

Abstract
To date, over 70 mutations in the TGFBR2 gene have been reported in patients with Loeys–Dietz syndrome (LDS), Marfan syndrome type 2 (MFS2), or other hereditary thoracic aortic aneurysms and dissections. Whereas almost all of mutations analyzed thus far are predicted to disrupt the constitutively active C-terminal serine/threonine kinase domain of TGFBR2, mounting evidence suggests that the molecular mechanism underlying these diseases is more complex than simple haploinsufficiency. Using exon-targeted oligonucleotide array comparative genomic hybridization, we identified an ~896 kb deletion of TGFBR2 in a 20-month-old female with microcephaly and global developmental delay, but no stigmata of LDS. FISH analysis showed no evidence of this deletion in the parental peripheral blood samples; however, somatic mosaicism was detected using PCR in the paternal DNA from peripheral blood lymphocytes and lymphoblasts. Our data suggest that TGFBR2 haploinsufficiency may cause a phenotype, which is distinct from LDS. Moreover, we propose that somatic mosaicism below the detection threshold of FISH analysis in asymptomatic parents of children with genomic disorders may be more common than previously recognized.
Keywords: microcephaly, somatic mosaicism, TGF-β signaling, Loeys–Dietz syndrome
INTRODUCTION
The TGFBR1 and TGFBR2 genes encode transmembrane kinase receptors involved in signal transduction of the transforming growth factor beta (TGF-β) family of ligands. Mutations in these genes have been identified in patients with Loeys–Dietz syndrome type 1A (LDS1A, OMIM 609192), type 1B (LDS1B, OMIM 609168), type 2A (LDS2A, OMIM 608967), type 2B (LDS2B, formerly Marfan syndrome type 2, OMIM 610168), and familial thoracic aortic aneurysms. The TGFBR2 gene consists of 7 exons that encode 567 amino acids organized into an N-terminal ligand binding domain, a transmembrane region, and a constitutively active C-terminal serine/threonine kinase domain [Shi and Massagué, 2003]. Binding of one of the TGF-β ligands leads to the formation of a heteroquatromeric complex composed of 2 TGFBR1 and 2 TGFBR2 subunits. TGFBR2 phosphorylates TGFBR1, which in turn phosphorylates Smad2 and Smad3, the downstream mediators of TGF-β signaling. TGF-β also activates the extracellular signal-regulated kinases (ERK).
To date, over 70 TGFBR2 mutations have been reported in association with vascular abnormalities [Mizuguchi et al., 2004; Ki et al., 2005; Loeys et al., 2005; Pannu et al., 2005; Disabella et al., 2006; Loeys et al., 2006; Mátyás et al., 2006; Singh et al., 2006; Lee et al., 2007; Waldmüller et al., 2007; Yetman et al., 2007]. The majority are missense substitutions or nonsense mutations in the penultimate or final exons predicted to disrupt the kinase domain; in the case of nonsense mutations, the transcripts likely escape nonsense mediated decay (NMD).
Tgfbr2−/− mice die in utero, likely secondary to defects in hematopoiesis and vasculogenesis, while heterozygous Tgfbr2+/− mice are reportedly developmentally normal [Oshima et al., 1996]. Transgenic mice containing floxed Tgfbr2 and Cre under the control of various promoters provided evidence that Tgfbr2 is vital for normal cardiac, axial skeleton, and skull vault development as well as for vascular integrity. Specifically, mice expressing inducible Cre in endothelial cells showed ventricular septal defects and cerebral hemorrhage [Robson et al., 2010]. Moreover, mice expressing Cre in osteoblasts are born without parietal or frontal bones [Seo and Serra, 2009].
Analyses of cell lines lacking endogenous TGFBR2 but expressing transgenic mutant TGFBR2 receptors isolated from patients suggested that these mutations result in impaired TGF-β signal transduction [Mizuguchi et al., 2004; Horbelt et al., 2010]. The same experiments reveal that mutant receptors also exert varying degrees of dominant negative effects on wild-type receptors. Paradoxically, biopsy and autopsy samples from patients with LDS have repeatedly shown increased nuclear accumulation of phosphorylated Smad2 [Loeys et al., 2005, 2006; Maleszewski et al., 2009], suggesting a toxic gain-of-function mechanism of pathogenesis. Thus, the exact molecular mechanism of LDS and the clinical significance of TGFBR2 haploinsufficiency remain to be elucidated.
