

Abstract
Significant advances in the use of metal complexes, precipitated by platinum, have fostered a renewed interest in harnessing their rich potential in the treatment of cancer. In addition to platinum-based complexes, the anticancer properties of other metals such as ruthenium have been realized, and ruthenium-based compounds are currently being investigated in clinical trials. Since the process of drug development can be expensive and cumbersome, finding new applications of existing drugs may provide effective means to expedite the regulatory process in bringing new drugs to the clinical setting. Encouraging findings from laboratory studies reveal significant anticancer activity from different classes of metal-chelating compounds, such as disulfiram, clioquinol, and dithiocarbamate derivatives that are currently approved for the treatment of various pathological disorders. Their use as coordination complexes with metals such as copper, zinc, and gold that target the ubiquitin-proteasome pathway have shown significant promise as potential anticancer agents. This review discusses the unique role of several selected metals in relation to their anti-cancer properties as well as the new therapeutic potential of several previously approved metal-chelating drugs. In vitro and in vivo experimental evidence along with mechanisms of action (e.g., via targeting the tumor proteasome) will also be discussed with anticipation of strengthening this exciting new concept.
Keywords: Disulfiram, Clioquinol, Dithiocarbamates, Copper, Zinc, Ubiquitin-Proteasome Pathway, Proteasome Inhibitor, Chymotrypsin-Like Activity, Review
2. INTRODUCTION
2.1. The use of metal complexes for cancer treatment
The development of metal-based complexes for the treatment of cancer began with the discovery of the anti-cancer properties of cisplatin in the early 1960s (Figure 1). Over 90% of testicular cancer cases have been cured by cisplatin, and it has also been important in the treatment of several other types of cancer, including ovarian, cervical, bladder, head and neck, melanoma, and lymphomas (1). Cisplatin interacts with DNA and forms adducts which interfere with the replication and transcription processes, and ultimately triggers apoptosis (2). These cisplatin-DNA interactions have been extensively studied, and it has been clearly shown that a (Pt)-GG intrastrand cross-link is responsible for the cytotoxicity of cisplatin (3). However, the toxicities associated with cisplatin, coupled with intrinsic and acquired drug resistance, have hampered its widespread clinical use (4–5).
Figure 1.
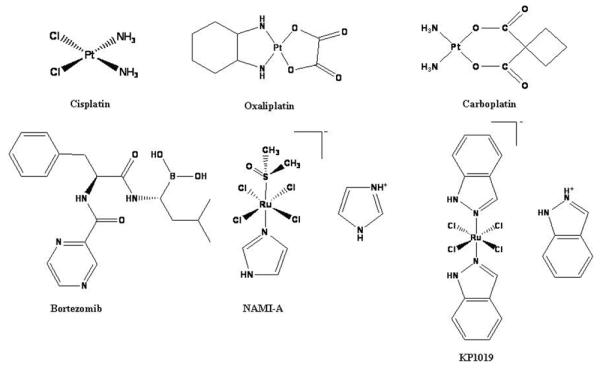
Chemical structures of platinum- and ruthenium-based drugs, as well as the proteasome inhibitor bortezomib, used in the treatment of human cancers.
Chemical structure of bortezomib, the first proteasome inhibitor clinically tested is shown. Cisplatin, oxaliplatin, and carboplatin are platinum-containing drugs used for the treatment of human cancers. NAMI-A and KP1019 are ruthenium-based drugs under investigation for the potential treatment of human cancers.
The limitations of cisplatin have stimulated the search for new, less toxic platinum-based drugs. Second and third generation platinum-based drugs, including carboplatin and oxaliplatin (Figure 1), have been developed as alternatives to increase efficacy and offset the toxicity associated with cisplatin (6). Carboplatin is an effective treatment for ovarian, lung, and head and neck cancers (7), and oxaliplatin is clinically approved for the treatment of cisplatin-resistant colorectal cancer (8). The investigation into other metal-based drugs began by the rational design of complexes that are structurally similar to cisplatin, the theory being that similar structure leads to similar function (Figure 1) (9). However, the activities of these metal compounds are not solely governed by the presence of the metal itself, but can be highly influenced by the oxidation state, number and type of ligand bound, and the coordination geometry of the complex. Other properties that can play a pivotal role in the biological activity of metal complexes include kinetic lability, redox behavior, and electric charge. These unique properties have prompted investigation into metal-based complexes as potential anticancer agents that present with various mechanisms of action.
2.2. Ruthenium-based complexes as potential anticancer agents
The severe side effects and resistance associated with platinum-based drugs have spurred a search for non-platinum metals that may decrease the toxicities associated with metal-based drugs. Because it is a transition metal of group 8B of the periodic table, the same group as platinum, ruthenium's anticancer effects were originally believed to be exerted by direct binding to DNA, the same mechanism by which platinum agents cause cell death. However, it has been demonstrated that ruthenium exhibits many distinct properties at the cellular level compared to platinum. First, ruthenium seems to preferentially accumulate in malignant cells rather than normal cells, possibly by utilizing a transferrin-mediated mode of transport (10). Additionally, prior to reaching the tumor mass, ruthenium remains in its inactive Ru (III) oxidation state. The lower oxygen level and higher acidity of the tumor environment reduce the ruthenium to its more reactive Ru (II) state (11). It has also been found that some ruthenium complexes display greater efficacy toward tumor cell metastases rather than acting on primary tumors. This is believed to be due to inhibition of tumor cell detachment, invasion and migration, and re-adhesion to new growth substrate (12). In light of these properties, ruthenium is predicted to show distinct patterns of anti-tumor activity that diverge from those demonstrated with platinum.
Currently, two ruthenium-containing complexes are undergoing clinical trials, NAMI-A and KP1019 (Figure 1) (13–14). Despite their structural similarities, these ruthenium (III) complexes differ drastically in their antitumor activities. Preclinical studies have demonstrated that NAMI-A is able to inhibit the formation of metastases in various animal tumor models but shows no direct cytotoxic effects (15–16). KP1019 however, has shown direct antitumor effects against a wide variety of tumor xenografts through induction of apoptosis (17–18). Despite encouraging preclinical and clinical data, the complete mechanisms of action still remain unresolved.
3. IMPORTANCE OF METALS IN THE GROWTH AND PROGRESSION OF CANCER
3.1. Copper
The discovery that copper levels in tumor-bearing mice and humans are altered (19–20) led to extensive studies regarding the role of copper in carcinogenesis. High serum and tissue levels of copper have been observed in a variety of human tumors including brain (21), breast (22–23), colon (24), lung (25), and prostate (24, 26). In 1980, it was first noticed that Cu played a critical role in angiogenesis (27). Gullino et al found that in the corneas of rabbits' eyes, new capillaries developed when angiogenesis effectors became rich in Cu ions (28). Results from cell culture studies showed that Cu could stimulate proliferation and migration of human endothelial cells (29). Vascular endothelial growth factor (VEGF) is a key regulator of angiogenesis. VEGF can stimulate growth promotion, migration and differentiation of endothelial cells from existing blood vessels (30). Studies in cell cultures and animal models have demonstrated that Cu is able to induce VEGF mRNA transcription and protein expression (31–32). Copper, but not other metals, is a co-factor required for several angiogenic mediators including VEGF (32), basic fibroblast growth factor (bFGF) (33), interleukin 1 (IL-1) and interleukin 8 (IL-8) (34), all of which are essential regulators for tumor angiogenesis (35–38). Tumors are dependent on angiogenesis for their growth, invasion and metastasis (39–40), and Cu plays an important role in this process. Due to the importance of angiogenesis and copper to tumor development, the use of copper chelators for antiangiogenic therapy has emerged as an interesting concept in cancer therapeutics (41–42).
3.2. Zinc
Like copper, zinc also plays an important role in many cellular processes, including proliferation and differentiation, as well as defense against free radicals (43–44). Zinc is also a structural component in various proteins and enzymes such as transcription factors, cell signaling proteins, and DNA repair enzymes (45–46). Additionally, a critical role has been suggested for zinc in apoptosis (47–49). However, this effect appears to be complex, and no firm conclusions have been established. For example, in prostate and ovarian epithelial, as well as glial cells, zinc is pro-apoptotic, while in breast, HeLa, renal, and lung epithelial cells, as well as macrophages, zinc is anti-apoptotic (49–50). Altered Zn levels have been found to be associated with certain systemic abnormalities such as the development of cancer (51). Although Zn levels are often compromised in cancer patients, a firm relationship between cancer development and Zn has not been proven, and seems dependent on tumor type (49, 52–53). Low levels of zinc have been observed in several malignancies, such as those of the liver, gallbladder, digestive tract, and prostate (54–56). Conversely, both high and low levels of zinc have been found in breast cancers (54, 57–58).
