

Abstract
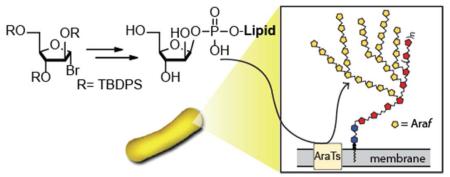
Mycobacteria and corynebacteria use decaprenylphosphoryl-β-D-arabinofuranose (DPA) as a critical cell wall building block. Arabinofuranosyltransferases that process this substrate to mediate cell wall assembly have served as drug targets, but little is known about the substrate specificity of any of these enzymes. To probe substrate recognition of DPA analogs, we developed a general and efficient synthetic route to β-D-arabinofuranosyl phosphodiesters. In this approach, the key glycosyl phosphodiester bond-forming reaction proceeds with high β-selectivity. In addition to its stereoselectivity, our route provides the means to readily access a variety of different lipid analogs, including aliphatic and polyprenyl substrates.
Introduction
Mycobacteria and corynebacteria have a robust cell wall that is crucial for their survival and pathogenesis. In the case of Mycobacterium tuberculosis, the enzymes responsible for cell wall biosynthesis have been identified as promising targets for the development of new drugs to treat tuberculosis.1 A significant proportion of the mycobacterial cell envelope is composed of arabinofuranose (Araf), which is found in the form of a large branched heteropolysaccharide known as the arabinogalactan (AG, Figure 1a) as well as the lipoarabinomannan (LAM).1-3 The assembly of the arabinan polysaccharide is catalyzed by a series of arabinofuranosyltransferases, which utilize the lipid-linked donor substrate, decaprenylphosphoryl-β-D-arabinofuranose (DPA, Figure 1b), to add single Araf residues to a growing arabinan polysaccharide.4 Some of these arabinofuranosyltransferases are targets of the first line antitubercular agent ethambutol.5 Despite their known potential value as therapeutic targets, little is known about the mechanism of substrate preferences of any of these enzymes.
Figure 1.

(a) Schematic depiction of a portion of the mycobacterial cell wall showing the galactofuranose (red) and arabinofuranose (yellow) polysaccharide components, and (b) decaprenylphosphoryl-β-D-arabinofuranose (DPA) is the building block used to incorporate arabinofuranose residues.
Paramount to the study of glycan biosynthesis is the ability to readily obtain renewable quantities of glycosyl donor and acceptor substrates or functional analogs thereof.6,7 DPA can be accessed by total synthesis; however, the required decaprenol precursor is costly and only available in small quantities.5,8 Additionally, the long decaprenyl lipid of DPA confers poor aqueous solubility upon the glycosyl donor, thereby complicating the study of arabinofuranosyltransferases. A more readily available, shorter lipid analog of DPA could facilitate a wide range of mechanistic studies. A recent report provides impetus to generate DPA analogs, as proteoliposomes containing the mycobacterial arabinofuranosyltransferase AftC could use a (Z,Z)-farnesyl lipid analog to transfer a single Araf residue to a synthetic acceptor.9 To explore arabinose incorporation into the cell wall, we sought to prepare various analogs of DPA with both saturated and unsaturated lipid substituents. Using the known synthetic methods, we were unable to generate a wide range of β-D-arabinofuranosyl phosphodiesters. Some methods have restrictions on the type of lipid and can be appended, and many approaches give rise to mixtures of glycosylphosphate derivatives that are difficult to separate. Because only β-glycosyl phosphodiesters are candidate substrates for the arabinofuranosyltransferases, the most valuable synthetic method would be one that generates the β-anomer selectively. Here we describe a route to β-D-arabinofuranosyl phosphodiesters that provides both saturated and unsaturated products with high stereoselectivity for the β-anomer.
Results and Discussion
To date, there have been two different approaches to the synthesis of DPA and shorter polyprenyl analogs. One approach shown previously by Lee and coworkers uses a phosphoramidite coupling method to join 2,3,5-silyl protected Araf (1) with a corresponding polyprenol (Figure 2a).5,10 After oxidation of the intermediate phosphite, the desired phosphodiester is formed as a 5:1 mixture of α- and β-anomers (2 and 3). This approach is advantageous in that any number of DPA analogs, both saturated and unsaturated, can be prepared by simply changing the alcohol coupling partner. A disadvantage, however, is that the desired β-anomer is the minor component of the product mixture. Another approach developed by Liav and Brennan relies on first installing a phosphoryl group at the C1 position of arabinose. In a subsequent step, the resulting Araf phosphate (4) is coupled to a lipid-linked trichloroacetimidate intermediate (Figure 2b).8,9,11,12 This approach favors formation of the desired β-anomer (2:1-4:1 β/α), but it is applicable only to activated lipid acetimidates (i.e. allylic or benzylic) and not aliphatic substrates.13 Furthermore, this approach requires an extra synthetic step to convert the lipid moiety into the corresponding trichloroacetimidate derivative.
Figure 2.

Synthetic approaches to DPA and analogs.
