arh−/−;Dab2flox/flox;hLDLR+/+ mice were infected with adenoviruses encoding the cre-recombinase and clonally selected for cells that lack Dab2 expression. Cells lacking Dab2 were infected with retroviruses encoding no ARH (Vector), WT ARH (WT), ARH-C199A (C199A), ARH-C286A (C286A), or ARH-C199A/C286A (CC>AA). All introduced ARH cDNAs used the human ARH sequence. The mRNA produced by the viral vector is bicistronic and encodes both ARH and GFP with an internal ribosome entry site separating the two. Thus, ARH-expressing cells also express GFP, allowing transgenic cells to be FACS sorted by GFP fluorescence. (B) Immunoblots showing Dab2, ARH, LDLR, and α-tubulin expression by the parental Arh−/−;Dab2flox/flox;hLDLR+/+ fibroblasts (Dab2) and by the Vector, WT, C199A, C286A, and CC>AA transgenic cells.
Generation of expression plasmids encoding ARH variants and transfection into 293 cells
ARH variants were generated using Quickchange II XL site-directed mutagenesis kit (Stratagene) and cloned into pCDNA3 mammalian expression vector. Then 293 cells were transfected with 1 μg of plasmid per well in 6-well dishes. Cells were cultured for two days and then lysed with lysis buffer [50 mM Tris-HCl (pH 7.4), 1% Triton X-100]. Lysates were run on SDS-PAGE and immunoblotted using either the V5 mAb (Invitrogen) or the ARH mAb (Santa Cruz Biotechnology).
Generation of ARH retroviruses and ARH-expressing cell lines
Human ARH variants were generated using Quickchange II XL site-directed mutagenesis kit (Stratagene), and cloned into the pMX bicistronic retroviral vector (21). These vectors use the 5′UTR of the virus to drive mRNA expression. The multicloning site is 5′ to the internal ribosome entry site (IRES), while GFP is encoded 3′ to the IRES element. Retroviral vectors were cotransfected with the pAmpho vector (Clontech) into 293T cells to produce infectious, replication-defective retroviruses. These viruses were used to infect Arh−/−;Dab2−/−;hLDLR+/+ mouse fibroblasts. Because both ARH and GFP are encoded on the same viral mRNA, GFP expression correlates with ARH expression, allowing ARH-expressing cells to be isolated by fluorescence-activated cell sorting (FACS). MOI for ARH virus infection was kept low (<5% cell infection) to enrich for cells with only single genomic integration events. Single integration yields populations of cells with similar mRNA production and hence similar protein expression (21).
LDL initial rate endocytosis assay
Endocytic rates of lipoprotein internalization were determined as previously described (10, 22). Briefly, cells were incubated with 10 μg/ml 125I-LDL for 1 h at 4°C in MEM medium [MEM supplemented with 10% fetal lipoprotein-poor serum (FLPPS)]. Media was changed for the times indicated with warm DMEM medium (high-glucose D-MEM supplemented with 10% FLPPS) also containing 10 μg/ml 125I-LDL. Cells were extensively washed with ice-cold PBS and incubated with 1 mg/ml protease K in protease buffer [PBS + 1 mM EDTA (pH 8.0] for 2 h at 4°C. The cell suspension was then centrifuged at 5000 g for 10 min over a cushion of 10% sucrose in PBS. The tubes were frozen in liquid nitrogen, cut to separate the cells (internal) from the solution (surface-bound material released by protease K), and counted on a γ counter. Nonspecific activity was assessed in parallel experiments in the presence of 250 μg/ml unlabeled lipoprotein. Nonspecific activities were subtracted from mean values for each data point. Data are means ± SEM of four replicate trials from four experiments (n = 16).
Lipoprotein uptake assay
LDL and β-VLDL uptake assays used previously published protocols (23). Briefly, cells were first treated with FLPPS medium (D-MEM supplemented with 10% fetal lipoprotein-poor serum) overnight to induce LDLR expression. Alexa546-labeled LDL (10 μg/ml) or Alexa546-labeled β-VLDL (5 μg/ml) in LPPS medium were incubated with the cells for 1–4 h. Cells were harvested hourly, washed with PBS, fixed with 3% paraformaldehyde, and held on ice for flow cytometry. Mean cellular fluorescence from 10,000 cells per time point was determined using a BD FACScalibur. As a negative control, all assays included cells without FLPPS treatment. Uptake of both LDL and β-VLDL by cells expressing wild-type (WT) ARH increased ∼20-fold following LPPS treatment and was consistent with the fold induction of LDLR expression. In all reported data, the uptake by cells without FLPPS treatment was subtracted from FLPPS-treated cells at each time point. Relative rates of uptake were determined by linear regression analysis using Prism 4.0 software.
LDL-binding assay
LDL was labeled with 125I using the Bolton-Hunter protocol (24). Binding assays were performed as previously described (10, 25).
Surface LDLR expression assay
Surface expression was measured by flow cytometry using the C7 monoclonal antibody to the LDLR as previously described (23). Briefly, cells were treated with LPPS medium overnight, fixed with 3% paraformaldehyde, and blocked with PBS containing 0.1% BSA. Cells were then incubated with 10 μg/ml C7 antibody for 1 h at room temperature, washed, and incubated for 1 h at room temperature with a secondary antibody coupled to allophycocyanin. Cells were lifted from the dishes, and cellular fluorescence determined by flow cytometry.
Biotin switch assay for protein nitrosylation
Nitrosylated proteins were identified by replacing S-nitrosyl groups with biotin using the S-nitrosylated protein detection assay kit (Cayman Chemical Co., Cat. No. 10006518), which is based upon the protocol developed by Jaffrey and Snyder (26). Biotinylated proteins were then purified using neutravidin-agarose, separated on SDS-PAGE, and immunoblotted for ARH.
Immunoprecipitation
Cells were lysed in RIPA buffer [50 mM Tris, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS (pH 7.5)] with proteinase inhibitors (Calbiochem). Protein concentration of cell lysate was measured by BCA assay (Thermo Scientific) and equalized prior to precipitation. Immunoprecipitation was carried out with monoclonal antibodies against ARH (Santa Cruz Biotechnology) or AP-2 (BD Biosciences). Bound proteins were separated by 8% SDS-PAGE and immunoblotted with the indicated polyclonal antibodies.
