

Abstract
Introduction
DOCK8 mutations are responsible for a rare primary combined immunodeficiency syndrome associated with severe cutaneous viral infections, elevated IgE, autoimmunity, and malignancy. Natural killer (NK) cells are essential for tumor surveillance and defense against virally infected cells. NK cell function relies on Wiskott-Aldrich syndrome protein (WASp) for filamentous actin (F-actin) accumulation at the lytic NK cell immunologic synapse (IS). DOCK8 activates Cdc42, which, together with WASp, coordinates F-actin reorganization. While abnormalities in T and B cell function have been described in DOCK8-deficient patients, the role of NK cells in this disease is unclear.
Objectives
Understand the role of DOCK8 in NK cell function in order to determine if NK cell abnormalities explain the pathogenesis of the clinical syndrome of DOCK8 deficiency.
Methods
A cohort of DOCK8-deficient patients was assembled and patient NK cells as well as NK cell lines with stably reduced DOCK8 expression were studied. NK cell cytotoxicity, F-actin content, and lytic immunological synapse formation were measured.
Results
DOCK8-deficient patient NK cells and DOCK8 knockdown cell lines all had decreased NK cell cytotoxicity, which could not be restored after IL-2 stimulation. Importantly, DOCK8 deficiency impaired F-actin accumulation at the lytic immunological synapse without affecting overall NK cell F-actin content.
Conclusions
DOCK8 deficiency results in severely impaired NK cell function owing to an inability to form a mature lytic IS via targeted synaptic F-actin accumulation. This defect may underlie and explain important attributes of the DOCK8 deficiency clinical syndrome including the unusual susceptibility to viral infection and malignancy.
Keywords: DOCK8 deficiency, NK cells, actin, cytotoxicity, immunologic synapse
INTRODUCTION
Patients with autosomal recessive hyper IgE syndrome (HIES) can be affected by recalcitrant cutaneous herpes-viral and papillomavirus infections as well as immune dysregulation.1, 2 The major genetic abnormality found in these patients is deletion or loss of function mutations in the gene encoding dedicator of cytokinesis 8 (DOCK8).3, 4 Mutagenized mice with an autoimmune phenotype were also identified as possessing DOCK8 mutations.5 Both DOCK8-deficient patients and mice are documented to have combined immunodeficiency of the B and T cell compartments.5–8 The high incidence of recurrent cutaneous viral infections, however, distinguishes them from other patients with combined immunodeficiencies and a mechanistic explanation for this phenotype is lacking. Currently the only definitive therapy for patients with DOCK8 deficiency is hematopoietic stem cell transplant.9–11
One immunological characteristic found in patients with susceptibility to herpesviruses and papillomaviruses can be abnormalities of natural killer (NK) cells.12, 13 This is illustrated by a number of human NK cell deficiency states, NK cell depleted murine models, as well as some of the specific aspects of the virology.13–15 NK cells are lymphocytes of the innate immune system that can inherently recognize many virally infected or malignantly transformed cells, especially those which escape the adaptive T cell response owing to down-regulated MHC (as in herpesvirus infections). NK cells have not been previously evaluated in DOCK8 deficiency other than absolute numbers, which were normal.1, 3, 4 Specifically, functional assessments of NK cells in DOCK8 deficiency have not been reported.
DOCK8 is a member of the DOCK180 super family of atypical guanine-nucleotide exchange factors16, 17 and activates cell division cycle 42 (Cdc42), a Rho GTPase critical for reorganization of the filamentous actin (F-actin) cytoskeleton in NK and dendritic cells.18, 19 Cdc42 functions along with Wiskott-Aldrich syndrome (WAS) protein (WASp) to regulate NK cell function.20 Defects in WASp are associated with the well known primary immunodeficiency WAS, and these patients exhibit several shared characteristics with DOCK8 deficiency including susceptibility to cutaneous viral infections and malignancy. NK cells isolated from WAS patients are functionally deficient.20–22 Given the clinical phenotype of DOCK8 patients and its integral role in Cdc42 activation, we hypothesized there was a substantive defect in NK cell function in patients with DOCK8 deficiency.
METHODS
Patients and human subjects
Ten patients with DOCK8 mutations were diagnosed by either targeted gene sequence or genomic approaches. DOCK8-4, DOCK8-5, DOCK8-8 and DOCK8-9 have previously been reported.4, 23, 24 There were two sibling pairs, DOCK8-4 and DOCK8-5 and DOCK8-3 and DOCK8-6, respectively. Three patients with signal transducer and activator of transcription 3 (STAT3) deficiency were included as disease controls. Patient and healthy donor blood samples were obtained after patient or parental informed consent under the approval of the local Institutional Review Board for the Protection of Human Subjects of the Children’s Hospital of Philadelphia, Baylor College of Medicine, or Ludwig Maximilian University, Munich, Germany.