To access both saturated and unsaturated substrates, we envisioned using an alternative strategy in which the phosphodiester moiety is generated directly from monophosphate salt 4 and a lipid alcohol (Figure 1c). Of the known coupling agents for linking alcohols and phosphoryl groups to form phosphodiesters (e.g., carbodiimides, sulfonyl chlorides, Mitsunobu conditions, etc.)13, we were attracted to the method of Cramer and coworkers, which uses trichloroacetonitrile to activate the phosphoryl group.14-17 Applying this approach to our targets involves generating iminophosphate intermediate 5, which can undergo nucleophilic attack by the alcohol. When we attempted to carry out this reaction on a TBS-protected substrate, we found the phosphorylated Araf derivative 4 to be quite unstable. We traced this instability to the lability of its silyl groups, which underwent cleavage even during storage at −20 °C. When phosphodiester coupling was attempted using freshly prepared 4 and dodecanol, only the minor α-anomer underwent coupling whereas the desired β-anomer afforded the corresponding C1,C2 cyclic phosphate 7.18 Although it was not noted, others appear to have encountered this problem previously.19 The production of this undesired product is a consequence of the deleterious removal of the C2 tert-butyldimethylsilyl (TBS) ether and subsequent intramolecular cyclization via the electrophilic iminophosphate intermediate. Thus, the unwanted desilylation not only decreased the yield of our target compound but also precluded the production of the requisite β-anomer.
To obtain our desired DPA analogs, a protecting group strategy for Araf was needed that circumvented the propensity of the TBS ether at the 2-position to undergo cleavage. It has previously been shown that non-participating protecting groups such as benzyl groups provide high selectivity for the β-anomer during phosphorylation of the corresponding protected α-glycosyl bromide intermediate.20 Still, the conditions needed to remove the benzyl protecting groups would interfere with accessing substrates that possess alkene-containing lipids. Alternatively, ester protecting groups are compatible with installing a wide variety of lipids, but offer poor selectivity in the glycosylation reaction due to competing neighboring group participation.20-22 For the synthesis of DPA derivatives, silyl groups offer an ideal combination of selectivity and functional group tolerance. We therefore reasoned that a potential solution to the observed instability of the TBS protecting group would be to use a more hindered tert- butyldiphenylsilyl (BPS) protecting group.
On the surface, the use of the BPS rather than TBS group seemed to be a straightforward perturbation, but there were uncertainties. The first was whether this exchange would indeed allow us to generate the β-linked Araf derivatives (vide infra). Perhaps the most pressing unknown, however, was whether the steric demands of this protecting group would prevent generating the target glycosyl donor. For example, a recent attempt by Dureau and coworkers to persilylate galactose with BPSCl led only to a mixture of partially silylated products.23 Fortunately, when D-arabinose was treated with BPSCl and imidazole, the desired tetra-BPS-protected Araf intermediate 8 was obtained in 70% yield. This product was obtained as a 10:1 mixture of anomers favoring the α-configuration (Scheme 1). Selective removal of the anomeric BPS group was accomplished by treating 7 with excess trifluoroacetic acid for 5 minutes, followed by carefully pouring the reaction mixture into a solution of ammonium hydroxide in methanol at −15 °C. The desired product, 2,3,5-(BPS)-Araf (9), was obtained as a 3:1 mixture of α- and β-anomers. Acetylation of 9 resulted in glycosyl acetate 10, which was formed as a 9:1 ratio of anomers, again favoring the α-anomer.
Scheme 1.

Synthesis of β-D-arabinofuranosyl phosphodiesters 12-18.
The C1 phosphate moiety was installed in two steps: first, by conversion of acetate 10 into the corresponding glycosyl bromide intermediate was effected using bromotrimethylsilane, and second, displacement of the anomeric bromide substituent was carried out with dibenzyl phosphate serving as a nucleophile to afford 11. This two-step sequence was highly selective for the desired β-anomer (>10:1 β/α).24 This selectivity is notable. When the same reaction sequence was performed on the analogous TBS-protected intermediate, the process was much less selective (a 2:1-4:1 mixture of β- and α-anomers). When BPS protection was employed, therefore, not only did the desired β-linked phosphotriester predominate but it was obtained in high yield.
We considered the origin of the higher selectivity obtained with the substrate bearing BPS relative to that possessing TBS protecting groups. Since the glycosylation reaction of the phosphodiester is anticipated to proceed through an SN2 mechanism, one might anticipate that the ratio of glycosyl bromide anomers would be reflected in the ratio of phosphorylated products.20 NMR analysis of the crude glycosyl bromides, however, revealed that the identity of the silyl group has little influence on the ratio of glycosyl bromides; both substrates had a preference for the α-configuration (92:8 α/β for TBS and >95:5 α/β for BPS).25,26 More flexibility in the TBS protected substrate may allow the reaction to proceed through not only the SN2 reaction, but also a competing SN1-like pathway involving an oxocarbenium intermediate.27 The increased steric demands in the BPS-protected substrate may make it difficult for this species to adopt a conformation that can accommodate an oxocarbenium intermediate. Whatever the origins of the selectivity, this phosphorylation reaction is a remarkable case in which the identity of the silyl group has a large effect on selectivity in a furanose ring system. It should be noted that bulky silicon protecting groups have been exploited in pyranose systems to control ring conformation and anomeric selectivity in glycosylation reactions.28-30 Moreover, Bols and coworkers demonstrated that conformational preferences in pyranose glycosyl donors enforced by bulky silicon protecting groups can dramatically enhance their reactivity in glycosylation reactions.31-34 No such effects had been reported in furanose sugars. We postulate that the dramatic influence of the bulky silyl substituents that we observed may be exploited to produce other furanosides with high stereoselectivity.