Consequently, it is no surprise that an association between zinc transporter levels and cancer progression has also been proposed (51, 59). Multiple zinc transporters, including ZIP4, ZIP6, ZIP10, and ZIP1, have been identified as factors in the progression of various types of cancer. ZIP4 has been reported to increase cell proliferation through zinc transport, resulting in tumor growth, most specifically in pancreatic cancer (44, 55). Both ZIP6 and ZIP10 have roles in the progression and metastasis of breast cancer (46, 57–58) and ZIP1 has been suggested as a tumor suppressor of prostate cancer (56). Thus, by disrupting the distribution of zinc in tissues, altered levels of zinc transporters may enhance the development of various tumors, indicating the potential of zinc as an anticancer agent.
4. UBIQUITIN-PROTEASOME PATHWAY
The ubiquitin-proteasome pathway (UPP) is so important to normal cellular function that, in 2004, the Nobel Prize in Chemistry was awarded to its discoverers (60–61). The ubiquitin-proteasome pathway (Figure 2) is responsible for selective proteolytic processing of proteins involved in various biological processes, such as development, differentiation, proliferation, signal transduction, and apoptosis (62). There are two critical steps in the ubiquitin-proteasome pathway: (i) conjugation of multiple ubiquitin molecules to the target protein, and (ii) degradation of the tagged protein by the 26S proteasome (63). The 26S proteasome is a large (2.5 MDa), multi-subunit complex found in the nucleus and cytosol of cells and consists of a catalytic core, the 20S proteasome, and two recognition sites, the 19S regulatory caps (Figure 2) (64–65). The 20S core is made up of four stacked rings, two non-catalytic alpha rings (seven subunits each) outside of two catalytic beta rings (seven subunits each), that form a barrel-like structure, consisting of 28 subunits total (66–68). While the function of the alpha subunits is to block direct access to the proteasomal active site by allowing access only to unfolded proteins, the beta subunits are responsible for the proteolytic activities of the proteasome (68). The active beta subunits are beta-1, beta-2, and beta-5, which are responsible for the caspase or peptidyl-glutamyl peptide-hydrolyzing (PGPH)-like, trypsin-like, and chymotrypsin (CT)-like activities, respectively (68–69). Each active subunit contains a Thr1 active residue at the amino terminal, which is responsible for catalysis. It is this active site that can be targeted by some proteasome inhibitors (such as Bortezomib) (Figure 1) through nucleophilic attack (67, 70). Additionally, the 19S regulatory particle (700 kDa) contains six ATPase and at least eight non-ATPase subunits, which are required for recognition, deubiquitination, unfolding, and translocation of tagged proteins prior to degradation by the 20S proteasome (71–72).
Figure 2.

Schematic representation of the ubiquitin-proteasome pathway (UPP).
The UPP is a highly regulated ATP-dependent pathway, which is vital for the processing of intracellular proteins. It is also a promising target for anticancer therapeutics, and many metal-based or metal-binding compounds, such as CQ, DSF and Au-complexes, have been shown to be potent inhibitors of the proteasomal activity.
The ubiquitination step of the UPP is typified by three different enzymes, E1, E2, and E3. The UPP pathway is initiated by ATP-dependent E1-mediated activation of ubiquitin, a 76 amino acid protein that is expressed ubiquitously and serves as a tag for target proteins destined for degradation by the UPP (Figure 2). Transfer of activated ubiquitin from E1 to E2, which is responsible for conjugation of ubiquitin, and E3, the ubiquitin-ligating enzyme, which then facilitates the transfer of active ubiquitin to lysine residues of the target protein (73–74). The ubiquitin-tagged target protein is then transported to the 26S proteasome, where degradation occurs and the ubiquitin is released for recycling (Figure 2) (75). This is a tightly regulated process and is important for the regulation of several cellular processes, including those involved in tumorigenesis (76).
Because of the essential role that unbalanced protein homeostasis plays in the development, growth, and survival of cancer (77), targeting factors responsible for the synthesis and degradation of proteins as an anticancer strategy has been investigated (78). Increased proteasome activity has been observed in various malignancies, such as prostate (79), colon (80), and leukemia (81), indicating that cancer cells are more dependent on the ubiquitin-proteasome pathway than normal cells and that targeting the UPP is a viable option in the treatment of human cancer. Importantly, inhibition of the CT-like activity of the tumor proteasome is associated with cell cycle arrest and induction of apoptosis (82–83), suggesting that proteasome inhibition may be effective in not only selectively killing cancer cells with minimal effect on healthy cells but also in sensitizing resistant cancer cells to chemotherapy or radiotherapy (84).
Bortezomib (Velcade, PS-341), a dipeptide boronic acid derivative, was the first proteasome inhibitor approved for clinical use by the FDA (Figure 1). It demonstrates potent apoptosis-inducing ability in various cancer cell lines and animal models (85–87) and is currently used for the treatment of multiple myeloma and mantle cell lymphoma as well as other cancers (88–89). Bortezomib is a slowly reversible inhibitor that induces cell death through direct inhibition of the beta-5 proteasomal subunit (90), which leads to suppression of NF-kB activity, causing down-regulation of its target genes (85, 91). In a series of animal studies, bortezomib was shown to inhibit tumor growth and angiogenesis in various solid tumors, including prostate (92), lung, breast (93), mesothelioma (94), and neuroblastoma (95). Phase I, II, and III clinical trials showed favorable responses in NHL, AML, and MM patients to treatment with bortezomib alone or in combination with various chemotherapeutics and indicated a significant clinical benefit (96–98). While bortezomib is highly effective against several hematological malignancies, it has exhibited little activity toward solid tumors, and development of resistance has been observed. Furthermore, most recently, it has been found that the proteasome-inhibitory and anticancer activity of bortezomib and other boronic acid-based proteasome inhibitors can be blocked by green tea polyphenols via direct drug-drug interactions (99–100). These observations, along with the toxicities associated with bortezomib, have prompted the development of next generation proteasome inhibitors with a more favorable therapeutic profile and a broader spectrum of activity. Given the significant achievement of platinum-based anticancer therapy, the use of different metals, especially when complexed with previously approved medicinal compounds that target the ubiquitin-proteasome pathway has received considerable attention as a viable route in cancer treatment.
5. DITHIOCARBAMATES
One class of medicinally important metal-chelating compounds is dithiocarbamates. This class of medicine includes several drugs that have previously been approved for the treatment of various ailments, such as bacterial and fungal infections, as well as AIDS (101–102).
5.1. Gold dithiocarbamates
The medicinal applications of gold date back thousands of years, but its rational use did not begin until the early twentieth century when Robert Koch found that K (Au (CN)2) could kill the tuberculosis bacteria. However, serious side effects were observed and the treatment for tuberculosis was changed to the less-toxic Au (I)-thiolate complexes. Jacque Forestier also used these gold complexes for the treatment of rheumatoid arthritis, and they remained the drug of choice for many years (103). The severe toxicity of these gold-based drugs prompted the search for novel, less toxic gold compounds. Because the coordination of gold (I) with phosphine ligands stabilizes the 1+ oxidation state, Au (I)-phosphine complexes have been investigated. This led to the discovery of auranofin (Figure 3), which, until only recently, was the drug of choice to treat rheumatoid arthritis (104).
Figure 3.

Chemical structures of gold-based compounds.
Auranofin, one of the first clinically used gold-based drugs, prompted the search for novel gold- compounds that may be effective in the treatment of various pathological disorders such as cancer. Au(DMDT)Br2, AUL12, and AUL15 are gold compounds with proteasome-inhibitory activity.
In an effort to broaden the medicinal applications of gold, gold compounds have been investigated for potential anticancer activity. Gold (I) complexes, including auranofin analogs, were synthesized and potent cytotoxic activity against B16 melanoma and P388 leukemia cells was observed (105). Phosphine-gold (I) thiosugars were the most potent, and while anti-tumor activity against leukemia was seen in vivo, these analogs were completely inactive against solid tumors (106). Digold (I) phosphine complexes were also found to confer cytotoxic activity in cisplatin-resistant cells in vitro. This activity is thought to be due to the ability of these complexes to alter mitochondrial function and inhibit protein synthesis (107). Like the previous gold (I)-phosphine thiolate sugars, these complexes were also inactive against solid tumors. Digold (I)-phosphines did not enter clinical trials due to their severe cardiotoxicity (108).