NK cells and cell lines
Peripheral blood mononuclear cells (PBMCs) or ex vivo NK (eNK) cells were isolated from whole blood via centrifugation through Ficoll-Paque Plus (Amersham Biosciences) with or without negative selection via RosetteSep human NK cell enrichment cocktail (StemCell Technologies) as previously described.25, 26 eNK cells and PBMCs were used immediately after preparation or were cryopreserved for later use and purity determined via flow cytometry. The human NK cell line YTS was used to establish the in vitro DOCK8 knockdown model. Briefly, HEK293T cells were co-transfected with lentiviral packaging plasmids (pPACK, System Biosciences) and pLKO.1 lentiviral vector containing puromycin resistance, and either DOCK8 or scrambled DOCK8 sequence short-hairpin RNA (shRNA) (MissionRNAi, Sigma-Aldrich) to produce replication-incompetent viral particles, which were used to infect YTS cells. 721.221 B-lymphoblastoid, K562 erythroleukemia, and K562 cells stably expressing CD86 (KT86)27 were used as target cells and maintained as previously described.
Cytotoxicity
Standard 51Cr-release assays were utilized to measure cytolytic activity of PBMCs isolated from human samples and NK cell lines as previously described.21 DOCK8-1 and DOCK8-4 were assessed 3 distinct times. Controls included both shipping and local donor control blood samples. Higher effector:target cell ratios (E:T) were used for PBMCs than for YTS cell lines. Lytic units were calculated as previously described.21
Flow cytometry
NK cells among PBMCs were quantified by flow cytometry as described21 using fluorophore-conjugated mAbs (BD Biosciences) for CD56 and CD3. NK cell F-actin content was also measured by flow cytometry using our published methods.22, 27 DOCK8-1 and DOCK8-4 were assessed 3 distinct times.
Microscopy and image analysis
Patient and control donor NK cells as well as YTS cells were prepared for evaluation of fixed effector:target cell conjugates by immunofluorescence microscopy as previously described22, 27, 28 and images acquired using Zeiss-Z1 microscope outfitted with a Yokogawa CSU10 spinning disc and 63× 1.45 NA objective. Images were acquired and quantified using Volocity software (PerkinElmer) and data was exported to GraphPad Prism (GraphPad Software). Previously published quantitative algorithms were applied to measure F-actin and CD18 accumulation28 and pericentrin distance from the immunologic synapse (IS).27
Quantitative PCR
RNA was harvested using Qiagen RNeasy Mini kit and reverse transcribed into cDNA using Taqman Reverse Transcription kit (Applied Biosystems). DOCK8 expression was analyzed by quantitative PCR (7900HT Analyzer, Applied Biosystems) utilizing forward (5′-ACGCGCCGTGTAACTGTGAA-3′) and reverse (5′-CCCCGAGCTCCTGGGCAA-3′) primers as previously reported3 with each assay performed in triplicate. Expression of GAPDH (forward 5′-CTCATTTCCTGGTATGACAACG-3′, reverse 5′-TTACTCCTTGGAGGCCATGT-3′) was used as a control for normalization.
Statistics
Data were compared using unpaired Student’s 2-tailed t tests or exact Wilcoxon-Mann-Whitney U tests with significance defined as p < 0.05.
RESULTS
DOCK8-deficient patients have impaired NK cell cytotoxicity that is not rescued by IL-2 stimulation
We collected an international cohort of 10 DOCK8-deficient patients to examine NK cell function. Our patients ranged from 1.5 to 26 years of age at evaluation and 90% had a history of herpesvirus, papillomavirus or molluscum contagiousum. Specific DOCK8 mutations varied, except for two sets of siblings who shared the same homozygous splice site mutation or homozygous deletion as indicated (Table 1). Other clinical findings, including elevated IgE levels and eosinophil counts, presence of asthma, autoimmunity, eczema and allergies are consistent with previously reported DOCK8-deficient patients.3, 4, 23 Patients DOCK8-4, DOCK8-5, DOCK8-8 and DOCK8-9 have previously been reported,4, 23, 24 while all others are unique to this series. Percentages of CD4+ and CD8+ T cells were within normal ranges, but absolute values were decreased in 7 of 10 and 5 of 10 patients, respectively. B cell numbers were normal in 8 of 10 of our DOCK8-deficient patients. In those patients where mitogen (9 patients) and/or antigen (6 patients) induced lymphocyte proliferation assays were performed, only DOCK8-2 and DOCK8-5 derived cells responded normally. Only DOCK8-2 maintained normal pneumococcal and tetanus antibodies, however, DOCK8-1 and DOCK8-6 also produced sustained tetanus antibodies.
Table I. DOCK8 patient characteristics.