With the C1 phosphoryl group installed in the desired β-configuration, we focused on forming the requisite phosphodiester. First, the benzyl substituents on the phosphotriester were removed by hydrogenolysis to provide the intermediate monophosphate. As expected, this entity was stable; we found no evidence of the silyl group lability that plagued our reactions when TBS was used as a protecting group. We could promote phosphodiester formation using a variety of alcohols (polyprenyl, saturated n-alkanes, and naphthyl) by heating the alcohol and protected Araf monophosphate in pyridine at 55 °C in the presence of trichloroacetonitrile. The crude phosphodiesters were then treated with ammonium fluoride in a 15% solution of aqueous ammonium hydroxide in methanol to afford the corresponding β-D-arabinofuranosyl phosphodiesters 12-18 in 62-79% yield over two steps.
Conclusion
The synthetic sequence described here can provide β-D-arabinofuranosyl phosphodiesters in 8 synthetic steps with an overall yield of 13-16%. This approach is not only highly selective for the β-anomer, but it also offers flexibility in that essentially any derivative can be prepared from the corresponding alcohol and Araf monophosphate salt. We envision that this route will facilitate the study of the mycobacterial arabinofuranosyltransferases as it efficiently and reliably provides access to shorter and more aqueous soluble analogs of the endogenous Araf donor, DPA.
Experimental Section
General Experimental Procedures, Materials, and Instrumentation
Solvents were purified according to the guidelines in Purification of Common Laboratory Chemicals.35 All reactions were run under nitrogen atmosphere unless otherwise stated. Analytical thin layer chromatography (TLC) was carried out on E. Merck (Darmstadt) TLC plates pre-coated with silica gel 60 F254 (250 Im layer thickness). Analyte visualization was accomplished using a UV lamp and by charring with p-anisaldehyde solution. Flash column chromatography was performed with Silicycle Flash Silica Gel (40-63Im, 60 Å pore size) using reagent grade hexanes and ACS grade ethyl acetate (EtOAc), or methanol and CH2Cl2. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a 300 MHz spectrometer (acquired at 300 MHz for 1H NMR and 75 MHz for 13C NMR) a 400 MHz spectrometer (acquired at 400 MHz for 1H NMR and 101 MHz for 13C NMR), or a 500 MHz spectrometer (acquired at 500 MHz for 1H NMR and 126 MHz for 13C NMR). Chemical shifts are reported relative to tetramethylsilane or residual solvent peaks in parts per million (CHCl3: 1H: 7.27, 13C: 77.23; MeOH: 1H: 3.31, 13C: 49.15). Peak multiplicity is reported as singlet (s), doublet (d), doublet of doublets (dd), doublets of doublets of doublets (ddd), triplet (t), doublet of triplets (dt), etc. High resolution electrospray ionization-time of flight mass spectra (HRESI-TOF MS) were obtained on a mass spectrometer.
1,2,3,5-tetra-O-tert-butyldiphenylsilyl-D-arabinofuranose (8)
A mixture of D-(-)-arabinose (200 mg, 1.33 mmol) and imidazole (633 mg, 9.31 mmol) were azeotropically dried by evaporation with toluene (3 × 3 mL). The mixture was taken-up in DMF (9 mL) and tert-butyldiphenylsilyl chloride (2.1 mL, 8.0 mmol) was added. The reaction mixture was stirred for 48 h at 70 °C, and was then quenched by the addition of diethanolamine (0.38 mL, 4.0 mmol). Stirring was continued for 2 h and the reaction mixture was then poured into a stirring mixture of 15% EtOAc/hexanes (25 mL) and water (25 mL). The phases were separated and the organic phase was washed with water (3 × 25 mL) and brine (30 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification was accomplished by flash column chromatography on silica gel, eluting with 4% EtOAc/hexanes. The product containing fractions were combined and concentrated under reduced pressure to provide 8 (1.03g, 70%) as a white foam: Rf = 0.53 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.71–7.49 (m, 16H), 7.41–7.07 (m, 34H), 5.23 (s, 1H), 4.51 (ddd, J = 7.4, 5.2, 2.3 Hz, 1H), 4.29 (s, 1H), 4.10 (d, J = 2.3 Hz, 1H), 3.53 (dd, J = 10.4, 7.2 Hz, 1H), 3.41 (dd, J = 10.5, 5.2 Hz, 1H), 1.00 (s, 9H) 0.97–0.95 (m, 18H), 0.70 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 136.1, 136.1, 136.1, 136.0, 135.8, 135.8, 135.7, 134.2, 133.8, 133.7, 133.5, 133.4, 132.9, 129.8, 129.8, 129.7, 129.7, 129.6, 129.5, 129.2, 128.4, 127.8, 127.8, 127.8, 127.8, 127.7, 127.7, 127.6, 127.5, 125.5, 103.9, 88.9, 84.8, 81.2, 65.5, 27.1, 27.1, 27.0, 26.8, 19.4, 19.4, 19.4, 19.1; HRMS (ESI-TOF+) calcd for C69H86NO5Si4 (M+NH4+) 1120.5578, found 1120.5588.