Although their use as anticancer drugs was originally questioned due to high redox activity and poor stability, gold (III) complexes were also investigated. Because the cellular environment is generally reducing, Au (III) is expected to be reduced to Au (I) and metallic Au, making Au (III) complexes less effective (109). Interest in these complexes increased after Pt (II) complexes showed positive results, because Au (III) and Pt (II) are isoelectronic and tetracoordinate gold (III) complexes share the same square planar geometry as cisplatin (110). Many complexes were synthesized and tested against a variety of human cancers, including cisplatin-resistant cell lines (108). Importantly, recent evidence suggests that the cellular proteasome is a molecular target for gold complexes (see the next section) and the mechanism of action underlying their activity is only beginning to emerge.
Recently, various gold (III) compounds exhibiting greater stability have been synthesized using ligand platforms containing nitrogen atoms as donor groups (111). These newer compounds exhibit a superior chemotherapeutic index than cisplatin due to greater bioavailabilty, increased cytotoxicity, and fewer toxic side effects (109). Dithiocarbamates have been evaluated for their efficacy as inhibitors of cisplatin-induced nephrotoxicity (112–113) and have since been tested for in vitro cytotoxicity toward a variety of human tumor cell lines. Most, particularly derivatives of N,N-dimethyldithiocarbamate and ethylsarcosinedithiocarbamate such as (Au (DMDT)Cl2), (Au (DMDT)Br2), (Au (ESDT)Cl2), and (Au (ESDT)Br2), were demonstrated to be 1–4 fold more potent than cisplatin and were able to overcome intrinsic and acquired cisplatin resistance (109). These dithiocarbamates act fast to inhibit RNA and DNA synthesis and show only minimal cross-resistance with cisplatin (110), suggesting a different mechanism of action.
In an attempt to discern a possible mechanism of action, our lab selected Au (DMDT)Br2 (Figure 3) and tested its proteasome-inhibitory potential. We reported that the CT-like activity of the purified 20S proteasome (IC50= 7.4 microM) and 26S proteasome in intact MDA-MB-231 breast cancer cells (10–20 microM) was significantly inhibited by Au (DMDT)Br2. PGPH-like and trypsin-like activities were also inhibited, but the CT-like inhibition was the most significant, indicating that this complex preferentially binds to and inhibits the CT-like beta-5 subunit of the proteasome. Associated with proteasomal inhibition, an accumulation of ubiquitinated proteins and p27 as well as induction of apoptosis was observed in the breast cancer cells. Considerable inhibition of proliferation (80–90%) was also seen in several breast cancer cell lines, including MCF10AT1K.c12, MCF10dcis.com, MCF-7, and MDA-MD-231. Additionally, Au (DMDT)Br2 was able to potently inhibit tumor growth (~50%) associated with inhibition of proteasomal CT-like activity (40%) in breast cancer xenografts (114).
Our lab has also investigated the effect of two gold compounds with different oxidation states (Figure 3) toward the cellular proteasome, and endeavored to gain insight into their potential mechanism of action. We first compared the effects of a gold (I) compound (Au (ESDT)2), AUL15, to a gold (III) compound (AuBr2 (ESDT)), AUL12, toward growth inhibition of breast cancer cells (Figure 3). We found that while both inhibited the growth of MDA-MB-231 breast cancer cells, AUL12 was much more potent (IC50= 4.5 microM, 70% inhibition) than AUL15 (IC50= 13.5 microM, 35% inhibition). We also observed that both complexes were able to inhibit purified 20S proteasome (AUL12 IC50= 1.13 microM; AUL15 IC50= 17.7 microM) as well as intact 26S proteasome, again with AUL12 exhibiting much higher activity (115). Additionally, we observed that AUL15 inhibited the cellular proteasome much later (> 24 hr) compared to AUL12 (4 hr) in intact breast cancer cells. Associated with these effects was the accumulation of ubiquitinated proteins and IkB-alpha as well as induction of cell death as demonstrated by PARP cleavage and increased levels of p36 Bax protein. These death-associated changes appeared much later in AUL15-treated cells compared to in AUL12-treated cells. In an effort to gain insight into the mechanism of action responsible for their biological effects, we investigated whether these gold compounds could induce the production of reactive oxygen species. Interestingly, we found that treatment with AUL12 (Au (III)), but not AUL15 (Au (I)), was associated with redox processes, suggesting that induction of oxidative stress may be partially responsible for the cytotoxic activity of gold (III) compounds (115).
5.2. Copper and zinc-containing dithiocarbamates
5.2.1. Disulfiram, EtDTC, and PyDTC
Copper and zinc are not only indispensable metals involved in critical biological processes such as tumorigenesis, they have also gained considerable interest as potential anticancer drug targets. An exciting new concept is ongoing in the field that takes advantage of the regulatory approval of drugs used for treatment of different pathological conditions which have also been shown to function as suitable metal chelators. The formation of metal complexes from previously approved drugs, such as Disulfiram (DSF), diethyldithiocarbamate (EtDTC), and pyrrolidinedithiocarbamates (PyDTC) (Figure 4), has been rigorously investigated as potential novel anticancer agents that target the ubiquitin-proteasome pathway.
Figure 4.
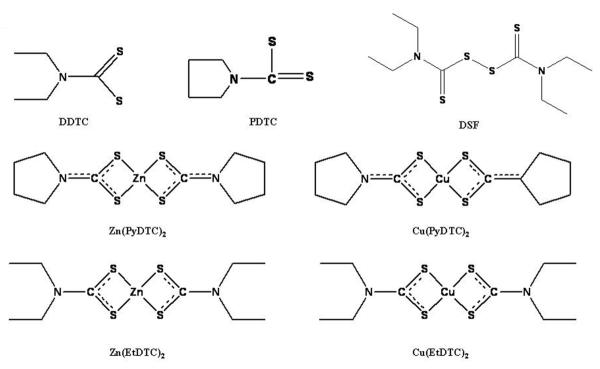
Chemical structures of dithiocarbamates.
DDTC, PDTC, and DSF have all been used previously in the clinical setting for a variety of diseases. These complexes are able to chelate copper, and DSF-Cu complexes are active against human tumor cells. EtDTC and PyDTC are metal chelating compounds from the dithiocarbamate family that have been investigated for their proteasome-inhibitory activity.
One such drug selected and tested is an irreversible inhibitor of aldehyde dehydrogenase, disulfiram (tetraethylthiuram disulfide, DSF) (Figure 4). DSF is one of only two drugs approved for the treatment of alcoholism, due to its efficacy without toxicity (116–118). The structure of DSF contains an R1R2NC (S)SR3 functional group, with sulfhydryl groups that afford it the ability to react with Cu (II) (119). This reaction was confirmed through the observation of an intense color change when DSF and CuCl2 were mixed at a 1:1 ratio (120). Although DSF is not suitable for binding various other biological metal ions such as Fe (II or III) or Mn (III) (119), it has been reported that DSF is able to interact with Zn (II). One group reported that DSF treatment of melanoma and hepatic cancer could be potentiated by Zn (II) supplementation (121) and we have also found that a DSF-Zn complex is able to inhibit the proteasome, though at a weaker potency than DSF-Cu (unpublished data). Studies have shown that DSF is rapidly converted to its copper complex during its absorption into the gastrointestinal system (116).
A potential target of disulfiram is superoxide dismutase, the inhibition of which may be associated with the inhibition of angiogenesis (122). Our lab has demonstrated that the DSF-Cu complex was also able to inhibit purified 20S proteasome (IC50=7.5 microM) and 26S proteasome in intact MDA-MB-231 breast cancer cells (20 microM). The CT-like activity was inhibited by >95% and proliferation was inhibited by up to 85%. An increase in levels of ubiquitinated proteins, as well as apoptotic PARP cleavage and morphological changes were apparent. DSF alone had no visible effect in cultured cells, but an increase in efficacy was observed in breast cancer cells cultured in copper-enriched conditions. Importantly, normal breast MCF-10A cells exhibited no response to DSF, indicating a lack of toxicity, as well as a therapeutic strategy that utilizes heightened levels of copper as a tumor-targeting mechanism (120).