DOCK8 patient cohort
| DOCK8-1 | DOCK8-2 | DOCK8-3 | DOCK8-4 | DOCK8-5 | DOCK8-6 | DOCK8-7 | DOCK8-8 | DOCK8-9 | DOCK8-10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at evaluation | 13 years | 12 years | 6 years | 17 years | 18 years | 3 years | 8 years | 26 years | 17 months | 12 years |
| Sex | F | M | M | F | F | F | F | M | F | F |
| Mutation | homozygous deletion upstream of exon 1 including exon 26 | homozygous deletion upstream of exon 1 including exon 2 | homozygous deletion (2 bp) in exon 8: c850_851 delCT; pL284fsX293 | homozygous splice donor site mutation: c3120+1 g>t [exon 25+1 GTA>TTA; on cDNA-Ebene: exon 25 skipped] | homozygous splice donor site mutation: c3120+1 g>t [exon 25+1 GTA>TTA; on cDNA-Ebene: exon 25 skipped] | homozygous deletion (2 bp) in exon 8: c850_851 delCT; pL284fsX293 | homozygous deletions 9p24.3 323,819 324,708 |
Heterozygous deletion g(371, 489_380, 404)_(462,145 _468,814) del plus 1266delC | c.[1-?_404+?del]+[1-?_404+?del] [deletion of exons 1-5] | homozygous deletion of exons 22-25; heterozygous deletion of exon 3-21 and 26-32 |
| IgE levels (IU/mL) | 10,970 | 35,720 | 2,303 | 25,987 | 62,429 | 38,908 | NA | 1162 | 1143 | 6270 |
| eosinophils (cells/μL) | 8,968 | 4,365 | 2,635 | 11,033 | 693 | 12,096 | NA | 260 | 430 | 1100 |
| eczema | yes | yes | yes | yes | yes | yes | yes | no | yes | yes |
| viral infections | HSV, MCV | HSV | HSV, MCV | VZV, HSV | none | MCV | CMV, BKV | HSV, HPV | HSV, VZV, MCV | HPV, MCV |
| bacterial infections | recurrent pneumonia, skin abscesses | pneumonia, skin abscesses | recurrent pneumonia, skin abscesses | recurrent pneumonia, skin abscesses | pneumonia, skin abscesses | recurrent pneumonia, skin abscesses | chronic salmonella & recurrent sinopulmonary infections, skin abscesses | recurrent sinopulmonary infections | recurrent sinopulmonary infections | meningitis, bacteremia |
| fungal infections | no | skin | CMC | CMC | skin | skin | no | no | no | skin |
| autoimmunity | no | no | no | hemolytic anemia | no | no | sclerosing cholangitis | no | no | vasculopathy |
| asthma | yes | yes | no | yes | no | no | yes | no | no | yes |
| food allergies | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| malignancy | no | no | no | no | no | no | no | SCC | no | no |
| previously published | no | no | no | yes | yes | no | no | yes | yes | no |
BKV: BK virus
CMC: cutaneous mucosal candidiasis
HPV: human papillomavirus
HSV: herpes simplex virus
MCV: molluscum contagiousum virus
NA: not available
SCC: squamous cell carcinoma
VZV: varicella zoster virus
To assess NK cell cytolytic function, PBMCs isolated from individual patients were used in 51Cr-release assays against K562 erythroleukemia target cells. All patients assessed had decreased killing, as measured by lytic units per NK cell (Figure 1A). The deficit in NK cell cytotoxicity relative to control could not be rescued by short-term stimulation of PBMCs with IL-2 (Figure 1B), a known potent activator of human NK cells.29 To ensure that the deficiency in NK cell cytotoxicity was not simply a feature of the HIES clinical presentation,30 three patients with autosomal dominant HIES due to STAT3 mutation were studied. In contrast to cells from patients with DOCK8 deficiency, those from the STAT3-deficient patients had normal NK cell cytotoxicity when evaluated as lytic units per NK cell (Figure 1A). The STAT-3 deficient patient cells also robustly responded to IL-2 with increased K562 cell killing (Figure 1B). Thus, NK cell cytotoxicity was deficient in ex vivo cells from patients with DOCK8 deficiency and could not be corrected by cytokine simulation.
Figure 1. DOCK8 deficiency impairs NK cell cytotoxicity, which is not restored by IL-2.

Control donor (n=15), DOCK8-defcient and STAT3 mutant patient PBMCs were used in chromium-release assays ex vivo, without (A) or with (B) 4h stimulation using 1000U IL-2. Cytotoxic function was expressed as LU10/NK cell. Error bars represent mean ±SEM for patients analyzed three times.