2,3,5-tri-O-tert-butyldiphenylsilyl-D-arabinofuranose (9)
To a stirred solution of 8 (1.27g, 1.15 mmol) in CH2Cl2 (11.5 mL) was added trifluoroacetic acid (2.90 mL, 37.9 mmol). The reaction mixture was stirred at rt for 5 min and was then slowly poured into a 15% solution of concentrated ammonium hydroxide in MeOH (38 mL) at −15 °C. The mixture was stirred at −15 °C for 30 min and was then warmed to rt. The mixture was partitioned between 1:1 CH2Cl2/water (50 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 30 mL). The combined organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification was accomplished by flash column chromatography on silica gel, eluting with 4% EtOAc/hexanes. The product containing fractions were combined and concentrated under reduced pressure to provide 9 (681 mg, 69%) as a clear, highly viscous oil: Rf = 0.31 (10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.63–7.12 (m, 30H), 5.28 (dd, J = 12.6, 3.4 Hz, 0.25H, H-1β), 5.10 (d, J = 12.1, 0.75H, H-1α), 4.56 (dd, J = 6.9, 6.9 Hz, 0.75H, H-4α), 4.26 (dd, J = 1.7, 1.7 Hz, 0.25H, H-2β), 4.22 (s, 0.75H, H-2α), 4.05 (s, 1H, H-3α,β), 3.97 (dd, J = 5.8, 5.8 Hz, 0.25H, H-4β), 3.72 (dd, J = 10.2, 6.7 Hz, 0.75H, H-5α), 3.68 (d, J = 12.1 Hz, 0.75H, OHα), 3.57–3.49 (m, 0.75H, H-5-α), 3.39 (dd, J = 10.5, 6.1 Hz, 0.25H, H-5β), 3.29 (dd, J = 10.5, 5.7 Hz, 0.25H, H-5-β), 0.98 (s, 2.3H, BPS-β), 0.96 (s, 6.7H, BPS-α), 0.95 (s, 6.7H, BPS-α), 0.92 (s, 2.3H, BPS-β), 0.88 (s, 2.3H, BPS-β), 0.80 (s, 6.7H, BPS-α); 13C NMR (101 MHz, CDCl3) δ 136.0, 135.9, 135.9, 135.8, 135.8, 135.7, 135.7, 135.0, 133.8, 133.5, 133.5, 133.4, 133.2, 133.0, 132.9, 132.58, 132.5, 132.5, 132.4, 132.2, 130.2, 130.2, 130.1, 130.0, 129.9, 129.9, 129.9, 129.8, 129.8, 129.7, 128.1, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.8, 104.0, 98.7, 88.8, 85.5, 81.2, 79.4, 78.7, 78.1, 65.2, 65.0, 27.1, 27.0, 27.0, 26.9, 26.8, 19.3, 19.3, 19.3, 19.2, 19.1; HRMS (ESI-TOF+) calcd for C53H68NO5Si3 (M+NH +4) 882.4400, found 882.4431.
1-O-acetyl-2,3,5-O-tert-butyldiphenylsilyl-β-D-arabinofuranose (10)
To a stirred solution of 9 (0.64 g, 0.74 mmol) in pyridine (7.4 mL) were added 4-dimethylaminopyridine (9 mg, 0.07 mmol) and acetic anhydride (0.21 mL, 2.2 mmol). The reaction mixture was stirred at rt for 3.5 h and was then quenched by the addition of saturated aqueous NaHCO3 solution (10 mL). The mixture was partitioned between CH2Cl2 (15 mL) and water (15 mL), the phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification was accomplished by flash column chromatography on a 2.5 × 19 cm silica gel column, eluting with 4% EtOAc/hexanes, collecting 13 × 100 mm test tube fractions. The product containing fractions (19–29) were combined and concentrated under reduced pressure to provide 10 (473 mg, 70%) as a clear, highly viscous oil: Rf = 0.30 (6% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) δ 7.62–7.13 (m, 30H), 5.97 (s, 1H), 4.49 (ddd, J = 7.6, 6.2, 1.3 Hz, 1H), 4.33 (s, 1H), 4.16 (d, J = 0.9 Hz, 1H), 3.66 (dd, J = 10.3, 6.4 Hz, 1H), 3.49 (dd, J = 10.3, 7.3 Hz, 1H), 1.94 (s, 3H), 0.97 (s, 9H), 0.96 (s, 9H), 0.83 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 169.9, 135.9, 135.9, 135.9, 135.8, 135.7, 133.7, 133.5, 133.4, 133.3, 132.7, 132.6, 130.1, 130.0, 129.9, 129.9, 129.8, 129.7, 127.9, 127.9, 127.8, 127.8, 102.6, 90.5, 82.4, 79.1, 64.8, 27.0, 26.9, 21.3, 19.4, 19.4, 19.1; HRMS (ESI-TOF+) calcd for C69H86NO5Si4 (M+NH4+) 924.4506, found 924.4528.
Dibenzyl(2,3,5-O-tert-butyldiphenylsilyl-β-D-arabinofuranosyl)-1-phosphate (11)
To a stirred solution of 10 (0.46 g, 0.50 mmol) in CH2Cl2 (5 mL) was added trimethylsilyl bromide (133 μL, 1.01 mmol). The reaction mixture was stirred at rt for 6 h and was then concentrated under reduced pressure. The crude glycosyl bromide was azeotropically dried with toluene (3 × 2 mL) and then used immediately.