We have also reported the ability of disulfiram to inhibit the proteasome under in vivo conditions. Daily treatment of mice bearing MDA-MB-231 xenografts with 50 mg/kg DSF for 30 days resulted in significant tumor growth inhibition (74%). Associated with this growth inhibition, a significant decrease in proteasomal chymotrypsin-like activity (87%) and accumulation of ubiquitinated proteins, p27, and Bax were visible. Furthermore, apoptosis-associated increases in caspase-3 activity and PARP cleavage were also observed (120). Interestingly, phase I/II clinical trials investigating the effects of DSF on metastatic melanoma have been completed, but the results are not yet available (NCT00256230; Chao Family Comprehensive Cancer Center). Patients are also currently being recruited for a phase I study determining the safety and toxicity profile of DSF and copper gluconate co-treatment in refractory malignancies with liver metastasis (NCT00742911; Huntsman Cancer Institute). Taken together, these results demonstrate a novel application for the use of DSF in the presence of copper for the potential treatment of cancer.
5.2.2. Synthetic EtDTC copper and zinc complexes
Because DSF can be converted to diethyldithiocarbamate (EtDTC) in the body, it is believed that the ability of EtDTC to complex with copper gives DSF its anticancer activity (116). EtDTC complexes have previously been shown to promote T cell maturation and reduce lymphadenopathy in animal models (123–124), and for this reason we investigated the anti-tumor activities of Zn (II) and Cu (II) EtDTC complexes (Figure 4). We treated MDA-MB-231 breast cancer cells with Cu (EtDTC)2 and Zn (EtDTC)2 and found that both complexes caused cell death associated with 26S proteasome inhibition (125). Inhibitory activity against purified 20S proteasome was much lower (125), suggesting that the effect of these complexes may be due to inhibition of the JAMM domain in the 19S particle of the proteasome (126). The JAMM domain, a metalloisopeptidase with a coordinated zinc ion (126), is necessary for the deubiquitinating activity of the 19S particle and has been proposed as a possible target for anticancer drugs (127).
5.2.3. Synthetic PyDTC copper and zinc complexes
We have also examined the possible chemotherapeutic properties of another group of dithiocarbamate complexes, pyrrolidinedithiocarbamates (PyDTC), when coupled with copper and zinc (Figure 4). We found that both the Zn (PyDTC)2 and Cu (PyDTC)2 complexes exhibited proteasome inhibitory activity against purified 20S proteasome as well as intact 26S proteasome in MDA-MB-231 cells (128). Accumulation of ubiquitinated proteins and proteasomal target proteins IkB-alpha and p27, associated with proteasome inhibition, and apoptosis associated morphological changes and PARP cleavage occurred in cells treated with either Zn (PyDTC)2 or Cu (PyDTC)2. We also observed that the effects of these PyDTC complexes were time-dependent, with >50% inhibition at early time points (128). To further examine the results observed with PyDTC complexes, we synthesized PyDTC:metal complexes in a 2:1 ratio (Zn (PyDTC)2 and Cu (PyDTC)2). We determined that these synthetic complexes were much less potent toward purified 20S proteasome (40% inhibition at 50 μM), but more potent toward intact 26S proteasome in MDA-MB-231 cells, with Cu (PyDTC)2 exhibiting higher activity than Zn (PyDTC)2. These synthetic complexes were also effective in other cell lines, including breast cancer DCIS and MCF7, and prostate cancer PC-3 cells (128). Additionally, partial inhibition of Cu/Zn (PyDTC)2-induced cell death occurred when cells were pretreated with a calpain inhibitor. However, addition of a calpain inhibitor did not affect proteasome inhibition, suggesting that calpain involvement is important in apoptotic cell death induced by synthetic Cu/Zn (PyDTC)2 complexes (128).
6. HYDROXYQUINOLINES
6.1. Clioquinol
Clioquinol (5-chloro-7-iodo-8-hydroxyuinoline, CQ) (Figure 5) is a compound in the hydroxyquinoline family and has been shown to reduce or prevent the formation of amyloid plaques in the brains of Alzheimer's disease transgenic mice (129). This discovery led to the initiation of two clinical trials which showed that CQ is beneficial in treating Alzheimer's disease with no visible toxicity (130–131). Consequently, CQ is currently in use clinically for the treatment of Alzheimer's and Huntington's diseases (132–133). Prior to the discovery of its efficacy in treating Alzheimer's disease, CQ was used successfully to treat and prevent shigella and entamoeba histolytica infections (134).
Figure 5.

Chemical structures of hydroxyquinolines.
CQ and 8-OHQ are members of the hydroxyquinoline family, and are able to bind copper and actively inhibit the proteasome. Analogs of 8-OHQ with methyl group additions were investigated, and found to exhibit proteasome-inhibitory activity when complexed with copper.
Although CQ use was thought to be associated with occurrence of subacute myelo-optic neuropathy in Japan, this conclusion was not supported by the subsequent epidemiologic analysis. Instead, decreased levels of vitamin B12 may play a role in this syndrome. In fact, CQ may be used safely in humans with vitamin B12 supplementation.
Clioquinol is a lipohilic compound that is able to form stable complexes with copper (II) ions (135). To test this, we mixed CQ and CuCl2 in a 1:1 molar ratio and an observable color change occurred, indicating that a chemical reaction had taken place (136). This was further confirmed by the use of X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS), which showed that the CQ-Cu mixture had a different copper oxidation state than CQ or CuCl2 alone, which verified that a coordination complex with copper had indeed been formed (136).
We examined the effects of the CQ-Cu complex on the purified 20S proteasome, and inhibition of chymotrypsin-like activity was observed (IC50= 2.5 microM). Human LNCaP and C4-2B prostate cancer cells were treated with the CQ-Cu complex (20 microM) to determine proteasome inhibitory activity in intact cells. We discovered that the complex is able to potently inhibit proteasomal chymotrypsin-like activity (82% and 83%), suppress androgen receptor expression, suppress cell proliferation, and induce apoptosis. PARP cleavage and cellular morphologic changes, associated with apoptosis-induction, were also detected (136). As observed with DSF, CQ alone (not in complex with Cu) was ineffective in intact cultured cells, due to the low levels of Cu present in cultured cell lines, contradictory to clinical or animal tumors, which contain high copper levels. Therefore, copper-enriched LNCaP and C4-2B cells were examined and found to be sensitive to treatment with CQ alone (136). Additionally, C4-2B xenograft-bearing mice were used to investigate the in vivo effects of CQ. In addition to significant tumor growth inhibition (66%) as compared to controls, proteasome inhibition, apoptosis induction, suppression of AR expression, and inhibition of angiogenesis (indicated by decreased CD31 expression) were also observed (136). These results present a compelling rationale for further investigation into the use of CQ in clinical trials as a potential anticancer agent.
6.2. 8-hydroxyquinoline analogs
Based on our encouraging findings with CQ, we selected and tested an analog, 8-hydroxyquinoline (8-OHQ) (Figure 5). We found that 8-OHQ is able to inhibit the proteasomal chymotrypsin-like activity when complexed with copper at a 1:1 molar ratio in purified 20S proteasome as well as in intact Jurkat leukemia T cells (10 microM) (137). Additionally, loss of cell viability and PARP cleavage were observed in the treated Jurkat cells and accumulation of ubiquitinated proteins occurred after treatment with 1 microM 8-OHQ. Importantly, no apoptosis was observed in non-transformed immortalized natural killer (YT) cells (137). To simulate in vivo tumor conditions, PC-3 prostate cancer cells were grown in copper (CuCl2) enriched media followed by treatment with 8-OHQ, which resulted in proteasome inhibition and PARP cleavage associated with induction of apoptosis (137).
Based on these positive results, seven new 8-OHQ analogs were synthesized and labeled #10–#16 (Figure 5). We treated MDA-MB-231 breast cancer cells with complexes of each analog and copper (1:1 molar ratio) at 25 microM (138). Measurement by MTT showed that the copper mixtures of #11, #12, and #13 (Figure 5) were able to inhibit ~90% of proliferation, while the other compounds showed little inhibitory activity. This suggests that the attachment of a methyl group to the phenyl ring does not affect the activity of the copper complexes (138). The copper mixtures of analogs #11, #12, and #13 also inhibited proteasome activity by 45–55%. The highest potency analog, #13, was chosen and found to inhibit proliferation of MDA-MB-231 cells, when mixed with copper, in a dose-dependent manner. Inhibition of proteasomal activity also occurred in a dose- and time-dependent manner. Consistent with this effect, accumulation of ubiquitinated proteins and IkB-alpha were observed, as well as morphological changes and PARP cleavage associated with apoptosis induction (138).