NK cell numbers and F-actin content are normal in DOCK8-deficient patients
As our DOCK8-deficient patients had reduced NK cell cytotoxicity, we evaluated the presence of NK cells amongst PBMCs as reduction in activity could result from an overall decrease in NK cell number. Historically, reports of DOCK8 deficiency did not identify quantitative deficits in NK cells.1, 3, 4 In order to more accurately evaluate the patients under study in this cohort, CD56+CD3− lymphocytes were measured in ex vivo blood samples. While the percentage of NK cells within patient PBMCs was variable, in aggregate they were statistically indistinguishable from our controls (Figure 2) and fell within the range of age-specific published normal values.31 The STAT3 disease control cohort was also similar to the DOCK8-deficient patients with regards to the size of the NK cell population. Thus, it is unlikely that the decrease in NK cell cytotoxicity we observed in DOCK8-deficient patient cells was a pure feature of quantitative abnormalities in NK cells.
Figure 2. DOCK8-deficient patients have NK cells.

Flow cytometric analysis of the percent CD56+CD3− cells in PBMCs. Dotted lines display ranges for the control donors (n=15). Error bars represent mean ±SD for patients analyzed three times. Inset displays the mean NK cell percentages for DOCK8-deficient patients compared to control donors and STAT3 patients.
Given that NK cells were present in DOCK8-deficient patients, we next considered other activities of DOCK8 downstream targets. We focused on Cdc42, which is required for the induction of WASp and is essential for the maintenance of F-actin content, as well as focused F-actin accumulation at the lytic synapse in NK cells.22, 32 In this light WAS patients are defective in NK cell cytotoxicity, but have normal percentages of NK cells.21 To determine if DOCK8 deficiency might represent a functional phenocopy of WAS we first evaluated total NK cell F-actin content via flow cytometry. Surprisingly, F-actin content in DOCK8-deficient patient NK cells was statistically indistinguishable from that of control donor as well as STAT3 patient NK cells (Figure 3). There was some variability between individual patients, but again this is in contrast to what has been observed in WAS patient NK cells.21, 33 Thus, total F-actin content in DOCK8-deficient patient NK cells was normal.
Figure 3. F-actin content is normal in DOCK8-deficient patients.

Flow cytometry was performed for F-actin content within CD56+CD3− PBMCs from patients and controls (n=15). Values were normalized to each specific control used. Error bars represent mean ±SD for patients analyzed three times. Inset displays the mean NK cell F-actin content for DOCK8 patients compared to controls and STAT3 patients.
The lytic NK cell immunological synapse is abnormal in DOCK8 deficiency owing to impaired synaptic F-actin accumulation and granule polarization
While WASp is needed for maintenance of F-actin content as well as its reorganization in NK cells, the role of Cdc42 is more specific to activation-induced WASp function.20 If DOCK8 functions as a Cdc42 activator in NK cells, its deficiency might result in an inability to target F-actin accumulation in a WASp-dependent manner. Thus, we evaluated the NK cell lytic IS, since F-actin is known to accumulate at this site after activation. To obtain the most direct correlate to the patients, we isolated NK cells from ex vivo PBMCs via negative selection and used them immediately in experiments. NK cells were allowed to conjugate with K562 target cells for 30 minutes and then fixed and evaluated for the presence of F-actin by confocal fluorescence microscopy. We also evaluated conjugates for the presence of β2 integrin (CD18), the pore-forming component of NK cell lytic granules – perforin, and the epicenter of the microtubule-organizing center (MTOC) – pericentrin. CD18 was studied because it typically clusters at the lytic IS, but is dependent upon WASp function for redistribution.20 Perforin and MTOC localization were considered because by 30 minutes of activation they typically polarize towards the target cell in a WASp-dependent manner.32
Control donor NK cells demonstrated robust accumulation of F-actin at the lytic IS, as well as clustering of CD18 and polarization of both perforin and pericentrin (Figure 4A). The IS formed similarly in NK cells isolated from a STAT3 patient with noted F-actin accumulation, CD18 clustering and perforin/pericentrin polarization. NK cells from DOCK8-deficient patients, however, appeared quite different. While they demonstrated clear F-actin in the cell cortex, they failed to show accumulation of F-actin at the IS. Similar to what we had observed in WAS patient NK cells or NK cells in which F-actin had been inhibited,22, 32 DOCK8-deficient patient NK cells also failed to cluster CD18 or polarize perforin and the MTOC to the IS.
Figure 4. Abnormal F-actin accumulation at the DOCK8-deficient NK cell IS.

(A) Representative confocal immunofluorescent micrographs of eNK:K562 cell conjugates showing differential interference contrast (DIC) F-actin (red), CD18 (green), perforin (blue), pericentrin (pink) and an overlay. Measurement of (B) F-actin accumulation at, (C) CD18 accumulation and (D) MTOC polarization to the IS. Points represent single cell values (error bars=mean±SD, *=p<0.05).