Using clinical exon-targeted oligonucleotide array comparative genomic hybridization (aCGH), we identified a novel ~896 kb deletion on chromosome 3p24.1, including the entire TGFBR2 gene in a 20-month-old female with microcephaly, mildly dysmorphic features, and global developmental delay.
CLINICAL REPORT
Patient
The patient was born to a 42-year-old father and a 19-year-old G1P0–1 mother at term via spontaneous vaginal delivery without complications. The family history was unremarkable pertaining to cardiovascular disease, sudden cardiac death, stroke in the young or intellectual disability. She had mild hyperbilirubinemia on day 1 of life, requiring phototherapy. She failed a hearing screen on the left at birth and again at 6 months of age. She sat unassisted at 12 months of age and was non-ambulatory and non-verbal at 20 months of age. Her physical exam was significant for weight at the 25th centile, length at the 5th centile, and head circumference 3 SD below the mean (FOC 42.5 cm). She had fontanels that were closed to palpation, deep-set eyes, flat nasal bridge, and overfolded superior ear helices. She had a flat philtrum, thin vermillion of the upper lip and a normal palate and uvula (Fig. 1A). There were no cardiac murmurs or vascular bruits appreciated. She had no evidence of arachnodactyly or pectus deformity. There were no dystrophic scars or other cutaneous abnormalities. Axial hypotonia with mild hypertonia of the lower extremities and dystonic posturing of the feet were present. Deep tendon reflexes were normal and symmetric.
FIG. 1.
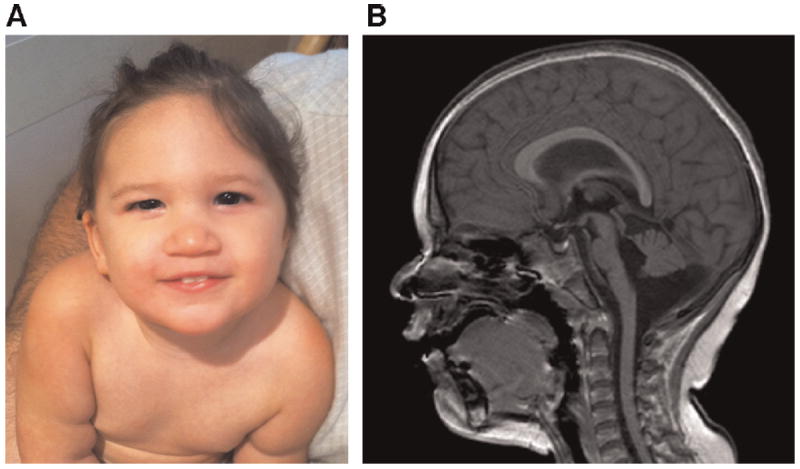
A: Patient at age 19 months. B: Sagittal FFE magnetic resonance image demonstrating a thin and abnormally shaped corpus callosum, hypoplasia of the brainstem, hypoplasia of the inferior cerebellar vermis, and a prominent cisterna magna [Color figure can be viewed in the online issue, which is available at www.wileyonlinelibrary.com].
Her ophthalmological exam was normal. A cardiac MRI demonstrated normal cardiac function without evidence of aortic root dilatation and an arterial tree in the neck, chest, and abdomen that was normal in configuration and branching without tortuosity, aneurysmal dilatation, or significant stenosis. The left internal jugular vein was not visualized. MRA of the head was unremarkable with no evidence of stenosis, occlusion, aneurysm, or vascular malformation. MRI of the brain was significant for paucity of white matter with a thin and abnormally shaped corpus callosum, hypoplasia of the brainstem, hypoplasia of the inferior cerebellar vermis, and a prominent cisterna magna (Fig. 1B). Laboratory investigation of plasma amino acids, urine organic acids, and the GJB2 gene were within normal limits. The parents had no skeletal, craniofacial, or cutaneous stigmata of LDS on physical examination.