To substantiate the anticancer properties of these analogs and to gain insight into the roles of the metal and ligand, we synthesized complexes 10, 13, and 16 with copper at a 2:1 molar ratio (S10-, S13-, S16-Cu) (Figure 5). We observed that S13-Cu induced >90% growth inhibition at 15 microM, and S10- and S16-Cu were almost as potent at 25 microM. At the highest concentration all three complexes inhibited only ~20% activity in purified 20S proteasome. In intact MDA-MB-231 cells, S13-Cu at 15 microM inhibited >70% proteasomal activity after four hours and almost 90% after 24 hours (138). S10- and S16-Cu, however, showed minimal activity toward MDA-MB-231 cells. Proteasome inhibition by S13-Cu was confirmed by Western blot, which showed accumulation of ubiquitinated proteins, IkB-alpha, and p36 Bax. Apoptotic morphological changes and PARP cleavage also occurred in S13-Cu treated cells. Importantly, S13-Cu had no effects in non-malignant MCF-10A cells (138). Additionally, to determine whether the position of the methyl group affects the potency, we compared activities of the three methyl-containing analogs, #11, #12, and #13. Complex 11 inhibited ~90% proliferation at 10 microM, but complex 12 and 13 showed very little activity at this concentration. Complex 11 was also the most potent inhibitor of 26S proteasome activity with observable apoptosis-related changes at 10 microM (138). These data indicate the possibility that both the ligand and the metal are important to the activity of these metal compounds, and that the use of analogs of old previously used drugs is a viable option in the search for novel anticancer agents.
Importantly, we have also investigated the necessity of copper to the anticancer properties of CQ and 8-OHQ. Synthetic chemical probe molecules that mimic the structures of 8-OHQ and CQ, but have no copper-binding capability, were tested for their anti-tumor activities (139) (Figure 6). In contrast to both CQ and 8-OHQ, these inactive analogs were unable to inhibit growth of human breast cancer DCIS cells, either alone or in combination with CuCl2. Similarly, neither cell death nor proteasome inhibition was observed in cells treated with these synthetic molecules plus copper (139). Additionally, accumulation of ubiquitinated proteins and Bax, associated with proteasome inhibition did not occur in cells treated with the analogs-copper mixtures. These data demonstrate that copper-binding is necessary for CQ and 8-OHQ to be transported into breast cancer cells, and to subsequently exert their proteasome-inhibiting and apoptosis-inducing activities (139).
Figure 6.

Chemical structures of synthetic non-copper binding CQ and 8-OHQ analogs.
Four molecules similar in structure to CQ and 8-OHQ without the ability to bind copper were synthesized. In contrast to CQ and 8-OHQ, these analogs exhibited no proteasome-inhibitory or apoptosis-inducing activity alone or in combination with copper.
7. CONCLUSIONS
The clinical use of platinum-containing drugs in the treatment of a variety of human tumors represented a landmark achievement in metal-based cancer chemotherapy. Efforts to develop novel anticancer agents based on different metals and ligand platforms have been prompted by exciting new preclinical and clinical evidence. Investigation of different metal complexes was stimulated not only by attempting to overcome shortcomings of platinum-based compounds, but that foster mechanisms of action not realized by these conventional drugs. In recent years, ruthenium complexes have been investigated as both anitumor and antimetastatic agents and are currently being investigated in clinical trials. The unique properties of ruthenium as it relates to tumor uptake and reduction potential, present the possibility of a more selective tumor targeting strategy.
The clinical use of proteasome inhibitiors, such as bortezomib was validation of the importance of the cellular proteasome as a critical molecular target to be exploited for therapeutic purposes. Different metals and metal compounds that target the ubiquitin-proteasome pathway have received considerable attention as potential anticancer drugs. Essential metals such as copper and zinc are not only critical components in cellular metabolism, but have displayed potential as anticancer drug targets via proteasome inhibition. Along this line, since the medicinal applications of gold and gold complexes have been known throughout human history, their potential as anticancer agents, with proteasome-inhibitory activity, are only beginning to be fully appreciated. The physiochemical properties of metal complexes are not only dictated by the nature of the metal, but also the type and number of ligands involved. Two prominent classes of metal-chelating compounds investigated for anticancer activity with different metals are dithiocarbamates and hydroxyquinolines. Their applications consist of being used as either coordination complexes or by targeting increased levels of tumor-associated copper, leading to tumor proteasome inhibition and subsequent tumor cell death. These represent significant findings since representative examples from these classes, i.e. disulfiram and clioquinol are already clinically approved for pathological disorders, so the findings that these drugs harbor anticancer activity in the presence of metals may help expedite the regulatory process as novel anticancer drugs. Since the drug development process can be burdensome replete with regulatory demands, this concept could represent a significant achievement in establishing positive momentum in generating further lead candidates in anticancer drug discovery. Overall, the interesting properties of metal-based complexes, coupled with the significant progress made in elucidating their mechanisms of action, will help facilitate these entities into the clinical setting as drug candidates.
ACKNOWLEDGEMENTS
This research was partially supported by a Karmanos Cancer Institute Pilot project funding (to QPD) and the National Cancer Institute grants (1R01CA120009, 3R01CA120009-04S1, 1R21CA139386-01, to QPD). We thank Di Chen, Ph.D. for critically reading this manuscript.
Abbreviations
- Pt
Platinum
- Ru
Ruthenium
- Cu
Copper
- Zn
Zinc
- VEGF
Vascular Endothelial Growth Factor
- UPP
Ubiquitin-proteasome pathway
- PGPH
peptidyl-glutamyl peptide-hydrolyzing
- CT
chymotrypsin
- DSF
Disulfiram
- PARP
poly-ADP ribose polymerase
- EtDTC
diethyldithiocarbamate
- PyDTC
pyrrolidinedithiocarbamate
- CQ
Clioquinol
- XANES
X-ray absorption near-edge spectroscopy
- EXAFS
extended X-ray absorption fine structure spectroscopy
- 8-OHQ
8-hydroxyquinoline
9. REFERENCES
- 1.Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev. 1999;99(9):2451–66. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 2.Eckhardt S. Recent progress in the development of anticancer agents. Curr Med Chem Anticancer Agents. 2002;2(3):419–39. doi: 10.2174/1568011024606389. [DOI] [PubMed] [Google Scholar]
- 3.Abrams MJ, Murrer BA. Metal compounds in therapy and diagnosis. Science. 1993;261(5122):725–30. doi: 10.1126/science.8102010. [DOI] [PubMed] [Google Scholar]
- 4.Galanski M, Arion VB, Jakupec MA, Keppler BK. Recent developments in the field of tumor-inhibiting metal complexes. Curr Pharm Des. 2003;9(25):2078–89. doi: 10.2174/1381612033454180. [DOI] [PubMed] [Google Scholar]
- 5.Galanski M, Jakupec MA, Keppler BK. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem. 2005;12(18):2075–94. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 6.Alama A, Tasso B, Novelli F, Sparatore F. Organometallic compounds in oncology: implications of novel organotins as antitumor agents. Drug Discov Today. 2009;14(9–10):500–8. doi: 10.1016/j.drudis.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Harrap KR. Preclinical studies identifying carboplatin as a viable cisplatin alternative. Cancer Treat Rev. 1985;12(Suppl A):21–33. doi: 10.1016/0305-7372(85)90015-5. [DOI] [PubMed] [Google Scholar]
- 8.de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, de Braud F, Wilson C, Morvan F, Bonetti A. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–47. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 9.Calamai P, Carotti S, Guerri A, Mazzei T, Messori L, Mini E, Orioli P, Speroni GP. Cytotoxic effects of gold(III) complexes on established human tumor cell lines sensitive and resistant to cisplatin. Anticancer Drug Des. 1998;13(1):67–80. [PubMed] [Google Scholar]