To quantitatively evaluate these differences, multiple conjugates were measured from control donors, 3 individual STAT3 patients and 4 DOCK8-deficient patients. Using a previously developed quantitative algorithm for F-actin accumulation28 we found that DOCK8-deficient patient NK cells had statistically decreased synaptic F-actin relative to that in control donor or STAT3 patient NK cells (Figure 4B). Furthermore the DOCK8-deficient patient NK cells failed to demonstrate any statistically significant synaptic F-actin accumulation. The same algorithm for F-actin accumulation was also applied to CD18 accumulation and similarly demonstrated that DOCK8-deficient, but not control donor or STAT3 patient NK cells had decreased synaptic CD18 (Figure 4C). The distance of pericentrin to the synapse was measured directly and was significantly greater on average in DOCK8-deficent as compared to control donor or STAT3 patient NK cells (Figure 4D). This demonstrates that the DOCK8-deficient patient cells failed to polarize their lytic machinery, which is a prerequisite for cytotoxicity. Overall these quantitative analyses define abnormal focal F-actin accumulation at the NK cell synapse and aberrant overall synapse maturity in DOCK8-deficient patient cells.
Stable knockdown of DOCK8 with shRNA in NK cells recapitulates the patient phenotype
The observations in DOCK8-deficient patient NK cells suggest a role for DOCK8 in normal cytolytic function through the promotion of F-actin accumulation at the IS in NK cells. In order to address this more directly, we modeled DOCK8 deficiency in human NK cells in vitro. Specifically we utilized the human YTS NK cell line and transduced shRNA expression constructs targeting DOCK8 packaged in lentiviruses. As controls we utilized a scrambled DOCK8 shRNA sequence as well as a vector containing no shRNA sequence at all (empty vector). Transduced cells were selected via puromycin resistance and evaluated in cytotoxicity assays against the 721.221 lymphoblastoid cell line. Parental unmanipulated YTS cells killed 721.221 target cells effectively and similarly to those containing empty vector or scrambled shRNA. In contrast, YTS cells stably expressing the DOCK8 shRNA had drastically reduced cytotoxicity at all effector to target cell ratios (Figure 5A). To confirm the effectiveness of the DOCK8 shRNA, real-time PCR was performed and demonstrated that YTS cells containing the specific shRNA had a >50% reduction in DOCK8 mRNA compared to all controls (Figure 5B). Thus, DOCK8 is required for NK cell cytotoxicity.
Figure 5. Stable knockdown of DOCK8 inhibits NK cell cytotoxicity.
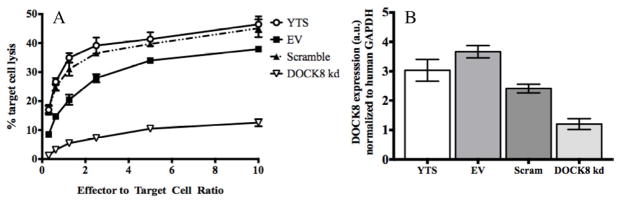
(A) NK cell lines (YTS) transduced with empty vector (EV), shRNA targeting DOCK8 (DOCK8 kd) or a scramble sequence were used in chromium-release cytotoxicity assays. (B) DOCK8 expression measured by real-time PCR, normalized to human GAPDH expression. Experiments performed in triplicate and representative graphs are depicted.
DOCK8 knockdown cell line cell conjugates between YTS and susceptible KT86 target cells were evaluated to further elucidate the proposed mechanism for DOCK8 in enabling NK cytotoxicity raised by our study of DOCK8-deficient patient NK cells. In these experiments KT86 target cells were utilized owing to a lower cortical actin content in this cell line. As is characteristic for a mature lytic synapse YTS cells expressing the scrambled shRNA accumulated F-actin at the synapse after 30 minutes of conjugation with targets (Figure 6). These control cells also demonstrated accumulation of CD18 at, and polarization of perforin to, the IS. In contrast, cells containing the DOCK8 shRNA failed to accumulate F-actin at the IS, and similar to DOCK8-deficient patient cells did not appear to have reduced overall cortical F-actin content. The decrease in F-actin accumulation in the DOCK8 shRNA containing cells relative to those containing the scrambled shRNA was statistically significant across multiple synapses (DOCK8 shRNA mean actin accumulation = −77.32±668, scrambled mean actin accumulation = 1438±603, p <0.05). DOCK8 shRNA-expressing YTS cells also failed to accumulate CD18 at, and polarize perforin to, the lytic synapse. Through specific and targeted DOCK8 knockdown, therefore, we recapitulated the mechanistic observations derived from DOCK8-deficient patient cells. Specifically, DOCK8 is required in NK cells for synaptic activation-induced F-actin accumulation and subsequent intracellular events needed in access to cytolytic function. This suggests that DOCK8 is an upstream regulator of the activation-induced actin reorganization machinery in NK cells, which is required for their ability to effectively participate in host defense functions.
Figure 6. DOCK8 knockdown abrogates synaptic F-actin accumulation and granule polarization.

Representative confocal immunofluorescence micrographs of conjugates between YTS cells containing DOCK8-targeting or control scrambled shRNA and KT86 target cells. Differential interference contrast (DIC) with localization of F-actin (red), CD18 (green), perforin (pink) and an overlay are shown.