Electronic microscopy
Colloidal gold-conjugated LDL (LDL-gold) was produced as previously described (27, 28). Surface labeling with LDL-gold was performed by incubating cells with 10 μg/ml LDL-gold in minimal essential media supplemented with 10% LPPS at 4°C for 2 h. The cells were washed three times with PBS and fixed with 3% paraformaldehyde followed by 0.8% glutaraldehyde. The cells were then embedded, sectioned, counter-stained, and visualized using an FEI Tecnai electron microscope operating at 120 kV as previously described (28). Micrographs of each cell type were coded, and the length of the noncoated pit membranes, the length of the coated pit membranes, and the number of gold particles associated with each class of membrane were determined using ImageJ software.
RT-PCR
RNA was isolated from white adipose tissue of a C57BL/6 mouse or from WT cells using the RNA STAT-60 kit (TEL-TEST Inc.) according to the manufacturer's instructions. RNA was then treated with DNA-free kit (Ambion) and reverse-transcribed into cDNA using the Taqman Reverse Transcription kit (Applied Biosystems). PCR was performed using the following primers: NOS1: 5′-GACCAAGCCCTGGTGGAGATTAAC, 5′-GCCTCTGCCAATTTCTTGAAGCCA; NOS2: 5′-CTGCTGGTGGTGACAAGCACATTTG, 5′-CGTTCTTTGCATGGATGCTGCTGAG; NOS3: 5′-CAGTTCCCGGAAAGAGGGATTGTG; 5′-GCATATGAAGAGGGCAGCAGGATG. All reactions used SpeedSTAR HS polymerase (Takara) with 50 ng of cDNA over 30 cycles. Expected product sizes are 332 bp for NOS1 (nNOS), 439 bp for NOS2 (iNOS), and 214 bp for NOS3 (eNOS).
Statistics
P-values were calculated by one-way ANOVA using PRISM 4.0.
RESULTS
ARH is nitrosylated
The reason to suspect that ARH might be subject to posttranslational modification originated from the first immunoblots of ARH, which showed that ARH exists in two forms of differing electrophoretic mobility (17). To ensure that the difference in mobility did not result from proteolysis, we introduced into HEK293 cells C-terminally V5-tagged human ARH variants encoding full-length ARH, an N-terminal fragment of ARH (residues 1–187), or a C-terminal fragment of ARH (residues 188–308). V5 immunoblots showed that cells expressing full-length and the C-terminal half of ARH produced two V5 immunoreactive bands, while the N-terminal fragment produced only a single band (Fig. 1B). Thus, proteolysis is not responsible for two ARH bands because hydrolysis at the N-terminal end of ARH would have produced a second band for the N-terminal fragment, while hydrolysis at the C-terminal end of ARH would have removed the V5 tag. To identify what part of the C-terminal half was responsible for the second ARH band, we introduced a series of untagged human ARH variants with C-terminal deletions into HEK293 cells. Deletions removing the last 17 amino acids had little effect on the presence of two ARH bands, but deletions of 25 or more residues eliminated the faster migrating band (Fig. 1C). We identified which residues were required for the fast mobility species by introducing ARH variants bearing single alanine substitutions. Cells expressing ARH variants with alanine mutation at T277, H282, Y283, or S288 produced both slow and fast mobility ARH forms, but cells expressing ARH-C286A produced only the slow mobility form (Fig. 1D). These observations indicate that C286 is required for posttranslational modification of ARH.
Most posttranslational modifications slow electrophoretic mobility; however, intramolecular disulfide bonds are an exception, because this modification reduces the hydrodynamic radius of SDS-denatured proteins. We tested whether C286 was involved in an intramolecular disulfide bond by treating lysates of normal human fibroblasts with increasing concentrations of DTT. Concentrations above 0.3 mM eliminated the faster mobility form of ARH (Fig. 1E). Disulfide bonds require two cysteines, and we tested whether the other cysteine in the C-terminal half of ARH, C199, was required for the faster mobility form of ARH. As with the C286A mutation, the C199A mutation eliminated the fast mobility form of ARH (Fig. 1F). Formation of disulfide bonds in cytosolic proteins is rare because the cytosol is normally a reducing environment; however, nitric oxide is an endogenously produced oxidant that has been shown to promote disulfide bond formation (29). Nitric oxide reacts with the sulfhydryl of cysteine to form S-nitroso cysteine, which can then react with other cysteines and nitrosylated cysteines to form disulfide bonds. To determine whether ARH is nitrosylated, we introduced into HEK293 cells WT ARH, ARH-C199A, ARH-C286A, or ARH-C199A/C286A and tested for nitrosylation using biotin switch assays. These assays showed that WT ARH, ARH-C199A, and ARH-C286A, but not ARH-C199A/C286A, are nitrosylated (Fig. 1F), indicating that ARH is nitrosylated at both C199 and C286 and that these cysteines can form a disulfide bond.
Nitrosylation of ARH is required for LDL uptake
To test whether the nitrosylation status of ARH influences lipoprotein uptake, we used fibroblasts from Arh−/−;Dab2flox/flox;hLDLR+/+ mice to make stable fibroblast cell lines that i) express Dab2 but not ARH (Dab2); ii) neither ARH nor Dab2 (Vector); iii) WT human ARH but not Dab2 (WT); iv) human ARH-C199A but not Dab2 (C199A); v) human ARH-C286A but not Dab2 (C286A); or vi) human ARH-C199A/C286A but not Dab2 (CC>AA) (Fig. 2A, B). These cells were then assayed for their ability to support lipoprotein uptake using a FACS-based assay that measures steady-state rates of lipoprotein accumulation. We observed that C199A and C286A supported rates of LDL accumulation that were similar to that of WT cells, while CC>AA cells supported a rate of LDL accumulation that was similar to that of Vector cells (Fig. 3A and supplementary Fig. I). Accumulation rates for the VLDL remnant, β-VLDL, were similar for all cell lines (Fig. 3B and supplementary Fig. I), consistent with the prior observation that β-VLDL uptake does not require ARH, dab2, or the FDNPVY sequence (10). These observations show that ARH-supported LDL uptake requires either of the two nitrosylated cysteines.
Fig. 3.