- 10.Sava G, Zorzet S, Giraldi T, Mestroni G, Zassinovich G. Antineoplastic activity and toxicity of an organometallic complex of ruthenium(II) in comparison with cis-PDD in mice bearing solid malignant neoplasms. Eur J Cancer Clin Oncol. 1984;20(6):841–7. doi: 10.1016/0277-5379(84)90223-2. [DOI] [PubMed] [Google Scholar]
- 11.Schluga P, Hartinger CG, Egger A, Reisner E, Galanski M, Jakupec MA, Keppler BK. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006;(14):1796–802. doi: 10.1039/b511792e. [DOI] [PubMed] [Google Scholar]
- 12.Bergamo A, Masi A, Dyson PJ, Sava G. Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int J Oncol. 2008;33(6):1281–9. [PubMed] [Google Scholar]
- 13.Pieper T, Borsky K, Keppler BK. Non-platinum antitumor compounds. Top Biol Inorg Chem. 1999;1:171–199. [Google Scholar]
- 14.Sava G, Alessio E, Bergamo A, Mestroni G. Sulfoxide ruthernium complexes. Top Biol Inorg Chem. 1999;1:143–169. [Google Scholar]
- 15.Sava G, Pacor S, Mestroni G, Alessio E. Na[trans-RuCl4(DMSO)Im], a metal complex of ruthenium with antimetastatic properties. Clin Exp Metastasis. 1992;10(4):273–80. doi: 10.1007/BF00133563. [DOI] [PubMed] [Google Scholar]
- 16.Sava G, Pacor S, Bergamo A, Cocchietto M, Mestroni G, Alessio E. Effects of ruthenium complexes on experimental tumors: irrelevance of cytotoxicity for metastasis inhibition. Chem Biol Interact. 1995;95(1–2):109–26. doi: 10.1016/0009-2797(94)03350-1. [DOI] [PubMed] [Google Scholar]
- 17.Jakupec MA, Arion VB, Kapitza S, Reisner E, Eichinger A, Pongratz M, Marian B, Graf von Keyserlingk N, Keppler BK. KP1019 (FFC14A) from bench to bedside: preclinical and early clinical development--an overview. Int J Clin Pharmacol Ther. 2005;43(12):595–6. doi: 10.5414/cpp43595. [DOI] [PubMed] [Google Scholar]
- 18.Hartinger CG, Jakupec MA, Zorbas-Seifried S, Groessl M, Egger A, Berger W, Zorbas H, Dyson PJ, Keppler BK. KP1019, a new redox-active anticancer agent--preclinical development and results of a clinical phase I study in tumor patients. Chem Biodivers. 2008;5(10):2140–55. doi: 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- 19.Apelgot S, Coppey J, Fromentin A, Guille E, Poupon MF, Roussel A. Altered distribution of copper (64Cu) in tumor-bearing mice and rats. Anticancer Res. 1986;6(2):159–64. [PubMed] [Google Scholar]
- 20.Zowczak M, Iskra M, Torlinski L, Cofta S. Analysis of serum copper and zinc concentrations in cancer patients. Biol Trace Elem Res. 2001;82(1–3):1–8. doi: 10.1385/BTER:82:1-3:001. [DOI] [PubMed] [Google Scholar]
- 21.Turecky L, Kalina P, Uhlikova E, Namerova S, Krizko J. Serum ceruloplasmin and copper levels in patients with primary brain tumors. Klin Wochenschr. 1984;62(4):187–9. doi: 10.1007/BF01731643. [DOI] [PubMed] [Google Scholar]
- 22.Kuo HW, Chen SF, Wu CC, Chen DR, Lee JH. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol Trace Elem Res. 2002;89(1):1–11. doi: 10.1385/BTER:89:1:1. [DOI] [PubMed] [Google Scholar]
- 23.Rizk SL, Sky-Peck HH. Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Res. 1984;44(11):5390–4. [PubMed] [Google Scholar]
- 24.Nayak SB, Bhat VR, Upadhyay D, Udupa SL. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J Physiol Pharmacol. 2003;47(1):108–10. [PubMed] [Google Scholar]
- 25.Diez M, Arroyo M, Cerdan FJ, Munoz M, Martin MA, Balibrea JL. Serum and tissue trace metal levels in lung cancer. Oncology. 1989;46(4):230–4. doi: 10.1159/000226722. [DOI] [PubMed] [Google Scholar]
- 26.Habib FK, Dembinski TC, Stitch SR. The zinc and copper content of blood leucocytes and plasma from patients with benign and malignant prostates. Clin Chim Acta. 1980;104(3):329–35. doi: 10.1016/0009-8981(80)90390-3. [DOI] [PubMed] [Google Scholar]
- 27.McAuslan BR, Reilly W. Endothelial cell phagokinesis in response to specific metal ions. Exp Cell Res. 1980;130(1):147–57. doi: 10.1016/0014-4827(80)90051-8. [DOI] [PubMed] [Google Scholar]
- 28.Gullino PM. Considerations on the mechanism of the angiogenic response. Anticancer Res. 1986;6(2):153–8. [PubMed] [Google Scholar]
- 29.Hu GF. Copper stimulates proliferation of human endothelial cells under culture. J Cell Biochem. 1998;69(3):326–35. doi: 10.1002/(sici)1097-4644(19980601)69:3<326::aid-jcb10>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 30.Veeravagu A, Hsu AR, Cai W, Hou LC, Tse VC, Chen X. Vascular endothelial growth factor and vascular endothelial growth factor receptor inhibitors as anti-angiogenic agents in cancer therapy. Recent Pat Anticancer Drug Discov. 2007;2(1):59–71. doi: 10.2174/157489207779561426. [DOI] [PubMed] [Google Scholar]
- 31.Frangoulis M, Georgiou P, Chrisostomidis C, Perrea D, Dontas I, Kavantzas N, Kostakis A, Papadopoulos O. Rat epigastric flap survival and VEGF expression after local copper application. Plast Reconstr Surg. 2007;119(3):837–43. doi: 10.1097/01.prs.0000252000.59231.5e. [DOI] [PubMed] [Google Scholar]
- 32.Sen CK, Khanna S, Venojarvi M, Trikha P, Ellison EC, Hunt TK, Roy S. Copper-induced vascular endothelial growth factor expression and wound healing. Am J Physiol Heart Circ Physiol. 2002;282(5):H1821–7. doi: 10.1152/ajpheart.01015.2001. [DOI] [PubMed] [Google Scholar]
- 33.Nasulewicz A, Mazur A, Opolski A. Role of copper in tumour angiogenesis--clinical implications. J Trace Elem Med Biol. 2004;18(1):1–8. doi: 10.1016/j.jtemb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Moriguchi M, Nakajima T, Kimura H, Watanabe T, Takashima H, Mitsumoto Y, Katagishi T, Okanoue T, Kagawa K. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin-8 production. Int J Cancer. 2002;102(5):445–52. doi: 10.1002/ijc.10740. [DOI] [PubMed] [Google Scholar]
- 35.Brem S. Angiogenesis and Cancer Control: From Concept to Therapeutic Trial. Cancer Control. 1999;6(5):436–458. [PubMed] [Google Scholar]
- 36.Brewer GJ. Copper control as an antiangiogenic anticancer therapy: lessons from treating Wilson's disease. Exp Biol Med (Maywood) 2001;226(7):665–73. doi: 10.1177/153537020222600712. [DOI] [PubMed] [Google Scholar]
- 37.Lowndes SA, Harris AL. The role of copper in tumour angiogenesis. J Mammary Gland Biol Neoplasia. 2005;10(4):299–310. doi: 10.1007/s10911-006-9003-7. [DOI] [PubMed] [Google Scholar]
- 38.Theophanides T, Anastassopoulou J. Copper and carcinogenesis. Crit Rev Oncol Hematol. 2002;42(1):57–64. doi: 10.1016/s1040-8428(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 39.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 40.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175(3):409–16. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C, De Carvalho M, Mesri EA, Robins DM, Dick RD, Brewer GJ, Merajver SD. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62(17):4854–9. [PubMed] [Google Scholar]
- 42.Yoshii J, Yoshiji H, Kuriyama S, Ikenaka Y, Noguchi R, Okuda H, Tsujinoue H, Nakatani T, Kishida H, Nakae D, Gomez DE, De Lorenzo MS, Tejera AM, Fukui H. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int J Cancer. 2001;94(6):768–73. doi: 10.1002/ijc.1537. [DOI] [PubMed] [Google Scholar]
- 43.Chang KL, Hung TC, Hsieh BS, Chen YH, Chen TF, Cheng HL. Zinc at pharmacologic concentrations affects cytokine expression and induces apoptosis of human peripheral blood mononuclear cells. Nutrition. 2006;22(5):465–74. doi: 10.1016/j.nut.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Franklin RB, Costello LC. The important role of the apoptotic effects of zinc in the development of cancers. J Cell Biochem. 2009;106(5):750–7. doi: 10.1002/jcb.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Provinciali M, Di Stefano G, Fabris N. Dose-dependent opposite effect of zinc on apoptosis in mouse thymocytes. Int J Immunopharmacol. 1995;17(9):735–44. doi: 10.1016/0192-0561(95)00063-8. [DOI] [PubMed] [Google Scholar]
- 46.Murakami M, Hirano T. Intracellular zinc homeostasis and zinc signaling. Cancer Sci. 2008;99(8):1515–22. doi: 10.1111/j.1349-7006.2008.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Federico A, Iodice P, Federico P, Del Rio A, Mellone MC, Catalano G. Effects of selenium and zinc supplementation on nutritional status in patients with cancer of digestive tract. Eur J Clin Nutr. 2001;55(4):293–7. doi: 10.1038/sj.ejcn.1601157. [DOI] [PubMed] [Google Scholar]
- 48.Prasad AS, Beck FW, Doerr TD, Shamsa FH, Penny HS, Marks SC, Kaplan J, Kucuk O, Mathog RH. Nutritional and zinc status of head and neck cancer patients: an interpretive review. J Am Coll Nutr. 1998;17(5):409–18. doi: 10.1080/07315724.1998.10718787. [DOI] [PubMed] [Google Scholar]