DISCUSSION
Human DOCK8 deficiency is a relatively severe primary immunodeficiency characterized by susceptibility to infections and immune dysregulation. The immunologic impact of DOCK8 deficiency has been documented in both innate and adaptive arms of the immune system.19, 34 Given the susceptibility to herpesviruses and papillomaviruses in affected patients, we extended previous assessment of DOCK8 patients to include NK cell function.1, 3, 4 The reason for this is that the primary immunodeficiencies known to affect NK cell function share as a common feature, namely, susceptibility to these infectious agents.12, 13 Genetic immunodeficiencies resulting in NK cell defects are also associated with malignancies, presumably because NK cells are held to serve an important role in tumor surveillance.14, 15 Aside from the occurrence of recalcitrant viral infections, DOCK8-deficient patients also have susceptibility to cancers, which in this case has been hypothesized to be linked to viral infections.23
Initial evaluations of NK cells in human DOCK8 deficiency have been limited to flow cytometric quantification.3, 4 In DOCK8-deficient mice, NK cells have similarly only been quantified.8 Thus, we decided to approach NK cells in DOCK8 deficiency from a functional perspective. Since there are numerous differences between murine and human NK cells, including lack of ex vivo cytotoxicity in mouse NK cells,35 we opted to focus our efforts upon patients and in vitro human NK cell modeling. Our results agreed with those previously published in that NK cells were present in patient peripheral blood,3, 4 albeit at variable percentages, but herein we identify a pervasive functional deficiency regarding cytotoxicity.
There are a variety of potential explanations for a functional NK cell deficiency in DOCK8-deficient patients. In other immune cells, cell cycle, proliferative and even migratory defects have been identified.5–7, 19 The presence of mature NK cells in the peripheral blood of our cohort of DOCK8-deficient patients that are CD56dim and express perforin, however, suggests that NK cell development and proliferation are at least occurring. Thus we focused upon the previously reported role of DOCK8 as a Cdc42 activator.19 Cdc42 has been described to serve a number of functions in lymphocytes and NK cells. For example, it is required for actin organization and MTOC polarization in T cells and NK cells.18, 27, 36, 37 Mature NK cells utilize Cdc42 to activate WASp and direct activation-induced actin reorganization to enable their spontaneous cytotoxic activity.20 While we identified an inability of DOCK8-deficient cells to accumulate F-actin at the IS, we were surprised to find normal overall F-actin content, thus distinguishing our patients from those with WAS. Thus an ability to generate actin filaments must be intact in DOCK8-deficient patient NK cells, but the induction of actin reorganization after receptor signaling is likely to be deficient. This is consistent with our observed defect in focused actin accumulation in both patient and DOCK8 knockdown NK cells. This suggests that DOCK8 is not required for maintenance of the filamentous actin network, but for its activation-induced reorganization.
Unlike WAS patient NK cells, we found that the cytolytic activity of DOCK8-deficient patient cells could not be restored by IL-2 stimulation. We also attempted classical human NK cell bulk culture and expansion according to established protocols (which rely upon IL-2)25 with NK cells derived from DOCK8-deficient patients on three separate occasions. Unlike patients with WAS, we were unable to expand NK cells from the three DOCK8-deficient patients for whom this was attempted. Since we have previously defined that IL-2 can bypass WASp and utilize a homologous actin effector, WAVE2, to productively reorganize actin,22 the lack of IL-2 response in patient NK cells may be telling. Specifically, this observation suggests that DOCK8 may serve as a more general and upstream activator of actin reorganization machinery. This could be due to multiple pathways dependent upon Cdc42, but might also suggest that DOCK8 lies upstream of other Rho GTPases necessary for inducing actin branching complexes. Further experiments will be needed to define a potential role for DOCK8 as a master regulator of activation-induced actin function.
The present observations also provide further insight into the overall regulation of the NK cell lytic IS and subsequent access to cytotoxicity. Previously, F-actin was shown to be required for lytic granule polarization and integrin clustering after synapse formation.21, 22 Specifically, the use of actin depolymerizing agents that deplete the cell of cortical filaments altogether block both. WASp-deficient NK cells had a defect akin to NK cells treated with chemical inhibitors, but could not be used to distinguish any role for actin accumulation since they have an overall reduced F-actin content. DOCK8 deficiency, however, suggests that it is the targeted reorganization of F-actin at the IS that is needed for both CD18 clustering as well as ultimately lytic granule polarization.32, 38 This is likely to be a feature of the physical movement of the receptor in the case of CD18 and resulting amplification of cell signaling in the case of granule polarization. The study of DOCK8-deficient cells therefore advances these basic immunological mechanisms as likely reliant upon synaptic actin accumulation.