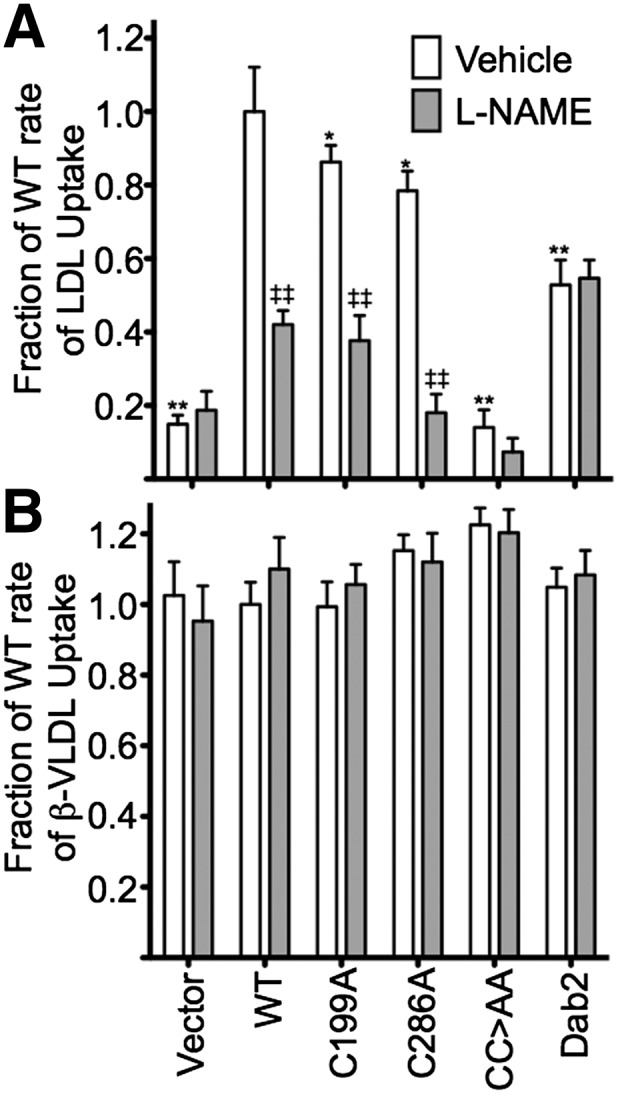
ARH nitrosylation is necessary for LDL uptake but not β-VLDL uptake. Vector, WT, C199A, C286A, CC>AA, and Dab2 cells were pretreated or not with 4 mM L-NAME for 30 min and then assayed by FACS for lipoprotein uptake at 1, 2, 3, and 4 h using 10 μg/ml Alexa546-labeled LDL or 5 μg/ml Alexa546-labeled β-VLDL. Data for each time point are shown in supplementary Fig. I. Linear regression of uptake data was used to determine rate constants for LDL uptake and β-VLDL uptake. Rate data are plotted as mean rates ± SD (n = 3 trials; 10,000 cells per time point per trial). Significance determined by one-way ANOVA. ‡P < 0.05, ‡‡P < 0.005 for untreated cells versus L-NAME-treated cells; *P < 0.05, **P < 0.005 for untreated cells compared with untreated WT cells.
To confirm the role of nitric oxide, we tested whether inhibition of nitric oxide synthase activity reduced LDL uptake in our fibroblast cell lines. Mammalian genomes encode three nitric oxide synthases (NOS1–3), and our fibroblasts express primarily NOS2 (iNOS) with lower levels of both NOS1 (nNOS) and NOS3 (eNOS) (supplementary Fig. II). This expression pattern is consistent with prior reports of NOS expression in fibroblasts (30). To reduce nitric oxide synthase activity, we used l-NG-nitroarginine methyl ester (L-NAME), a cell-permeable arginine analog that inhibits all three NOS enzymes (31). Pretreatment of WT, C199A, or C286A cells with 4 mM L-NAME sharply reduced their rates of LDL uptake (Fig. 3A and supplementary Fig. I), indicating that nitrosylation of ARH by nitric oxide is necessary for ARH to support LDL uptake. Importantly, L-NAME had no effect on dab2-supported LDL uptake or β-VLDL uptake (Fig. 3A and supplementary Fig. I). These findings indicate that the role of nitric oxide on LDLR function is specific for ARH-supported LDL uptake.
During LDL uptake, the LDLR cycles between the cell surface where it binds LDL and endosomes where it releases LDL. In fibroblasts at steady state, half of the total LDLR pool is exposed on the cell surface, while the remainder is in transit through the endosomal and recycling systems (4). In fibroblasts, which lack both ARH and dab2 or which express only LDLRs that lack a functional FDNPVY sequence, the internal pool of LDLRs is lost and all receptors are present on the cell surface (10, 13, 18, 28). Consistent with these prior findings, our Vector cells, which lack both adaptors, had twice as many surface LDLRs and bound twice as much LDL as WT cells (Fig. 4), despite having a similar total LDLR content as WT cells (Fig. 2). C199A and C286A cells had LDLR surface expression and LDL binding that was similar to WT cells; however, CC>AA cells had levels of surface LDLR and LDL binding that were similar to Vector cells (Fig. 4), despite WT levels of total LDLR (Fig. 2). These findings suggest that nitrosylation is required for ARH to engage the endocytic machinery of coated pits.
Fig. 4.

Loss of ARH function increases surface LDLRs. (A) Vector and CC>AA cells have twice as much surface LDL-binding capacity as WT, C199A, and C286A cells. Surface binding of 125I-labeled LDL was performed as described in Materials and Methods and is reported as nanograms of LDL bound per milligram of cellular protein. Experiments were performed in quadruplicate and data is shown as means ± SD. (B) Vector and CC>AA cells have more surface LDLRs than WT, C199A, and C286A cells. Relative surface number of LDLRs was determined by flow cytometry using the C7 antibody against the LDLR. Cellular fluorescence data are reported as a fraction of the mean fluorescence of WT cells. Experiments were performed in triplicate, and data is shown as means ± SD. *P < 0.05 compared with WT.
To test whether nitrosylation is necessary for ARH-dependent targeting of LDL-LDLR complexes to coated pits, we loaded surface LDLRs of the fibroblast cell lines with LDL-gold and then visualized the location of LDL-LDLR complexes by thin section electron microscopy (Fig. 5A). Quantification of the LDL-gold distribution showed that Vector cells had little enrichment of LDL-gold in coated pits, while WT cells had a 10-fold enrichment. C199A, C286A, and Dab2 cells had coated pit enrichments that were equal to or better than that of WT cells; however, CC>AA cells had enrichment that was nearly as poor as Vector cells (Fig. 5B). These observations show that ARH requires the ability to be nitrosylated for coated pit targeting of LDL.