- 49.Chakravarty PK, Ghosh A, Chowdhury JR. Zinc in human malignancies. Neoplasma. 1986;33(1):85–90. [PubMed] [Google Scholar]
- 50.Franklin RB, Costello LC. Zinc as an anti-tumor agent in prostate cancer and in other cancers. Arch Biochem Biophys. 2007;463(2):211–7. doi: 10.1016/j.abb.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta SK, Singh SP, Shukla VK. Copper, zinc, and Cu/Zn ratio in carcinoma of the gallbladder. J Surg Oncol. 2005;91(3):204–8. doi: 10.1002/jso.20306. [DOI] [PubMed] [Google Scholar]
- 52.Margalioth EJ, Schenker JG, Chevion M. Copper and zinc levels in normal and malignant tissues. Cancer. 1983;52(5):868–72. doi: 10.1002/1097-0142(19830901)52:5<868::aid-cncr2820520521>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz AE, Leddicotte GW, Fink RW, Friedman EW. Trace elements in noraml and malignant human breast tissue. Surgery. 1974;76(2):325–9. [PubMed] [Google Scholar]
- 54.Zhao H, Eide D. The yeast ZRT1 gene encodes the zinc transporter protein of a high-affinity uptake system induced by zinc limitation. Proc Natl Acad Sci U S A. 1996;93(6):2454–8. doi: 10.1073/pnas.93.6.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M, Zhang Y, Liu Z, Bharadwaj U, Wang H, Wang X, Zhang S, Liuzzi JP, Chang SM, Cousins RJ, Fisher WE, Brunicardi FC, Logsdon CD, Chen C, Yao Q. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc Natl Acad Sci U S A. 2007;104(47):18636–41. doi: 10.1073/pnas.0709307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costello LC, Franklin RB. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Mol Cancer. 2006;5:17. doi: 10.1186/1476-4598-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manning DL, Robertson JF, Ellis IO, Elston CW, McClelland RA, Gee JM, Jones RJ, Green CD, Cannon P, Blamey RW, et al. Oestrogen-regulated genes in breast cancer: association of pLIV1 with lymph node involvement. Eur J Cancer. 1994;30A(5):675–8. doi: 10.1016/0959-8049(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 58.Taylor KM, Morgan HE, Smart K, Zahari NM, Pumford S, Ellis IO, Robertson JF, Nicholson RI. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol Med. 2007;13(7–8):396–406. doi: 10.2119/2007-00040.Taylor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66(17):8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 60.Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81(4):1100–5. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- 61.Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A. 1980;77(4):1783–6. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5(7):596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 63.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17(24):7151–60. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 65.Peters JM, Cejka Z, Harris JR, Kleinschmidt JA, Baumeister W. Structural features of the 26 S proteasome complex. J Mol Biol. 1993;234(4):932–7. doi: 10.1006/jmbi.1993.1646. [DOI] [PubMed] [Google Scholar]
- 66.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92(3):367–80. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 67.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–71. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 68.Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A. 1999;96(20):10976–83. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeMartino GN, Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem. 1999;274(32):22123–6. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- 70.Goldberg AL, Cascio P, Saric T, Rock KL. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol. 2002;39(3–4):147–64. doi: 10.1016/s0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 71.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–47. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 72.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31(1):137–55. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 73.Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev. 2003;29(Suppl 1):3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 74.Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22(5):442–51. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 75.Ciechanover A. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66(2 Suppl 1):S7–19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- 76.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5(5):417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 77.Smith L, Lind MJ, Drew PJ, Cawkwell L. The putative roles of the ubiquitin/proteasome pathway in resistance to anticancer therapy. Eur J Cancer. 2007;43(16):2330–8. doi: 10.1016/j.ejca.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 78.Dou QP, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updat. 1999;2(4):215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- 79.Li B, Dou QP. Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A. 2000;97(8):3850–5. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3(2):231–4. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 81.Kumatori A, Tanaka K, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, Ichihara A. Abnormally high expression of proteasomes in human leukemic cells. Proc Natl Acad Sci U S A. 1990;87(18):7071–5. doi: 10.1073/pnas.87.18.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998;5(12):1062–75. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- 83.Lopes UG, Erhardt P, Yao R, Cooper GM. p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272(20):12893–6. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 84.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14(6):1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 85.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–6. [PubMed] [Google Scholar]
- 86.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59(11):2615–22. [PubMed] [Google Scholar]
- 87.Frankel A, Man S, Elliott P, Adams J, Kerbel RS. Lack of multicellular drug resistance observed in human ovarian and prostate carcinoma treated with the proteasome inhibitor PS-341. Clin Cancer Res. 2000;6(9):3719–28. [PubMed] [Google Scholar]
- 88.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8(6):508–13. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 89.Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13(18 Pt 1):5291–4. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 90.Crawford LJ, Walker B, Ovaa H, Chauhan D, Anderson KC, Morris TC, Irvine AE. Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO, PS-341, and MG-132. Cancer Res. 2006;66(12):6379–86. doi: 10.1158/0008-5472.CAN-06-0605. [DOI] [PubMed] [Google Scholar]
- 91.Orlowski RZ, Baldwin AS., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8(8):385–9. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 92.Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther. 2003;2(9):835–43. [PubMed] [Google Scholar]
- 93.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5(9):2638–45. [PubMed] [Google Scholar]
- 94.Sartore-Bianchi A, Gasparri F, Galvani A, Nici L, Darnowski JW, Barbone D, Fennell DA, Gaudino G, Porta C, Mutti L. Bortezomib inhibits nuclear factor-kappaB dependent survival and has potent in vivo activity in mesothelioma. Clin Cancer Res. 2007;13(19):5942–51. doi: 10.1158/1078-0432.CCR-07-0536. [DOI] [PubMed] [Google Scholar]
- 95.Michaelis M, Fichtner I, Behrens D, Haider W, Rothweiler F, Mack A, Cinatl J, Doerr HW, Cinatl J., Jr. Anti-cancer effects of bortezomib against chemoresistant neuroblastoma cell lines in vitro and in vivo. Int J Oncol. 2006;28(2):439–46. [PubMed] [Google Scholar]
- 96.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 97.Jagannath S, Durie BG, Wolf J, Camacho E, Irwin D, Lutzky J, McKinley M, Gabayan E, Mazumder A, Schenkein D, Crowley J. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129(6):776–83. doi: 10.1111/j.1365-2141.2005.05540.x. [DOI] [PubMed] [Google Scholar]
- 98.Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105(8):3058–65. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- 99.Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen TC, Schonthal AH. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113(23):5927–37. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- 100.Kim TY, Park J, Oh B, Min HJ, Jeong TS, Lee JH, Suh C, Cheong JW, Kim HJ, Yoon SS, Park SB, Lee DS. Natural polyphenols antagonize the antimyeloma activity of proteasome inhibitor bortezomib by direct chemical interaction. Br J Haematol. 2009;146(3):270–81. doi: 10.1111/j.1365-2141.2009.07752.x. [DOI] [PubMed] [Google Scholar]