To a stirred solution of azeotropically dried dibenzylphosphate (281 mg, 1.01 mmol) in toluene (3 mL) were added powdered 4 Å molecular sieves (400 mg) and triethylamine (182 μL, 1.31 mmol). The mixture was cooled to 0 °C and a solution of the aforementioned glycosyl bromide in toluene (1 mL) was added slowly via cannula. The transfer was completed by rinsing twice with toluene (2 × 0.5 mL). Stirring was continued at 0 °C for 1 h and then at rt overnight. The reaction mixture was filtered through a pad of sand and celite, rinsing with EtOAc, and the solvent was removed under reduced pressure. Purification was accomplished by flash column chromatography on a 2.5 × 19 cm silica gel column, eluting with 15% EtOAc/hexanes, collecting 13 × 100 mm test tube fractions. The product containing fractions (10–29) were combined and concentrated under reduced pressure to provide 11 (356 mg, 63% over two steps) as a clear viscous oil: Rf = 0.26 (15% EtOAc/hexanes); 1H NMR (300 MHz, CDCl3) δ 7.60–7.41 (m, 13H), 7.40–7.10 (m, 27H), 5.78 (dd, J = 5.0, 3.0 Hz, 1H), 4.98 (dd, J = 11.9, 6.9 Hz, 1H), 4.87 (dd, J = 11.9, 7.7 Hz, 1H), 4.77 (d, J = 6.5 Hz, 2H), 4.32 (s, 1H), 4.24 (td, J = 7.0, 1.5 Hz, 1H), 4.12 (dd, J = 2.4, 2.4 Hz, 1H), 3.72 (dd, J = 10.4, 7.2 Hz, 1H), 3.61 (dd, J = 10.4, 6.8 Hz, 1H), 0.92 (s, 9H), 0.92 (s, 9H), 0.87 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 136.2, 136.1, 136.1, 136.0, 136.0, 135.9, 135.9, 135.7, 133.7, 133.5, 133.2, 132.8, 132.8, 130.0, 129.9, 129.9, 129.8, 129.7, 128.5, 128.5, 128.4, 128.3, 128.0, 127.9, 127.9, 127.9, 127.9, 127.8, 127.7, 101.1(d), 87.0, 78.3, 78.2(d), 69.1(d), 69.0(d), 64.7, 27.1, 27.0, 26.9, 19.5, 19.3; HRMS (ESI-TOF+) calcd for C67H81NO8Si3 (M+NH +4) 1142.5003, found 1142.5027.
Bis-triethylammonium(2,3,5-O-tert-butyldiphenylsilyl-β-D-arabinofuranosyl)-1-phosphate (S3)
To a stirred solution of 11 (259 mg, 0.230 mmol) in 10% EtOH/EtOAc (8.2 mL) were added triethylamine (0.8 mL, 5.75 mmol) and 10% palladium on carbon (17 mg, 0.016 mmol). The reaction vessel was purged with N2 and then equipped with an H2 filled balloon. The reaction mixture was stirred at rt overnight and was then filtered through a plug of sand and celite with 10% EtOH/EtOAc. The solvent was removed under reduced pressure to provide monophosphoryl salt S3 (259 mg, 98%) as a white powder: Rf = 0.5 (15% MeOH/CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.75 – 7.67 (m, 2H), 7.56 (m, 4H), 7.53 – 7.48 (m, 2H), 7.46 – 7.40 (m, 2H), 7.40 – 7.21 (m, 14H), 7.15 (m, 4H), 7.07 (t, J = 7.5 Hz, 2H), 5.86 (dd, J = 7.1, 3.0 Hz, 1H), 4.22 (s, 1H), 4.10 (brt, J = 2.4 Hz, 1H), 4.05 (t, J = 7.2 Hz, 1H), 3.70 (dd, J = 10.4, 7.6 Hz, 1H), 3.47 (m, 1H), 2.97 (q, J = 7.3 Hz, 12H), 1.25 (t, J = 7.3 Hz, 18H), 0.93 (s, 9H), 0.92 (s, 9H), 0.85 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 136.7, 136.0, 135.8, 135.2, 135.7, 134.1, 133.7, 133.7, 133.6, 133.0, 132.7, 129.7, 129.7, 129.6, 129.4, 127.8, 127.7, 127.7, 127.6, 99.8(d), 85.3, 78.9, 78.5(d), 65.2, 45.6, 27.2, 27.0, 27.0, 19.6, 19.3, 19.2, 8.7. HRMS (ESI-TOF+) calcd for C53H69NO8PSi3 (M+NH +4) 962.4064, found 962.4080.
(Z,Z)-Farnesylphosphoryl-β-D-arabinofuranose (12)9
A mixture of monophosphate salt S3 (86 mg, 0.075 mmol) and (Z,Z)-farnesol36 (67 mg, 0.30 mmol) were azeotropically dried with anhydrous pyridine (3 × 0.5 mL). The mixture was taken-up in pyridine (1 mL) and trichloroacetonitrile (75 μL, 0.75 mmol) was added. The reaction mixture was stirred at 55 °C for 12 h. The solvent was removed under reduced pressure and the crude phosphodiester was carried onto deprotection without purification.