Fig. 5.

ARH nitrosylation promotes association with AP-2 and is necessary for efficient targeting of LDL to coated pits. (A) Representative images of coated pits with and without LDL-gold labeling. The indicated cells were incubated with 10 μg/ml colloidal gold-labeled LDL for 2 h on ice, and then washed, fixed and processed for thin-section EM. Quantification of enrichment is shown below representative examples of coated pits. (B) CC>AA cells have poor ability to target LDL-LDLR complexes to coated pits. Coated pit enrichment is reported using the summation of 10 random micrographs ± SEM of enrichments calculated from each micrograph separately. The P-value relative to WT is indicated above the error bar. (C) CC>AA cells have poor ability to support LDL internalization. Surface receptors were saturated with 10 μg/ml 125I-LDL at 4°C and then shifted to 37°C in the presence of 10 μg/ml 125I-LDL. At the indicated times, internalization was stopped and surface-bound and internalized pools of LDL were assayed as described in Materials and Methods. Data is shown as the mean ratio of internal/surface ± SEM, n = 16.
The ability of ARH to be nitrosylated also correlated with the ability of ARH to support LDL internalization. Assays of LDL internalization were performed by loading surface LDLR with 125I-LDL at 4°C, shifting to 37°C for various periods of time, and measuring surface and internal pools of LDL. Fibroblasts expressing endogenous levels of both ARH and dab2 show uptake rates of 0.065–0.085 min−1 (10, 32). WT and Dab2 cells were both in this range with LDL internalization rates of 0.081 and 0.067 min−1, respectively (Fig. 5C). Vector cells, which express neither an ARH nor dab2, only slowly internalized LDL with a rate of 0.0059 min−1. CC>AA cells were similar to Vector cells with a rate of 0.0099 min−1. C199A and C286A cells were intermediate with rates of 0.030 and 0.029 min−1, respectively. These results indicate that loss of both cysteines prevents ARH from supporting LDL internalization, while loss of either cysteine slows LDL internalization.
Nitrosylation is necessary for normal interaction of ARH with AP-2
Published experiments have shown that β-arrestin2, an endocytic adaptor that promotes G-protein coupled receptor (GPCR) uptake, is nitrosylated and that this modification promotes association of β-arrestin2 with AP-2 (33). Like β-arrestin2, ARH binds to the β2-adaptin component of AP-2 (5), suggesting that nitrosylation may promote binding of ARH to AP-2. We tested this possibility using immunoprecipitation and found that ARH efficiently coprecipitated AP-2 from lysates of WT, C199A, and C286 cells, but not from lysates of CC>AA cells (Fig. 6). Clathrin also coprecipitated better with WT ARH, ARH-C199A, and ARH-C286A than with ARH-CC>AA. Pretreatment of cells with L-NAME reduced the amount of AP-2 and clathrin coprecipitating with ARH in WT, C199A, and C286A cells to the level observed with CC>AA cells. We confirmed the preferential interaction of AP-2 with nitrosylated ARH by immunoprecipitating AP-2 and comparing coprecipitation of ARH. Immunoprecipitation of AP-2 coprecipitated more ARH from lysates of WT, C199A, and C286 cells than from CC>AA cells or from cells treated with L-NAME. AP-2 has its own binding sites for clathrin heavy chain, and coprecipitation of clathrin with AP-2 was unaffected by L-NAME treatment or mutation of ARH. No differences were observed among WT ARH, ARH-C199A, and ARH-C286A with regards to association with AP-2 in either set of immunoprecipitation. These observations indicate that nitrosylation of either C199 or C286 is sufficient to facilitate the binding of ARH to AP-2.
Fig. 6.
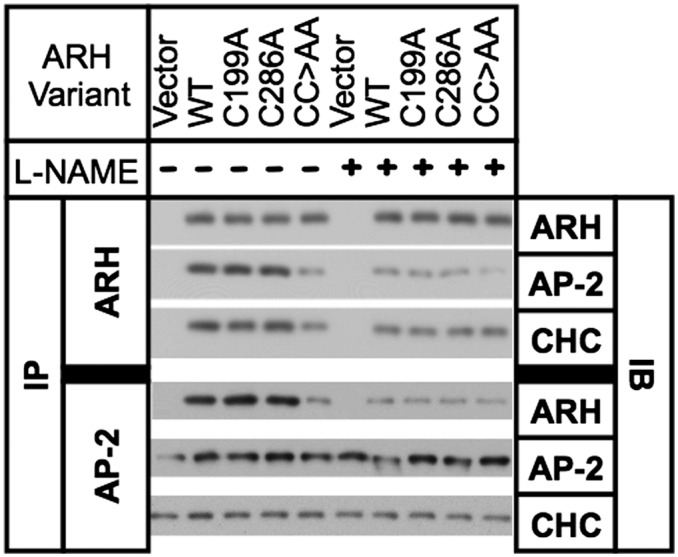
Nitrosylation is required for normal interaction of ARH with AP-2. Cells were treated or not with 4 mM L-NAME for 30 min, and then lysed and immunoprecipitated with antibodies to ARH or AP-2. Immunoprecipitants were run on SDS-PAGE and immunoblotted for the presence of ARH, AP-2, and clathrin heavy chain (CHC).
DISCUSSION
The central finding of this study is that nitric oxide regulates the ability of ARH to support LDL uptake. LDL uptake by the LDLR requires either ARH or dab2, which binds to AP-2, clathrin, and the FDNPVY sequence of the LDLR cytoplasmic domain. The combination of these three interactions is necessary for efficient targeting LDL-LDLR complexes to coated pits for endocytosis (10, 13, 34). The ability of ARH to associate strongly with AP-2 requires that ARH be nitrosylated at either C199 or C286 (Figs. 1F and 6). Failure to nitrosylate ARH cripples the ability of ARH to cluster LDL-LDLR complexes in coated pits (Fig. 5A, B) and impairs LDL uptake (Figs. 3 and 5). C199 and C286 can form a disulfide bond in cells (Fig. 1), and nitrosylation may catalyze this disulfide bond formation. Together, these findings suggest that ARH cycles between an inactive un-nitrosylated state, an active nitrosylated state, and a disulfide-bonded state (Fig. 7). Interchange between these states may depend upon NOS activity, denitrosylation activities, and the cellular redox state.