- 101.Malaguarnera L, Pilastro MR, DiMarco R, Scifo C, Renis M, Mazzarino MC, Messina A. Cell death in human acute myelogenous leukemic cells induced by pyrrolidinedithiocarbamate. Apoptosis. 2003;8(5):539–45. doi: 10.1023/a:1025550726803. [DOI] [PubMed] [Google Scholar]
- 102.Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175(5):1181–94. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Forestier J. Rheumatoid arthritis and its treatment with gold salts - results of six years' experience. J Lab Clin Med. 1935;20:827–840. [Google Scholar]
- 104.Tiekink ER. Gold derivatives for cancer treatment. Bioinorg Chem Applns. 2003;53:1–9. [Google Scholar]
- 105.Mirabelli CK, Johnson RK, Hill DT, Faucette LF, Girard GR, Kuo GY, Sung CM, Crooke ST. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J Med Chem. 1986;29(2):218–23. doi: 10.1021/jm00152a009. [DOI] [PubMed] [Google Scholar]
- 106.Milacic V, Fregona D, Dou QP. Gold complexes as prospective metal-based anticancer drugs. Histol Histopathol. 2008;23(1):101–8. doi: 10.14670/HH-23.101. [DOI] [PubMed] [Google Scholar]
- 107.Sadler PJ, Sue RE. The chemistry of gold drugs. Met Based Drugs. 1994;1(2–3):107–44. doi: 10.1155/MBD.1994.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fricker SP. A Screening Strategy for Metal Antitumor Agents as Exemplified by Gold(III) Complexes. Met Based Drugs. 1999;6(4–5):291–300. doi: 10.1155/MBD.1999.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ronconi L, Giovagnini L, Marzano C, Bettio F, Graziani R, Pilloni G, Fregona D. Gold dithiocarbamate derivatives as potential antineoplastic agents: design, spectroscopic properties, and in vitro antitumor activity. Inorg Chem. 2005;44(6):1867–81. doi: 10.1021/ic048260v. [DOI] [PubMed] [Google Scholar]
- 110.Ronconi L, Marzano C, Zanello P, Corsini M, Miolo G, Macca C, Trevisan A, Fregona D. Gold(III) dithiocarbamate derivatives for the treatment of cancer: solution chemistry, DNA binding, and hemolytic properties. J Med Chem. 2006;49(5):1648–57. doi: 10.1021/jm0509288. [DOI] [PubMed] [Google Scholar]
- 111.Messori L, Marcon G, Orioli P. Gold(III) compounds as new family of anticancer drugs. Bioinorg Chem Appl. 2003:177–87. doi: 10.1155/S1565363303000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bodenner DL, Dedon PC, Keng PC, Borch RF. Effect of diethyldithiocarbamate on cisdiamminedichloroplatinum(II)-induced cytotoxicity, DNA cross-linking, and gamma-glutamyl transpeptidase inhibition. Cancer Res. 1986;46(6):2745–50. [PubMed] [Google Scholar]
- 113.Huang H, Zhu L, Reid BR, Drobny GP, Hopkins PB. Solution structure of a cisplatin-induced DNA interstrand cross-link. Science. 1995;270(5243):1842–5. doi: 10.1126/science.270.5243.1842. [DOI] [PubMed] [Google Scholar]
- 114.Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res. 2006;66(21):10478–86. doi: 10.1158/0008-5472.CAN-06-3017. [DOI] [PubMed] [Google Scholar]
- 115.Zhang X, Frezza M, Milacic V, Ronconi L, Fan Y, Bi C, Fregona D, Dou QP. Inhibition of tumor proteasome activity by gold-dithiocarbamato complexes via both redox-dependent and -independent processes. J Cell Biochem. 2010;109(1):162–72. doi: 10.1002/jcb.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl. 1992;369:15–26. doi: 10.1111/j.1600-0447.1992.tb03310.x. [DOI] [PubMed] [Google Scholar]
- 117.Vallari RC, Pietruszko R. Human aldehyde dehydrogenase: mechanism of inhibition of disulfiram. Science. 1982;216(4546):637–9. doi: 10.1126/science.7071604. [DOI] [PubMed] [Google Scholar]
- 118.Meyer RE. Prospects for a rational pharmacotherapy of alcoholism. J Clin Psychiatry. 1989;50(11):403–12. [PubMed] [Google Scholar]
- 119.Cen D, Brayton D, Shahandeh B, Meyskens FL, Jr., Farmer PJ. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J Med Chem. 2004;47(27):6914–20. doi: 10.1021/jm049568z. [DOI] [PubMed] [Google Scholar]
- 120.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66(21):10425–33. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 121.Brar SS, Grigg C, Wilson KS, Holder WD, Jr., Dreau D, Austin C, Foster M, Ghio AJ, Whorton AR, Stowell GW, Whittall LB, Whittle RR, White DP, Kennedy TP. Disulfiram inhibits activating transcription factor/cyclic AMP-responsive element binding protein and human melanoma growth in a metal-dependent manner in vitro, in mice and in a patient with metastatic disease. Mol Cancer Ther. 2004;3(9):1049–60. [PubMed] [Google Scholar]
- 122.Marikovsky M, Nevo N, Vadai E, Harris-Cerruti C. Cu/Zn superoxide dismutase plays a role in angiogenesis. Int J Cancer. 2002;97(1):34–41. doi: 10.1002/ijc.1565. [DOI] [PubMed] [Google Scholar]
- 123.Reisinger EC, Kern P, Ernst M, Bock P, Flad HD, Dietrich M. Inhibition of HIV progression by dithiocarb. German DTC Study Group. Lancet. 1990;335(8691):679–82. doi: 10.1016/0140-6736(90)90802-c. [DOI] [PubMed] [Google Scholar]
- 124.Kaplan CS, Petersen EA, Yocum D, Hersh EM. A randomized, controlled dose response study of intravenous sodium diethyldithiocarbamate in patients with advanced human immunodeficiency virus infection. Life Sci. 1989;45(22):iii–ix. doi: 10.1016/0024-3205(89)90070-2. [DOI] [PubMed] [Google Scholar]
- 125.Cvek B, Milacic V, Taraba J, Dou QP. Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells. J Med Chem. 2008;51(20):6256–8. doi: 10.1021/jm8007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, 3rd, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298(5593):611–5. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 127.Gallery M, Blank JL, Lin Y, Gutierrez JA, Pulido JC, Rappoli D, Badola S, Rolfe M, Macbeth KJ. The JAMM motif of human deubiquitinase Poh1 is essential for cell viability. Mol Cancer Ther. 2007;6(1):262–8. doi: 10.1158/1535-7163.MCT-06-0542. [DOI] [PubMed] [Google Scholar]
- 128.Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP. Pyrrolidine dithiocarbamate-zinc(II) and -copper(II) complexes induce apoptosis in tumor cells by inhibiting the proteasomal activity. Toxicol Appl Pharmacol. 2008;231(1):24–33. doi: 10.1016/j.taap.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30(3):665–76. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 130.Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG. Treatment of Alzheimer's disease with clioquinol. Dement Geriatr Cogn Disord. 2001;12(6):408–14. doi: 10.1159/000051288. [DOI] [PubMed] [Google Scholar]
- 131.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60(12):1685–91. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 132.Ritchie CW, Bush AI, Masters CL. Metal-protein attenuating compounds and Alzheimer's disease. Expert Opin Investig Drugs. 2004;13(12):1585–92. doi: 10.1517/13543784.13.12.1585. [DOI] [PubMed] [Google Scholar]
- 133.Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc Natl Acad Sci U S A. 2005;102(33):11840–5. doi: 10.1073/pnas.0502177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gholz LM, Arons WL. Prophylaxis and Therapy of Amebiasis and Shigellosis with Iodochlorhydroxyquin. Am J Trop Med Hyg. 1964;13:396–401. doi: 10.4269/ajtmh.1964.13.396. [DOI] [PubMed] [Google Scholar]
- 135.Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P. Clioquinol, a drug for Alzheimer's disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg Chem. 2004;43(13):3795–7. doi: 10.1021/ic0494051. [DOI] [PubMed] [Google Scholar]
- 136.Chen D, Cui QC, Yang H, Barrea RA, Sarkar FH, Sheng S, Yan B, Reddy GP, Dou QP. Clioquinol, a therapeutic agent for Alzheimer's disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007;67(4):1636–44. doi: 10.1158/0008-5472.CAN-06-3546. [DOI] [PubMed] [Google Scholar]
- 137.Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP. Organic copper complexes as a new class of proteasome inhibitors and apoptosis inducers in human cancer cells. Biochem Pharmacol. 2004;67(6):1139–51. doi: 10.1016/j.bcp.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 138.Milacic V, Jiao P, Zhang B, Yan B, Dou QP. Novel 8-hydroxylquinoline analogs induce copper-dependent proteasome inhibition and cell death in human breast cancer cells. Int J Oncol. 2009;35(6):1481–91. doi: 10.3892/ijo_00000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhai S, Yang L, Cui QC, Sun Y, Dou QP, Yan B. Tumor cellular proteasome inhibition and growth suppression by 8-hydroxyquinoline and clioquinol requires their capabilities to bind copper and transport copper into cells. J Biol Inorg Chem. 2010;15(2):259–69. doi: 10.1007/s00775-009-0594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]