Our observations add a pervasive deficiency in NK cell cytotoxicity to the clinical syndrome resulting from DOCK8 deficiency. Although IL-2 does not induce function, other potentially therapeutic strategies for accessing NK cell function from patient cells should be explored. This would be of potential value to patients given the severe consequences of viral infections in affected individuals. We hypothesize, however, that NK cell cytotoxicity will be difficult to access owing to the upstream role of DOCK8. This suggests that other therapeutic options would more likely be appropriate in DOCK8-deficiency. It is unclear if gene corrected NK cells will have a proliferative advantage, but if there is at least some parallel to WAS the DOCK8 gene therapy may be advantageous. Since DOCK8 is expressed outside of the hematopoietic system, it is unclear whether immune reconstitution will solve all clinical issues that have been observed in patients. Irrespective, our results underscore the extensive immunodeficiency in DOCK8 deficiency and reiterate a clinical need and rationale for hematopoietic transplantation. Further experience will determine whether the reconstitution of NK cell defenses in DOCK8-deficient patients can provide increased resistance against cancers and viral infections. Overall our work defines an essential role for DOCK8 in human NK cell cytotoxic function and demonstrates a specific mechanism by which it enables activation-induced immune function.
Clinical Implications or Key Messages.
NK cells are functionally impaired in DOCK8 deficiency, consistent with patients’ infectious phenotypes.
DOCK8 deficiency prevents F-actin accumulation at the NK cell lytic immunological synapse without reducing overall F-actin content.
Acknowledgments
Funding sources:
NIH-NIAID R01067946 (to JSO)
German Research Foundation (DFG RE2799/3-1) and Fritz-Thyssen research foundation grant (Az. 10.07.1.159) (to EDR)
Dubai-Harvard Foundation of Medical Research, Jeffrey Modell Foundation, “Role of TACI Mutations in CVID”, 5P01AI076210-04, NIH, and “Combined SNP analysis and whole genome sequencing to discover immunodeficiency genes”, 5R03AI094017-02, NIH (to RSG)
The authors wish to thank the DOCK8 and STAT3 patients and their families for graciously providing blood samples and sharing their time participating in this analysis. Additionally, the authors thank Drs. Helen Su and Huie Jing for providing sequencing analysis of patient DOCK8-8.
Abbreviations used
- Cdc42
cell division cycle 42
- DOCK8
dedicator of cytokinesis 8
- eNK
ex vivo natural killer
- F-actin
filamentous actin
- HIES
hyper-IgE syndrome
- IS
immunologic synapse
- MTOC
microtuble organizing center
- NK
natural killer
- PBMCs
peripheral blood mononuclear cells
- shRNA
short-hairpin RNA
- STAT3
signal transducer and activator of transcription 3
- WAS
Wiskott-Aldrich syndrome
- WASp
WAS protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. The Journal of pediatrics. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 2.Freeman AF, Holland SM. Clinical manifestations of hyper IgE syndromes. Disease markers. 2010;29:123–30. doi: 10.3233/DMA-2010-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. The New England journal of medicine. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2009;124:1289–302. e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Domaschenz H, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nature immunology. 2009;10:1283–91. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. The Journal of experimental medicine. 2011;208:2305–20. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nature immunology. 2012;13:612–20. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambe T, Crawford G, Johnson AL, Crockford TL, Bouriez-Jones T, Smyth AM, et al. DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory. European journal of immunology. 2011;41:3423–35. doi: 10.1002/eji.201141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald DR, Massaad MJ, Johnston A, Keles S, Chatila T, Geha RS, et al. Successful engraftment of donor marrow after allogeneic hematopoietic cell transplantation in autosomal-recessive hyper-IgE syndrome caused by dedicator of cytokinesis 8 deficiency. The Journal of allergy and clinical immunology. 2010;126:1304–5. e3. doi: 10.1016/j.jaci.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bittner TC, Pannicke U, Renner ED, Notheis G, Hoffmann F, Belohradsky BH, et al. Successful long-term correction of autosomal recessive hyper-IgE syndrome due to DOCK8 deficiency by hematopoietic stem cell transplantation. Klinische Padiatrie. 2010;222:351–5. doi: 10.1055/s-0030-1265135. [DOI] [PubMed] [Google Scholar]
- 11.Barlogis V, Galambrun C, Chambost H, Lamoureux-Toth S, Petit P, Stephan JL, et al. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency. The Journal of allergy and clinical immunology. 2011;128:420–22. e2. doi: 10.1016/j.jaci.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 12.Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes and infection/Institut Pasteur. 2002;4:1545–58. doi: 10.1016/s1286-4579(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 13.Orange JS. Human natural killer cell deficiencies. Current opinion in allergy and clinical immunology. 2006;6:399–409. doi: 10.1097/ACI.0b013e3280106b65. [DOI] [PubMed] [Google Scholar]