Fig. 7.

Model for regulation of ARH activity. We propose that ARH exists in three states: a free sulfhydryl state, a nitrosylated state, and a disulfide-bonded state. The free sulfhydryl form can react with nitric oxide produced by NOS enzymes to generate the active, nitrosylated form of ARH. Nitrosylation catalyzes disulfide bond formation, leading to inactivation of ARH. Cytosolic reductases (e−) reduce disulfide-bonded ARH to the free sulfhydryl form. Active ARH is presented as a hexagon, and inactive ARH is presented as a square.
ARH binds to the β2-adaptin component of AP-2 through a sequence that has homology with the AP-2-binding sequence of β-arrestins. Structural studies have shown that the AP-2-binding sequences of both ARH and the β-arrestins adopt an α-helical conformation when bound to β2-adaptin (35–37). In the case of β-arrestins, the AP-2-binding activity is regulated by GPCRs. When β-arrestins are in the inactive/basal state, their AP-2-binding sequences are stretched into a β-strand conformation (38, 39). Interaction of β-arrestins with active, phosphorylated GPCRs induces a conformational change in β-arrestins that allows the AP-2-binding sequence to coil into the active, α-helical conformation. In β-arrestin2, the C-terminal cysteine is nitrosylated in response to GPCR activation, and this nitrosylation event facilitates the conformational change that activates AP-2 binding (33). ARH may use an analogous mechanism to control AP-2-binding activity. While β-arrestins and ARH have no sequence conservation outside their AP-2-binding sites, ARH may hold its AP-2-binding sequence in an inactive, nonhelical state. S-nitrosylation at C199 and C286 may release the AP-2-binding sequence, allowing it to adopt the active, helical state.
In addition to activating AP-2 binding, S-nitrosylation at C199 and C286 may play a role in LDL internalization. Mutation of either cysteine slowed LDL internalization more than 2-fold (Fig. 5C). The contribution of C199 and C286 to LDL uptake was not at the level of coated-pit targeting because both C199A and C286A cells displayed normal LDL-gold enrichment in coated pits and because binding of AP-2 and clathrin to ARH-C199A and ARH-C286A were normal (Figs. 5 and 6). The contribution of these cysteines does not appear to influence the internalization of nonlipoprotein-loaded LDLRs because the surface LDLR levels on C199A and C286A cells were normal (Fig. 4). Only a fraction of LDL-LDLR complexes that reach coated pits are internalized through the budding portion of coated pits (40), and nitrosylation of both cysteines may help to anchor LDL-LDLR complexes in budding portion of coated pits. Importantly, the decreases in LDL internalization rate caused by the C199A and C286A mutations had only a small effect on overall LDL accumulation rates (Fig. 3). LDL accumulation is a function of LDL binding, LDL-LDLR trafficking to coated pits, internalization, LDL release, receptor recycling, and LDL resecretion. The differences between LDL accumulation and LDL internalization by WT, C199A, and C286A cells suggest that internalization is not rate limiting for LDL accumulation in WT cells.
Of the two cysteines that are nitrosylated in human ARH, only C286 is conserved across vertebrate species (supplementary Fig. III). The strong conservation of C286 from fish to primates suggests that nitrosylation at C286 plays a conserved role in ARH-supported LDL uptake; however, the lack of a cysteine near residue position 199 in nonprimate ARH proteins suggests that the function of C199 is primate-specific. Other species may use different cysteines to form a disulfide bond with C286, because mouse ARH has a fast-migrating form (17), despite the absence of a cysteine near position 199.
The most likely role for disulfide bond formation is denitrosylation. Most protein denitrosylation involves transnitrosylation events that transfer the nitric oxide moiety from a nitrosylated protein to either glutathione or thioredoxin. The nitric oxide moiety of glutathione and thioredoxin is then removed through a process that involves disulfide bond formation followed by disulfide bond cleavage (41). The ability of ARH to form an intramolecular disulfide bond may provide an intrinsic mechanism that accelerates denitrosylation of ARH.
Importantly, the involvement of nitric oxide is specific to ARH-supported LDL uptake. Neither dab2-supported LDL uptake nor VLDL remnant uptake are impaired by inhibitors of nitric oxide synthase activity (Fig. 3). The ability of dab2 to support LDL uptake in the absence of NOS activity may allow cells that express dab2 to support LDLR-dependent uptake of LDL when nitric oxide levels are low. For example, the adrenal glands and ovaries of humans use LDL-derived cholesterol to produce steroid hormones (42–44). Nitric oxide inhibits steroid production (45), and the high levels of dab2 expressed in steroidogenic tissues (46, 47) may facilitate LDL uptake when nitric oxide levels are low and cholesterol demand for steroid production is high. More generally, activation of ERK induces LDLR expression in dividing cells to supply the cholesterol needed for membrane biogenesis (48). The moderate levels of dab2 that are expressed in most tissues may allow dividing cells to use the LDLR to supply LDL-derived cholesterol irrespective of nitric oxide levels.
Liver hepatocytes express ARH but not dab2, suggesting that changes in nitric oxide production may dictate whether hepatic LDLRs internalize LDL. In humans, reductions in the rate of whole-body nitric oxide production correlate strongly with increased circulating LDL-cholesterol in age-matched individuals (49). Whole-body nitric oxide production also decreases with age (50, 51), coincident with a decrease in LDL clearance rates (52, 53). The ability of lipoproteins to inhibit eNOS activity (54, 55) likely explains part of the correlation between LDL-cholesterol and nitric oxide production; however, the correlation is much stronger in individuals with two normal LDLR alleles than in individuals with a defective LDLR allele (49, 56), suggesting that nitric oxide also promotes LDLR function. In support of this conclusion, transgenic overexpression of eNOS in apoE-deficient mice decreases LDL-cholesterol levels (57, 58). Because apoE is not required for LDLR-dependent LDL uptake, the reduction in LDL-cholesterol observed in the transgenic animals suggests that elevated nitric oxide improves LDL uptake by the LDLR. This improvement in LDLR function may involve nitrosylation of ARH.
Under normal conditions, hepatocytes may use nitric oxide to regulate whether the LDLR internalizes LDL or remnant particles. Whole body nitric oxide production falls sharply following the consumption of a large meal (59, 60) and this drop may reduce ARH nitrosylation, thereby focusing the endocytic activity of hepatic LDLRs on uptake of chylomicron and VLDL remnants, which flood the circulation in the postprandial state. Remnant particles are more atherogenic than LDL (61) and focusing the endocytic activity of the LDLR on remnants may help prevent atherosclerosis both by more rapidly reducing the circulating number of remnants and by preventing the conversion of VLDL remnants into LDL.