MATERIALS AND METHODS
Written informed consent was obtained from the patient’s parents. The study was performed in accordance with the Institutional Guidelines for Human Research with approval by the Institutional Review Board at Baylor College of Medicine (BCM).
aCGH, FISH, and PCR Analyses
Genomic DNA was extracted from the patient’s and both parents’ peripheral blood lymphocytes and Epstein–Barr virus-transformed lymphoblastoid cells using a Genomic DNA purification kit (Puregene, Gentra Systems, Minneapolis, MN) according to the manufacturer’s protocols.
Array CGH was performed using a custom 180K oligonucleotide array, V8.1 OLIGO, designed in Medical Genetics Laboratories at BCM and manufactured by Agilent Technology (Santa Clara, CA). Digestion, labeling, and hybridization were completed following the manufacturers’ protocols. BCM web-based software was used for genomic copy number analysis. For fine mapping of the size and extent of the deletion, whole-genome high-resolution oligonucleotide microarray CGH analysis was performed with the NimbleGen array HG18_WG_CGH_v1 with 2.1M oligonucleotides (NimbleGen Systems, Madison, WI), in accordance with the manufacturer’s instructions. FISH analysis was performed on the patient’s and both parents’ peripheral blood lymphocytes using BAC clone RP11-1024P17, specific for chromosome 3p24.1.
Genomic sequence was determined based on the oligonucleotide coordinates from the aCGH experiments, compared with the UCSC genome browser (Build 36, UCSC genome browser, March 2006) and assembled with Sequencher (Gene Codes Corporation, Ann Arbor, MI). Long range PCR was employed to amplify the junction fragment of the deletion utilizing the manufacturer’s protocol (Takara Bio Inc, Otsu, Japan). PCR products were Sanger sequenced (Lone Star, Houston, TX).
RESULTS
Chromosomal microarray analyses revealed an 851–928 kb loss in copy number on chromosome 3p23p24.1. The proximal breakpoint was mapped between 31,214,839 and 31,176,203 and the distal breakpoint between 30,287,071 and 30,325,687 (Fig. 2A). This region harbors only two genes, TGFBR2 and GADL1, both of which are entirely encompassed by the deletion (Fig. 2D). To address whether the identified deletion was a benign CNV, we screened the BCM database of aCGH studies performed in over 12,000 patients and found no individuals with similar losses disrupting TGFBR2. Additionally, no CNVs in this genomic region were found in DECIPHER, the Database of Genomic Variants, and over 5,000 normal controls [Pinto et al., 2007; Shaikh et al., 2009; Conrad et al., 2010]. These data indicate that this genomic region is not polymorphic and further support its pathogenic role in the described child. FISH analysis confirmed presence of the deleted region in the patient and a lack of any detectable deletion in either parent in 150 interphase nuclei analyzed.
FIG. 2.
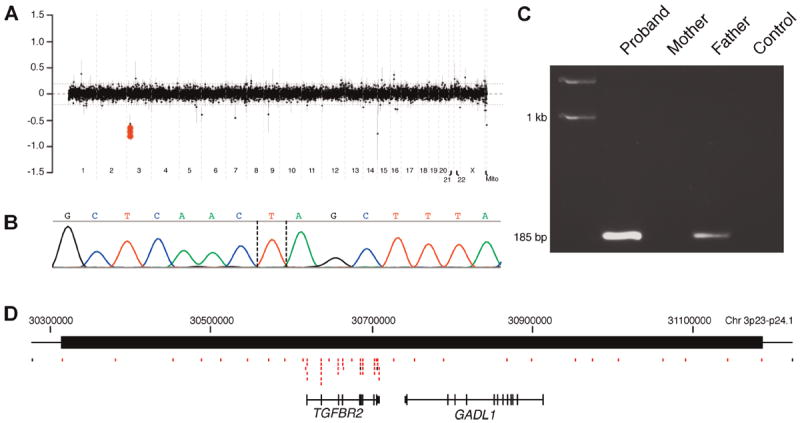
A: Clinical aCGH (CMA V8.1 OLIGO) plot showing a deletion on chromosome 3q24.1-q23. The proximal breakpoint was mapped between 31,214,839 and 31,176,203 and the distal breakpoint between 30,287,071 and 30,325,687. B: Chromatogram showing DNA sequence of the junction fragment. Dashed lines show possible breakpoints. C: 1% Agarose gel demonstrating the presence of the family-specific fragment amplified from the patient’s and the father’s peripheral blood lymphocytes. Note the weaker intensity of the PCR band for a given reaction volume in the father indicating a low level mosaicism. D: Schematic diagram of the genomic structure of the deleted region. Red boxes represent CGH oligonucleotides with log ratios below −0.30 [Color figure can be viewed in the online issue, which is available at www.wileyonlinelibrary.com].