To a solution of the aforementioned BPS protected phosphodiester in a 15% solution of conc. ammonium hydroxide in methanol (1.5 mL) was added ammonium fluoride (83 mg, 2.2 mmol). The reaction mixture was stirred at 55 °C for 10 h. After cooling to rt, CH2Cl2 (2 mL) was added and the precipitate that had formed was removed by filtration through celite. The solvent was removed under reduced pressure and purification was accomplished by flash column chromatography on a 1 × 6 cm silica gel column. The column was eluted with 10% MeOH/CH2Cl2 (fractions 1–10) then 1% H2O/20% MeOH/CH2Cl2 (fractions 11–60) while collecting 1.5 mL test tube fractions. The product containing fraction (14–65) were combined and concentrated under reduced pressure to provide 12 (27 mg, 79%) as a clear colorless oil: Rf = 0.27 (4% H2O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 5.48 (t, J = 4.5 Hz, 1H), 5.45–5.38 (m, 1H), 5.19–5.07 (m, 2H), 4.43 (t, J = 6.3 Hz, 2H), 4.13–4.04 (m, 1H), 3.90–3.93 (m, 1H), 3.82–3.69 (m, 2H), 3.63 (dd, J = 12.2, 5.7 Hz, 1H), 2.17–1.98 (m, 8H), 1.74 (s, 3H), 1.68 (s, 6H), 1.61 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 140.9, 136.7, 132.5, 125.9, 125.5, 123.5(d), 99.2(d), 85.1, 79.5(d), 75.3, 64.2, 63.4(d), 33.4, 33.1, 27.8, 27.7, 26.1, 23.9, 23.8, 17.9; HRMS (ESI-TOF−) calcd for C20H34O8P (M−) 433.1996, found 433.1991.
(Z)-Nerylphosphoryl-β-D-arabinofuranose (13)
Prepared in the same manner as described for compound 12 from intermediate monophosphate salt S3 (30 mg, 0.026 mmol) and nerol (20 μL, 0.11 mmol). Compound 13 (7.6 mg, 76%) was generated as a clear colorless oil: Rf = 0.26 (4% H O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 5.48 (t, J = 4.6 Hz, 1H), 5.41 (t, J = 6.9 Hz, 1H), 5.15-5.09 (m, 1H), 4.49-4.38 (m, 2H), 4.08 (dd, J = 8.0, 7.0 Hz, 1H), 3.96 (ddd, J = 8.1, 4.3, 2.1 Hz, 1H), 3.80-3.69 (m, 2H), 3.63 (dd, J = 12.2, 5.8 Hz, 1H), 2.17-2.01 (m, 4H), 1.74 (s, 3H), 1.67 (s, 3H), 1.61 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 141.0, 132.9, 125.2, 123.5(d), 99.2(d), 85.1, 79.5(d), 75.3, 64.1, 63.5(d), 33.2, 27.9, 26.1, 23.8, 17.9; HRMS (ESI-TOF−) calcd for C20H34O8P (M−) 365.1370, found 365.1373.
((R)-Citronellyl)phosphoryl-β-D-arabinofuranose (14)
Prepared in the same manner as described for compound 12 from intermediate monophosphate salt S3 (30 mg, 0.026 mmol) and (R)-citronellol (21 μL, 0.11 mmol). Compound 14 (6.2 mg, 62%) was generated as a clear colorless oil: Rf = 0.24 (4% H2O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 5.47 (t, J = 4.6 Hz, 1H), 5.14–5.08 (m, 1H), 4.08 (dd, J = 8.1, 6.9 Hz, 1H), 4.00–3.89 (m, 3H), 3.81–3.69 (m, 2H), 3.62 (dd, J = 12.1, 5.8 Hz, 1H), 2.09–1.91 (m, 2H), 1.75–1.63 (m, 1H), 1.67 (s, 3H), 1.61(s, 3H), 1.48–1.33 (m, 3H), 1.22–1.11 (m, 1H), 0.92 (d, J = 6.5 Hz, 3H); 13C NMR (101 MHz, CD3OD) δ 132.0, 126.1, 99.2(d), 85.1, 79.5(d), 75.3, 65.3(d), 64.2, 39.0(d), 38.5, 30.5, 26.6, 26.0, 19.9, 17.9, 17.8. HRMS (ESI-TOF−) calcd for C15H28O8P (M−) 367.1527, found 367.1534.
n-Octylphosphoryl-β-D-arabinofuranose (15)
Prepared in the same manner as described for compound 12 from intermediate monophosphate salt S3 (30 mg, 0.026 mmol) and n-octanol (18 μL, 0.11 mmol). Compound 15 (7.3 mg, 78%) was generated as a clear colorless oil: Rf = 0.27 (4% H2O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 5.47 (t, J = 4.5 Hz, 1H), 4.07 (dd, J = 8.3, 6.9 Hz, 1H), 3.99-3.93 (m, 1H), 3.89 (q, J = 6.4 Hz, 2H), 3.82-3.69 (m, 2H), 3.62 (dd, J = 12.1, 5.7 Hz, 1H), 1.63 (p, J = 6.8 Hz, 2H), 1.47-1.21 (m, 10H), 0.90 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CD3OD) δ 99.2(d), 85.1, 79.5(d), 75.3, 67.0(d), 64.2, 33.2, 32.0(d), 30.6, 27.1, 27.0, 23.9, 14.6; HRMS (ESI-TOF−) calcd for C13H26O8P (M−) 341.1370, found 341.1365.