Peripheral blood leukocytes also express ARH, but not dab2 (14), and the absence of dab2 may protect leukocytes from excessive LDL uptake in atherosclerotic lesions. Atherosclerotic lesions are inflammatory responses to lipoprotein accumulation in the intima of arteries (62). As part of the inflammatory response, peripheral blood leukocytes extravasate into the lesion (63). Once in a lesion, leukocytes shut down LDLR function via transcriptional pathways that are triggered by the elevated cholesterol and oxysterols in the lesion. Both cholesterol and oxysterols suppress LDLR mRNA expression through inactivation of sterol response-element binding proteins (SREBP), which are transcription factors required for LDLR mRNA transcription (64). Elevation of oxysterols also promote LDLR turnover by activating liver X receptors (LXR), which are transcription factors that drive production of Idol/MYLIP, an E3-ubiquitin ligase that promotes lysosomal degradation of the LDLR (65). Because both processes are dependent upon changes in transcription, they require time for new protein synthesis, and changes in ARH function may complement transcriptional inactivation of LDLR function. Atherosclerotic lesions have abundant reactive oxygen species (ROS), which react with nitric oxide and reduce nitric oxide availability (66). Loss of nitric oxide inactivates ARH (Figs. 2–7) and may halt LDLR-dependent LDL uptake while LDLR mRNA and protein are being destroyed.
In summary, the data presented here show that LDL uptake by the LDLR requires S-nitrosylation of ARH. Nitrosylation promotes interaction of ARH with AP-2 and is required for ARH to target LDL-LDLR complexes to coated pits for internalization. β-VLDL uptake and dab2-supported LDL uptake are not dependent upon nitric oxide, indicating that the influence of nitric oxide on LDLR function is specific to ARH-supported LDL uptake. Thus, cells that express ARH, but not dab2, can reduce LDL uptake in response to reductions in nitric oxide without affecting either LDLR expression or LDLR-dependent uptake of VLDL remnants.
Supplementary Material
Acknowledgments
The authors thank Drs. Michael Brown and Joseph Goldstein (UT Southwestern) for providing human LDL and rabbit β-VLDL; Dr. Helen Hobbs (UT Southwestern) for providing ARH antibodies and Arh−/−;Dab2flox/flox;hLDLR+/+ mouse fibroblasts; and Dr. Joachim Herz (UT Southwestern) for providing LDLR antibodies and cre-recombinase adenovirus. The authors also thank Dr. Frank Graham (McMaster University) for consenting to our use of the cre-recombinase virus.
Footnotes
Abbreviations:
- AP-2
- adaptor protein 2
- ARH
- autosomal recessive hypercholesterolemia protein
- dab2
- disabled-like protein 2
- FACS
- fluorescence-activated cell sorting
- GPCR
- G-protein coupled receptor
- LDL-gold
- gold-conjugated LDL
- LDLR
- LDL receptor
- L-NAME
- l-NG-nitroarginine methyl ester
- NOS
- nitric oxide synthase
- PTB
- phosphotyrosine binding
- WT
- wild-type
This work was supported by National Institutes of Health Grant HL-085218 (to P.M.).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven figures.
REFERENCES
- 1.Ishibashi S., Brown M. S., Goldstein J. L., Gerard R. D., Hammer R. E., Herz J. 1993. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest. 92: 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.James R. W., Martin B., Pometta D., Fruchart J. C., Duriez P., Puchois P., Farriaux J. P., Tacquet A., Demant T., Clegg R. J., et al. 1989. Apolipoprotein B metabolism in homozygous familial hypercholesterolemia. J. Lipid Res. 30: 159–169 [PubMed] [Google Scholar]
- 3.Chen W. J., Goldstein J. L., Brown M. S. 1990. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 265: 3116–3123 [PubMed] [Google Scholar]
- 4.Anderson R. G., Goldstein J. L., Brown M. S. 1977. A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature. 270: 695–699 [DOI] [PubMed] [Google Scholar]
- 5.He G., Gupta S., Yi M., Michaely P., Hobbs H. H., Cohen J. C. 2002. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J. Biol. Chem. 277: 44044–44049 [DOI] [PubMed] [Google Scholar]
- 6.Mishra S. K., Watkins S. C., Traub L. M. 2002. The autosomal recessive hypercholesterolemia (ARH) protein interfaces directly with the clathrin-coat machinery. Proc. Natl. Acad. Sci. USA. 99: 16099–16104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra S. K., Keyel P. A., Hawryluk M. J., Agostinelli N. R., Watkins S. C., Traub L. M. 2002. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 21: 4915–4926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dvir H., Shah M., Girardi E., Guo L., Farquhar M. G., Zajonc D. M. 2012. Atomic structure of the autosomal recessive hypercholesterolemia phosphotyrosine-binding domain in complex with the LDL-receptor tail. Proc. Natl. Acad. Sci. USA. 109: 6916–6921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown M. S., Goldstein J. L. 1976. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell. 9: 663–674 [DOI] [PubMed] [Google Scholar]
- 10.Michaely P., Zhao Z., Li W. P., Garuti R., Huang L. J., Hobbs H. H., Cohen J. C. 2007. Identification of a VLDL-induced, FDNPVY-independent internalization mechanism for the LDLR. EMBO J. 26: 3273–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia C. K., Wilund K., Arca M., Zuliani G., Fellin R., Maioli M., Calandra S., Bertolini S., Cossu F., Grishin N., et al. 2001. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 292: 1394–1398 [DOI] [PubMed] [Google Scholar]