The obtained junction fragment of the deletion was amplified using LR PCR with primers F: CCTCTAAGTTACTGAGGACTTGACCCTTCC and R: CCCCTTGTGTCCTTAGTTATTCTGCCATAG (Fig. 2C) and subsequently sequenced (Fig. 2B). The proximal breakpoint was mapped to chr3:31,220,816(7) and the distal breakpoint to 30,325,042(3); thus the deletion is 895,775 bp in size. DNA sequence analysis of the PCR product revealed a single thymidine base of homology between the junction fragments, most consistent with a non-homologous end joining mechanism of formation (Fig. 2B).
Interestingly, PCR analysis of the parental DNA samples revealed an identical, although lower intensity, product amplified from the father’s peripheral blood lymphocytes (Fig. 2C) and lymphoblasts (data not shown). We were unable to obtain other tissue samples (fibroblasts, hair roots, oral swabs, or nails) from the father. The PCR and FISH results suggest that the father is mosaic for the deletion at a level below the detection threshold of FISH.
DISCUSSION
Loeys–Dietz syndrome, a rare autosomal dominant disorder caused by heterozygous mutations in the TGFBR1 or TGFBR2 genes, is characterized by vascular, skeletal, and craniofacial abnormalities. Affected individuals demonstrate vascular fragility leading to aortic aneurysms and dissections similar to those seen in patients with Marfan syndrome and Ehlers–Danlos syndrome type IV (OMIM 130050). Owing to this predisposition to hemodynamic compromise, the median survival for patients with LDS is only 37 years, with the most common cause of death being thoracic aortic aneurysm rupture [Loeys et al., 2005, 2006]. Early diagnosis of LDS is essential as patients benefit from increased vascular surveillance and medical management to optimize hemodynamic parameters. In addition to life-threatening abnormalities of the vascular system, many patients with LDS also have characteristic findings of craniosynostosis, hypertelorism, downward slanting palpebral fissures, strabismus, highly arched palate, cleft palate or bifid uvula, micro-/retrognathia, malar hypoplasia, pectus deformity, scoliosis, arachnodactyly, and joint laxity. Less common findings that are rare in the general population include blue sclera, velvety or transparent skin, camptodactyly, patent ductus arteriosus, and atrial septal defects [Loeys et al., 2006]. A minority of affected individuals have developmental delay, which is more likely to be present in association with craniosynostosis and/or hydrocephalus [Loeys et al., 2005].
TGFBR2 haploinsufficiency as a mechanism for LDS has been argued against in the literature because immunohistochemical staining of pathological surgical sections revealed an increase in downstream effectors of TGF-β signaling in vascular tissue and because of the wide repertoire of documented missense mutations [Dietz, 2010].
Thus, the identification of a TGFBR2 deletion in a 20-month-old girl presented us with the clinical dilemma of having no precedent to guide clinical surveillance. In consultation with our cardiology colleagues, we chose to perform cardiac MRI and MRA studies of the arterial tree of the neck, chest, and abdomen, as well as MRA of the cerebral vasculature in our patient. Although, no arterial or cardiac abnormalities were detected, the risk of aortic dilatation and vascular fragility is undetermined in this individual. We have recommended repeat surveillance, as patients with LDS benefit from medical management and surgical repair of aortic aneurysms [Loeys et al., 2006].
We were unable to definitively verify a diagnosis of craniosynostosis because X-ray or CT imaging would not have altered our clinical management since surgical intervention was not a consideration.
Recently, a patient with a large (~14 Mb) duplication CNV, including the entire TGFBR1 gene was reported to have an LDS-like phenotype [Breckpot et al., 2010]. The 17-year-old boy presented with bifid uvula, pubertas tarda, camptodactyly, and dysmorphic features. Similarly, a patient harboring a large duplication of 3p21.3-3p25 (containing the TGFBR2 gene) was identified [Zhang and Wang, 1984]. The 7-year-old boy presented with developmental delay, brachycephaly, micrognathia, and other dysmorphic features. Both of these patients have CNVs including a multitude of potentially dosage sensitive genes. Nonetheless, it appears that mammals are exquisitely sensitive to dosage variations in TGF-β signaling pathways, and thus it is reasonable to suggest that both phenotypes are at least partially caused by variation in dosage of TGF-β receptors.