(8-Phenyloctyl)phosphoryl-β-D-arabinofuranose (16)
Prepared in the same manner as described for compound 12 from intermediate monophosphate salt S3 (30 mg, 0.026 mmol) and 8-phenyl-1-octanol (24 mg, 0.11 mmol). Compound 16 (7.4 mg, 65%) was generated as a clear colorless oil: Rf = 0.30 (4% H2O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 7.24 (t, J = 7.6 Hz, 2H), 7.19–7.08 (m, 3H), 5.47 (t, J = 4.6 Hz, 1H), 4.15–4.03 (m, 1H), 3.99–3.92 (m, 1H), 3.92–3.81 (m, 2H), 3.80–3.70 (m, 2H), 3.62 (dd, J = 12.1, 5.8 Hz, 1H), 2.59 (t, J = 7.7 Hz, 2H), 1.67-1.56 (m, 4H), 1.49–1.20 (m, 8H); 13C NMR (101 MHz, CD3OD) δ 144.1, 129.5, 129.4, 126.7, 99.2(d), 85.1, 78.9, 75.3, 67.0(d), 64.2, 37.1, 32.9, 31.9(d), 30.75, 30.6, 30.4, 27.0; HRMS (ESI-TOF−) calcd for C19H30O8P (M−) 417.1683, found 417.1693.
n-Dodecylphosphoryl-β-D-arabinofuranose (17)
Prepared in the same manner as described for 12 from intermediate monophosphate salt S3 (89 mg, 0.078 mmol) and n-dodecanol (70 μL, 0.31 mmol). Compound 17 (23 mg, 71%) was generated as a colorless wax: Rf = 0.30 (4% H2O/30% MeOH/66% CH2Cl2); 1H NMR (300 MHz, CD3OD) δ 5.47 (t, J = 4.6 Hz, 1H), 4.12–4.03 (m, 1H), 3.97 (ddd, J = 8.1, 4.3, 2.2 Hz, 1H), 3.90 (q, J = 6.5 Hz, 2H), 3.82–3.69 (m, 2H), 3.68-3.58 (m, 1H), 1.69–1.55(m, 2H), 1.48–1.17 (m, 18H), 0.90 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 99.2(d), 85.08, 79.4(d), 75.33, 67.0(d), 64.2, 33.2, 32.0, 31.9, 30.9, 30.9, 30.9, 30.7, 30.6, 27.0, 23.9, 14.6, 9.3; HRMS (ESI-TOF−) calcd for C17H34O8P (M−) 397.1996, found 397.1995.
(2-Naphthalenemethyl)phosphoryl-β-D-arabinofuranose (18)
Prepared in the same manner as described for compound 12 from intermediate monophosphate salt S3 (30 mg, 0.026 mmol) and 2-naphthalenemethanol (18 mg, 0.11 mmol). Compound 18 (6.6 mg, 65%) was generated as a clear colorless oil: Rf = 0.24 (4% H2O/25% MeOH/71% CH2Cl2); 1H NMR (400 MHz, CD3OD) δ 7.90 (s, 1H), 7.88–7.79 (m, 3H), 7.55 (dd, J = 8.5, 1.8 Hz, 1H), 7.50–7.41 (m, 2H), 5.58 (t, J = 4.7 Hz, 1H), 5.13 (d, J = 5.9 Hz, 2H), 4.11 (t, J = 7.6 Hz, 1H), 4.03-3.97 (m, 1H), 3.85–3.69 (m, 2H), 3.64 (dd, J = 12.1, 5.8 Hz, 1H); 13C NMR (101 MHz, CD3OD) δ 137.4, 137.3, 134.9, 134.6, 129.1, 129.1, 128.8, 127.18, 127.0, 126.8, 99.4(d), 85.2, 79.5, 75.3, 68.7(d), 64.3; HRMS (ESI-TOF−) calcd for C16H18O8P (M−) 369.0744, found 369.0743.
Supplementary Material
Acknowledgements
This paper is dedicated to the memory of our colleague Howard Zimmerman, who inspired us with his relentless pursuit of mechanistic understanding. This research was supported by National Institutes of Health (R01-AI063596). The UW-Madison Chemistry NMR facility is supported by the NSF (CHE-9208463 and CHE-9629688) and NIH (1 s10 RRO 8389). M.B.K. was supported by an NIH postdoctoral fellowship (F32 GM100729). M.A.M. was supported by an NIH predoctoral fellowship (F31 GM101953).