- 12.Morris S. M., Tallquist M. D., Rock C. O., Cooper J. A. 2002. Dual roles for the Dab2 adaptor protein in embryonic development and kidney transport. EMBO J. 21: 1555–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keyel P. A., Mishra S. K., Roth R., Heuser J. E., Watkins S. C., Traub L. M. 2006. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol. Biol. Cell. 17: 4300–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eden E. R., Patel D. D., Sun X. M., Burden J. J., Themis M., Edwards M., Lee P., Neuwirth C., Naoumova R. P., Soutar A. K. 2002. Restoration of LDL receptor function in cells from patients with autosomal recessive hypercholesterolemia by retroviral expression of ARH1. J. Clin. Invest. 110: 1695–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuliani G., Arca M., Signore A., Bader G., Fazio S., Chianelli M., Bellosta S., Campagna F., Montali A., Maioli M., et al. 1999. Characterization of a new form of inherited hypercholesterolemia: familial recessive hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 19: 802–809 [DOI] [PubMed] [Google Scholar]
- 16.Arca M., Zuliani G., Wilund K., Campagna F., Fellin R., Bertolini S., Calandra S., Ricci G., Glorioso N., Maioli M., et al. 2002. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet. 359: 841–847 [DOI] [PubMed] [Google Scholar]
- 17.Jones C., Hammer R. E., Li W. P., Cohen J. C., Hobbs H. H., Herz J. 2003. Normal sorting but defective endocytosis of the low density lipoprotein receptor in mice with autosomal recessive hypercholesterolemia. J. Biol. Chem. 278: 29024–29030 [DOI] [PubMed] [Google Scholar]
- 18.Jones C., Garuti R., Michaely P., Li W. P., Maeda N., Cohen J. C., Herz J., Hobbs H. H. 2007. Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia. J. Clin. Invest. 117: 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada-Shiba M., Takagi A., Marutsuka K., Moriguchi S., Yagyu H., Ishibashi S., Asada Y., Yokoyama S. 2004. Disruption of autosomal recessive hypercholesterolemia gene shows different phenotype in vitro and in vivo. Circ. Res. 95: 945–952 [DOI] [PubMed] [Google Scholar]
- 20.Rosenbauer F., Kallies A., Scheller M., Knobeloch K. P., Rock C. O., Schwieger M., Stocking C., Horak I. 2002. Disabled-2 is transcriptionally regulated by ICSBP and augments macrophage spreading and adhesion. EMBO J. 21: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Sun Y., Constantinescu S. N., Karam E., Weinberg R. A., Lodish H. F. 1997. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA. 94: 10669–10674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lombardi P., Mulder M., van der Boom H., Frants R. R., Havekes L. M. 1993. Inefficient degradation of triglyceride-rich lipoprotein by HepG2 cells is due to a retarded transport to the lysosomal compartment. J. Biol. Chem. 268: 26113–26119 [PubMed] [Google Scholar]
- 23.Zhao Z., Michaely P. 2008. The epidermal growth factor homology domain of the LDL receptor drives lipoprotein release through an allosteric mechanism involving H190, H562, and H586. J. Biol. Chem. 283: 26528–26537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolton A. E., Hunter W. M. 1973. The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acylating agent. Biochem. J. 133: 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein J. L., Basu S. K., Brown M. S. 1983. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98: 241–260 [DOI] [PubMed] [Google Scholar]
- 26.Jaffrey S. R., Snyder S. H. 2001. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE. 2001: pl1. [DOI] [PubMed] [Google Scholar]
- 27.Handley D. A., Arbeeny C. M., Witte L. D., Chien S. 1981. Colloidal gold–low density lipoprotein conjugates as membrane receptor probes. Proc. Natl. Acad. Sci. USA. 78: 368–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michaely P., Li W. P., Anderson R. G., Cohen J. C., Hobbs H. H. 2004. The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J. Biol. Chem. 279: 34023–34031 [DOI] [PubMed] [Google Scholar]
- 29.Arnelle D. R., Stamler J. S. 1995. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch. Biochem. Biophys. 318: 279–285 [DOI] [PubMed] [Google Scholar]
- 30.Jiang Z. L., Zhu X., Diamond M. P., Abu-Soud H. M., Saed G. M. 2008. Nitric oxide synthase isoforms expression in fibroblasts isolated from human normal peritoneum and adhesion tissues. Fertil. Steril. 90: 769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reif D. W., McCreedy S. A. 1995. N-nitro-L-arginine and N-monomethyl-L-arginine exhibit a different pattern of inactivation toward the three nitric oxide synthases. Arch. Biochem. Biophys. 320: 170–176 [DOI] [PubMed] [Google Scholar]
- 32.Brown M. S., Anderson R. G., Basu S. K., Goldstein J. L. 1982. Recycling of cell-surface receptors: observations from the LDL receptor system. Cold Spring Harb. Symp. Quant. Biol. 46: 713–721 [DOI] [PubMed] [Google Scholar]
- 33.Ozawa K., Whalen E. J., Nelson C. D., Mu Y., Hess D. T., Lefkowitz R. J., Stamler J. S. 2008. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol. Cell. 31: 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boucrot E., Saffarian S., Zhang R., Kirchhausen T. 2010. Roles of ap-2 in clathrin-mediated endocytosis. PLoS ONE. 5: e10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edeling M. A., Mishra S. K., Keyel P. A., Steinhauser A. L., Collins B. M., Roth R., Heuser J. E., Owen D. J., Traub L. M. 2006. Molecular switches involving the AP-2 beta2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev. Cell. 10: 329–342 [DOI] [PubMed] [Google Scholar]
- 36.Mishra S. K., Keyel P. A., Edeling M. A., Dupin A. L., Owen D. J., Traub L. M. 2005. Functional dissection of an AP-2 beta2 appendage-binding sequence within the autosomal recessive hypercholesterolemia protein. J. Biol. Chem. 280: 19270–19280 [DOI] [PubMed] [Google Scholar]
- 37.Schmid E. M., Ford M. G., Burtey A., Praefcke G. J., Peak-Chew S. Y., Mills I. G., Benmerah A., McMahon H. T. 2006. Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLoS Biol. 4: e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milano S. K., Pace H. C., Kim Y. M., Brenner C., Benovic J. L. 2002. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 41: 3321–3328 [DOI] [PubMed] [Google Scholar]
- 39.Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., Schubert C. 2001. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane Translocation. Structure. 9: 869–880 [DOI] [PubMed] [Google Scholar]