Another rational conclusion might be to attribute our patient’s microcephaly and prematurely closed fontanels to a failure of brain development (suggesting a secondary craniosynostosis), especially given the abnormalities detected on brain MRI. Whereas, mutation of the TGFBR2 gene has been associated with primary craniosynostosis, developmental abnormalities of the nervous system are usually not observed in the affected individuals.
The GADL1 gene, encoding a glutamate decarboxylase-like protein 1, is also located within this patient’s deleted region (Fig. 2D), but cytosolic enzymes are not typically dosage dependent [Phadnis and Fry, 2005].
The PCR finding of mosaicism in this patient’s father, undetectable by traditional diagnostic FISH, raises the question of the clinical significance of such somatic mosaic CNVs. Previous studies have reported TGFBR2 mosaicism; the father of a severely affected child was found to harbor low levels of a missense mutation, suggesting mosaicism [Loeys et al., 2006] and the father of another individual with LDS was more thoroughly investigated and found to have TGBFR2 mutations in multiple cell lineages [Watanabe et al., 2008]. In both of these cases, the fathers had clinical manifestations to provoke investigation. In contrast, our patient’s father appears asymptomatic.
The relatively recent observation of CNV somatic mosaicism suggests that a meaningful number of CNVs can occur during mitosis [Notini et al., 2008; Piotrowski et al., 2008]. In our patient’s father, cells with the genomic rearrangement were too rare to be detected by peripheral blood FISH, but the CNV was nonetheless transmitted to his daughter. The importance of this observation lies in its impact on genetic counseling. Standard interpretation of the CGH and FISH results presented here would have suggested a near-zero recurrence risk. The recurrence risk for this father is higher based on our studies, and these observations have broader applicability for patients and counselors alike.
In summary, we present a first case of TGFBR2 deletion in a patient with microcephaly, mild dysmorphic features, and global developmental delay. Our findings suggest that TGFBR2 haploinsufficiency is insufficient to cause LDS but may have clinical consequences. The identification of additional patients with TGFBR2 haploinsufficiency and documentation of their natural history will be critical to demonstrate the full spectrum of phenotypes associated with alterations in the TGFBR2 gene and to guide clinical care and surveillance.
Supplementary Material
Acknowledgments
We are grateful to the patient and her family for participating in this study and to our Cardiology and Radiology colleagues Dr. Tim Slesnick, Dr. Rajesh Krishnamurthy, and Dr. Bhairav Patel. P.S. was supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education. M.B.R. is grateful for the support of grant 5K08NS062711-03 from the NIH/NINDS.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Breckpot J, Budts W, De Zegher F, Vermeesch JR, Devriendt K. Duplication of the TGFBR1 gene causes features of Loeys–Dietz syndrome. Eur J Med Genet. 2010;53:408–410. doi: 10.1016/j.ejmg.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AWC, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Consortium WTCC, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz HC. TGF-beta in the pathogenesis and prevention of disease: A matter of aneurysmic proportions. J Clin Invest. 2010;120:403–407. doi: 10.1172/JCI42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disabella E, Grasso M, Marziliano N, Ansaldi S, Lucchelli C, Porcu E, Tagliani M, Pilotto A, Diegoli M, Lanzarini L, Malattia C, Pelliccia A, Ficcadenti A, Gabrielli O, Arbustini E. Two novel and one known mutation of the TGFBR2 gene in Marfan syndrome not associated with FBN1 gene defects. Eur J Hum Genet. 2006;14:34–38. doi: 10.1038/sj.ejhg.5201502. [DOI] [PubMed] [Google Scholar]