Footnotes
Supporting Information
1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Umesiri FE, Sanki AK, Boucau J, Ronning DR, Sucheck SJ. Med. Res. Rev. 2010;30:290–326. doi: 10.1002/med.20190. [DOI] [PubMed] [Google Scholar]
- (2).Tam PH, Lowary TL. Curr. Opin. Chem. Biol. 2009;13:618–625. doi: 10.1016/j.cbpa.2009.09.012. [DOI] [PubMed] [Google Scholar]
- (3).Berg S, Kaur D, Jackson M, Brennan PJ. Glycobiology. 2007;17:35–56R. doi: 10.1093/glycob/cwm010. [DOI] [PubMed] [Google Scholar]
- (4).Wolucka BA, McNeil MR, de Hoffmann E, Chojnacki T, Brennan PJ. J. Biol. Chem. 1994;269:23328–23335. [PubMed] [Google Scholar]
- (5).Lee RE, Mikusova K, Brennan PJ, Besra GS. J. Am. Chem. Soc. 1995;117:11829–11832. [Google Scholar]
- (6).Splain RA, Kiessling LL. Bioorg. Med. Chem. 2010;18:3753–3759. doi: 10.1016/j.bmc.2010.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brown CD, Rusek MS, Kiessling LL. J. Am. Chem. Soc. 2012;134:6552–6555. doi: 10.1021/ja301723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liav A, Huang H, Ciepichal E, Brennan PJ, McNeil MR. Tetrahedron Lett. 2006;47:545–547. [Google Scholar]
- (9).Zhang J, Angala SK, Pramanik PK, Li K, Crick DC, Liav A, Jozwiak A, Swiezewska E, Jackson M, Chatterjee D. ACS Chem. Biol. 2011;6:819–828. doi: 10.1021/cb200091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lee RE, Brennan PJ, Besra GS. Bioorg. Med. Chem. Lett. 1998;8:951–954. doi: 10.1016/s0960-894x(98)00147-4. [DOI] [PubMed] [Google Scholar]
- (11).Liav A, Brennan PJ. Tetrahedron Lett. 2005;46:2937–2939. [Google Scholar]
- (12).Liav A, Ciepichal E, Swiezewska E, Bobovska̵ A, Dianiskova̵ P, Blasko J, Mikusova̵ K, Brennan PJ. Tetrahedron Lett. 2009;50:2242–2244. [Google Scholar]
- (13).Shibaev VN, Danilov LL. Biochem. Cell Biol. 1992;70:429–437. doi: 10.1139/o92-066. [DOI] [PubMed] [Google Scholar]
- (14).Neumann JM, Herve M, Debouzy JC, Iglesias Guerra F, Gouyette C, Dupraz B, Huynh Dinh T. J. Am. Chem. Soc. 1989;111:4270–4277. [Google Scholar]
- (15).Cramer F, Weimann G. Chem. Ber. 1961;94:996–1007. [Google Scholar]
- (16).Cramer F, Böhm W. Angew. Chem. 1959;71:775–775. [Google Scholar]
- (17).Cramer F, Rittersdorf W, Böhm W. Justus Liebigs Annalen der Chemie. 1962;654:180–188. [Google Scholar]
- (18).Anastasi C, Buchet FF, Crowe MA, Helliwell M, Raftery J, Sutherland JD. Chem. Eur. J. 2008;14:2375–2388. doi: 10.1002/chem.200701351. [DOI] [PubMed] [Google Scholar]
- (19).Recently Shih and coworkers have reported microwave assisted conditions for phosphodiester formation. The authors report these condition on a 2:1 beta/alpha mixture of 4 to generate a solanesol analog of DPA. While reaction times are shorter using microwave assisted conditions, our analysis of the authors- NMR data suggests than mainly the alpha-anomer was formed. This is consistent with our own attempt to form the phosphodiester from intermediate 4: Shih H-W, Chen K-T, Cheng W-C. Tetrahedron Lett. 2012;53:243–246.
- (20).Maryanoff BE, Reitz AB, Tutwiler GF, Benkovic SJ, Benkovic PA, Pilkis SJ. J. Am. Chem. Soc. 1984;106:7851–7853. [Google Scholar]
- (21).Maryanoff BE, Reitz AB, Nortey SO. Tetrahedron. 1988;44:3093–3106. [Google Scholar]
- (22).Smellie IA, Bhakta S, Sim E, Fairbanks AJ. Org. Biomol. Chem. 2007;5:2257–2266. doi: 10.1039/b704788f. [DOI] [PubMed] [Google Scholar]
- (23).Dureau R, Legentil L, Daniellou R, Ferrières V. J. Org. Chem. 2011;77:1301–1307. doi: 10.1021/jo201913f. [DOI] [PubMed] [Google Scholar]
- (24).It should be noted that after prolonged storage (>6 mo.) at -20 °C compound 11 equilibrated to the alpha-anomer. It is therefore recommended that this intermediate is carried forward to avoid erosion of the anomeric stereochemistry.
- (25).Gillard JW, Israel M. Tetrahedron Lett. 1981;22:513–516. [Google Scholar]
- (26).See Supporting Information for 1H and 13C NMR spectra of the glycosyl bromides.
- (27).Bohe̵ L, Crich D. C. R. Chim. 2011;14:3–16. [Google Scholar]
- (28).Shuto S, Terauchi M, Yahiro Y, Abe H, Ichikawa S, Matsuda A. Tetrahedron Lett. 2000;41:4151–4155. [Google Scholar]
- (29).Ichikawa S, Shuto S, Matsuda A. J. Am. Chem. Soc. 1999;121:10270–10280. [Google Scholar]
- (30).Hosoya T, Ohashi Y, Matsumoto T, Suzuki K. Tetrahedron Lett. 1996;37:663–666. [Google Scholar]
- (31).Heuckendorff M, Pedersen CM, Bols M. J. Org. Chem. 2012;77:5559–5568. doi: 10.1021/jo300591k. [DOI] [PubMed] [Google Scholar]
- (32).Jensen HH, Pedersen CM, Bols M. Chem. Eur. J. 2007;13:7576–7582. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]
- (33).Pedersen CM, Nordstrøm LU, Bols M. J. Am. Chem. Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- (34).Pedersen CM, Marinescu LG, Bols M. Chem. Commun. 2008:2465–2467. doi: 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]
- (35).Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4th ed. Butterworth Heinemann; Oxford ; Boston: 1996. [Google Scholar]
- (36).Snyder SA, Treitler DS, Brucks AP. J. Am. Chem. Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.