- 40.Anderson R. G., Brown M. S., Goldstein J. L. 1977. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell. 10: 351–364 [DOI] [PubMed] [Google Scholar]
- 41.Lima B., Forrester M. T., Hess D. T., Stamler J. S. 2010. S-nitrosylation in cardiovascular signaling. Circ. Res. 106: 633–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carr B. R., Parker C. R. J., Milewich L., Porter J. C., MacDonald P. C., Simpson E. R. 1980. The role of low density, high density, and very low density lipoproteins in steroidogenesis by the human fetal adrenal gland. Endocrinology. 106: 1854–1860 [DOI] [PubMed] [Google Scholar]
- 43.Carr B. R., Sadler R. K., Rochelle D. B., Stalmach M. A., MacDonald P. C., Simpson E. R. 1981. Plasma lipoprotein regulation of progesterone biosynthesis by human corpus luteum tissue in organ culture. J. Clin. Endocrinol. Metab. 52: 875–881 [DOI] [PubMed] [Google Scholar]
- 44.Benahmed M., Reventos J., Saez J. M. 1983. Steroidogenesis of cultured purified pig Leydig cells: effects of lipoproteins and human chorionic gonadotropin. Endocrinology. 112: 1952–1957 [DOI] [PubMed] [Google Scholar]
- 45.Ducsay C. A., Myers D. A. 2011. eNOS activation and NO function: differential control of steroidogenesis by nitric oxide and its adaptation with hypoxia. J. Endocrinol. 210: 259–269 [DOI] [PubMed] [Google Scholar]
- 46.Fazili Z., Sun W., Mittelstaedt S., Cohen C., Xu X. X. 1999. Disabled-2 inactivation is an early step in ovarian tumorigenicity. Oncogene. 18: 3104–3113 [DOI] [PubMed] [Google Scholar]
- 47.Romero D. G., Yanes L. L., de Rodriguez A. F., Plonczynski M. W., Welsh B. L., Reckelhoff J. F., Gomez-Sanchez E. P., Gomez-Sanchez C. E. 2007. Disabled-2 is expressed in adrenal zona glomerulosa and is involved in aldosterone secretion. Endocrinology. 148: 2644–2652 [DOI] [PubMed] [Google Scholar]
- 48.Li C., Kraemer F. B., Ahlborn T. E., Liu J. 1999. Induction of low density lipoprotein receptor (LDLR) transcription by oncostatin M is mediated by the extracellular signal-regulated kinase signaling pathway and the repeat 3 element of the LDLR promoter. J. Biol. Chem. 274: 6747–6753 [DOI] [PubMed] [Google Scholar]
- 49.Maas R., Schwedhelm E., Kahl L., Li H., Benndorf R., Luneburg N., Forstermann U., Boger R. H. 2008. Simultaneous assessment of endothelial function, nitric oxide synthase activity, nitric oxide-mediated signaling, and oxidative stress in individuals with and without hypercholesterolemia. Clin. Chem. 54: 292–300 [DOI] [PubMed] [Google Scholar]
- 50.Virdis A., Ghiadoni L., Giannarelli C., Taddei S. 2010. Endothelial dysfunction and vascular disease in later life. Maturitas. 67: 20–24 [DOI] [PubMed] [Google Scholar]
- 51.Vanhoutte P. M., Shimokawa H., Tang E. H., Feletou M. 2009. Endothelial dysfunction and vascular disease. Acta Physiol. (Oxf.). 196: 193–222 [DOI] [PubMed] [Google Scholar]
- 52.Miller N. E. 1984. Why does plasma low density lipoprotein concentration in adults increase with age? Lancet. 1: 263–267 [DOI] [PubMed] [Google Scholar]
- 53.Kreisberg R. A., Kasim S. 1987. Cholesterol metabolism and aging. Am. J. Med. 82: 54–60 [DOI] [PubMed] [Google Scholar]
- 54.Andrews H. E., Bruckdorfer K. R., Dunn R. C., Jacobs M. 1987. Low-density lipoproteins inhibit endothelium-dependent relaxation in rabbit aorta. Nature. 327: 237–239 [DOI] [PubMed] [Google Scholar]
- 55.Hayashi T., Naito M., Ishikawa T., Kuzuya M., Funaki C., Tateishi T., Asai K., Hidaka H., Kuzuya F. 1989. Beta-migrating very low density lipoprotein attenuates endothelium-dependent relaxation in rabbit atherosclerotic aortas. Blood Vessels. 26: 290–299 [DOI] [PubMed] [Google Scholar]
- 56.Wever R., Boer P., Hijmering M., Stroes E., Verhaar M., Kastelein J., Versluis K., Lagerwerf F., van Rijn H., Koomans H., et al. 1999. Nitric oxide production is reduced in patients with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 19: 1168–1172 [DOI] [PubMed] [Google Scholar]
- 57.van Haperen R., de Waard M., van Deel E., Mees B., Kutryk M., van Aken T., Hamming J., Grosveld F., Duncker D. J., de Crom R. 2002. Reduction of blood pressure, plasma cholesterol, and atherosclerosis by elevated endothelial nitric oxide. J. Biol. Chem. 277: 48803–48807 [DOI] [PubMed] [Google Scholar]
- 58.Ozaki M., Kawashima S., Yamashita T., Hirase T., Namiki M., Inoue N., Hirata K., Yasui H., Sakurai H., Yoshida Y., et al. 2002. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Invest. 110: 331–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marchesi S., Lupattelli G., Siepi D., Roscini A. R., Vaudo G., Sinzinger H., Mannarino E. 2001. Oral L-arginine administration attenuates postprandial endothelial dysfunction in young healthy males. J. Clin. Pharm. Ther. 26: 343–349 [DOI] [PubMed] [Google Scholar]
- 60.Magne J., Huneau J. F., Delemasure S., Rochette L., Tome D., Mariotti F. 2009. Whole-body basal nitric oxide production is impaired in postprandial endothelial dysfunction in healthy rats. Nitric Oxide. 21: 37–43 [DOI] [PubMed] [Google Scholar]
- 61.Goldstein J. L., Ho Y. K., Brown M. S., Innerarity T. L., Mahley R. W. 1980. Cholesteryl ester accumulation in macrophages resulting from receptor-mediated uptake and degradation of hypercholesterolemic canine beta-very low density lipoproteins. J. Biol. Chem. 255: 1839–1848 [PubMed] [Google Scholar]
- 62.Tabas I. 2010. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 10: 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lahoute C., Herbin O., Mallat Z., Tedgui A. 2011. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat. Rev. Cardiol. 8: 348–358 [DOI] [PubMed] [Google Scholar]
- 64.Horton J. D., Goldstein J. L., Brown M. S. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109: 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zelcer N., Hong C., Boyadjian R., Tontonoz P. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 325: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kojda G., Harrison D. 1999. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res. 43: 562–571 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.