- 14.Bryceson YT, Ljunggren HG. Natural killer cells: biology, physiology and medicine--part 1. Journal of innate immunity. 2011;3:213–5. doi: 10.1159/000325332. [DOI] [PubMed] [Google Scholar]
- 15.Bryceson YT, Ljunggren HG. Natural killer cells: biology, physiology and medicine - part 2. Journal of innate immunity. 2011;3:327–8. doi: 10.1159/000327015. [DOI] [PubMed] [Google Scholar]
- 16.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. Journal of cell science. 2002;115:4901–13. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 17.Ruusala A, Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS letters. 2004;572:159–66. doi: 10.1016/j.febslet.2004.06.095. [DOI] [PubMed] [Google Scholar]
- 18.Sinai P, Nguyen C, Schatzle JD, Wulfing C. Transience in polarization of cytolytic effectors is required for efficient killing and controlled by Cdc42. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11912–7. doi: 10.1073/pnas.0913422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119:4451–61. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stabile H, Carlino C, Mazza C, Giliani S, Morrone S, Notarangelo LD, et al. Impaired NK-cell migration in WAS/XLT patients: role of Cdc42/WASp pathway in the control of chemokine-induced beta2 integrin high-affinity state. Blood. 2010;115:2818–26. doi: 10.1182/blood-2009-07-235804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orange JS, Ramesh N, Remold-O’Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11351–6. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orange JS, Roy-Ghanta S, Mace EM, Maru S, Rak GD, Sanborn KB, et al. IL-2 induces a WAVE2-dependent pathway for actin reorganization that enables WASp-independent human NK cell function. The Journal of clinical investigation. 2011;121:1535–48. doi: 10.1172/JCI44862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Archives of dermatology. 2012;148:79–84. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Herz W, Ragupathy R, Massaad MJ, Al-Attiyah R, Nanda A, Engelhardt KR, et al. Clinical, immunologic and genetic profiles of DOCK8-deficient patients in Kuwait. Clinical immunology. 2012;143:266–72. doi: 10.1016/j.clim.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanborn KB, Rak GD, Maru SY, Demers K, Difeo A, Martignetti JA, et al. Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. Journal of immunology. 2009;182:6969–84. doi: 10.4049/jimmunol.0804337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanborn KB, Mace EM, Rak GD, Difeo A, Martignetti JA, Pecci A, et al. Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood. 2011;118:5862–71. doi: 10.1182/blood-2011-03-344846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee PP, Pandey R, Zheng R, Suhoski MM, Monaco-Shawver L, Orange JS. Cdc42-interacting protein-4 functionally links actin and microtubule networks at the cytolytic NK cell immunological synapse. The Journal of experimental medicine. 2007;204:2305–20. doi: 10.1084/jem.20061893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee PP, Orange JS. Quantitative measurement of F-actin accumulation at the NK cell immunological synapse. Journal of immunological methods. 2010;355:1–13. doi: 10.1016/j.jim.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young HA, Ortaldo J. Cytokines as critical co-stimulatory molecules in modulating the immune response of natural killer cells. Cell research. 2006;16:20–4. doi: 10.1038/sj.cr.7310004. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, Su HC. Hyperimmunoglobulin E syndromes in pediatrics. Current opinion in pediatrics. 2011;23:653–8. doi: 10.1097/MOP.0b013e32834c7f65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comans-Bitter WM, de Groot R, van den Beemd R, Neijens HJ, Hop WC, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. The Journal of pediatrics. 1997;130:388–93. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 32.Orange JS, Harris KE, Andzelm MM, Valter MM, Geha RS, Strominger JL. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14151–6. doi: 10.1073/pnas.1835830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gismondi A, Cifaldi L, Mazza C, Giliani S, Parolini S, Morrone S, et al. Impaired natural and CD16-mediated NK cell cytotoxicity in patients with WAS and XLT: ability of IL-2 to correct NK cell functional defect. Blood. 2004;104:436–43. doi: 10.1182/blood-2003-07-2621. [DOI] [PubMed] [Google Scholar]
- 34.Randall KL, Lambe T, Goodnow CC, Cornall RJ. The essential role of DOCK8 in humoral immunity. Disease markers. 2010;29:141–50. doi: 10.3233/DMA-2010-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Stowers L, Yelon D, Berg LJ, Chant J. Regulation of the polarization of T cells toward antigen-presenting cells by Ras-related GTPase CDC42. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5027–31. doi: 10.1073/pnas.92.11.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tskvitaria-Fuller I, Seth A, Mistry N, Gu H, Rosen MK, Wulfing C. Specific patterns of Cdc42 activity are related to distinct elements of T cell polarization. Journal of immunology. 2006;177:1708–20. doi: 10.4049/jimmunol.177.3.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lub M, van Kooyk Y, van Vliet SJ, Figdor CG. Dual role of the actin cytoskeleton in regulating cell adhesion mediated by the integrin lymphocyte function-associated molecule-1. Molecular biology of the cell. 1997;8:341–51. doi: 10.1091/mbc.8.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]