- Horbelt D, Guo G, Robinson PN, Knaus P. Quantitative analysis of TGFBR2 mutations in Marfan-syndrome-related disorders suggests a correlation between phenotypic severity and Smad signaling activity. J Cell Sci. 2010;123:4340–4350. doi: 10.1242/jcs.074773. [DOI] [PubMed] [Google Scholar]
- Ki CS, Jin DK, Chang SH, Kim JE, Kim JW, Park BK, Choi JH, Park IS, Yoo HW. Identification of a novel TGFBR2 gene mutation in a Korean patient with Loeys–Dietz aortic aneurysm syndrome; no mutation in TGFBR2 gene in 30 patients with classic Marfan’s syndrome. Clin Genet. 2005;68:561–563. doi: 10.1111/j.1399-0004.2005.00535.x. [DOI] [PubMed] [Google Scholar]
- Lee RS, Fazel S, Schwarze U, Fleischmann D, Berry GJ, Liang D, Miller DC, Mitchell RS. Rapid aneurysmal degeneration of a Stanford type B aortic dissection in a patient with Loeys–Dietz syndrome. J Thorac Cardiovasc Surg. 2007;134:242–243. 243.e1. doi: 10.1016/j.jtcvs.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- Maleszewski JJ, Miller DV, Lu J, Dietz HC, Halushka MK. Histopathologic findings in ascending aortas from individuals with Loeys–Dietz syndrome (LDS) Am J Surg Pathol. 2009;33:194–201. doi: 10.1097/PAS.0b013e31817f3661. [DOI] [PubMed] [Google Scholar]
- Mátyás G, Arnold E, Carrel T, Baumgartner D, Boileau C, Berger W, Steinmann B. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat. 2006;27:760–769. doi: 10.1002/humu.20353. [DOI] [PubMed] [Google Scholar]
- Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notini AJ, Craig JM, White SJ. Copy number variation and mosaicism. Cytogenet Genome Res. 2008;123:270–277. doi: 10.1159/000184717. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- Phadnis N, Fry JD. Widespread correlations between dominance and homozygous effects of mutations: Implications for theories of dominance. Genetics. 2005;171:385–392. doi: 10.1534/genetics.104.039016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Hum Mol Genet. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- Piotrowski A, Bruder CE, Andersson R, Diaz de Ståhl T, Menzel U, Sandgren J, Poplawski A, von Tell D, Crasto C, Bogdan A, Bartoszewski R, Bebok Z, Krzyzanowski M, Jankowski Z, Partridge EC, Komorowski J, Dumanski JP. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- Robson A, Allinson KR, Anderson RH, Henderson DJ, Arthur HM. The TGFbeta type II receptor plays a critical role in the endothelial cells during cardiac development. Dev Dyn. 2010;239:2435–2442. doi: 10.1002/dvdy.22376. [DOI] [PubMed] [Google Scholar]
- Seo HS, Serra R. Tgfbr2 is required for development of the skull vault. Dev Biol. 2009;334:481–490. doi: 10.1016/j.ydbio.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O’Hara R, Casalunovo T, Conlin LK, D’Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, Zackai EH, Chiavacci RM, Grundmeier R, Rappaport EF, Grant SF, White PS, Hakonarson H. High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Singh K, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, Arslan-Kirchner M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys–Dietz syndrome. Hum Mutat. 2006;27:770–777. doi: 10.1002/humu.20354. [DOI] [PubMed] [Google Scholar]
- Waldmüller S, Müller M, Warnecke H, Rees W, Schöls W, Walterbusch G, Ennker J, Scheffold T. Genetic testing in patients with aortic aneurysms/dissections: A novel genotype/phenotype correlation? Eur J Cardiothorac Surg. 2007;31:970–975. doi: 10.1016/j.ejcts.2007.02.027. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Sakai H, Nishimura A, Miyake N, Saitsu H, Mizuguchi T, Matsumoto N. Paternal somatic mosaicism of a TGFBR2 mutation transmitting to an affected son with Loeys–Dietz syndrome. Am J Med Genet Part A. 2008;146A:3070–3074. doi: 10.1002/ajmg.a.32567. [DOI] [PubMed] [Google Scholar]
- Yetman AT, Beroukhim RS, Ivy DD, Manchester D. Importance of the clinical recognition of Loeys–Dietz syndrome in the neonatal period. Pediatrics. 2007;119:e1199–e1202. doi: 10.1542/peds.2006-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SZ, Wang Q. Partial serial duplication of the short arm of chromosome 3. Chin Med J (Engl) 1984;97:425–428. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.