

Abstract
Obesity is now a leading cause of preventable death in the industrialised world. Understanding its genetic influences can enhance insight into molecular pathogenesis and potential therapeutic targets. A non-synonymous polymorphism (rs35859249, p.Arg125Trp) in the N-terminal TBC1D1 phosphotyrosine-binding (PTB) domain has shown a replicated association with familial obesity in women. We investigated these findings in the Avon Longitudinal Study of Parents and Children (ALSPAC), a large European birth cohort of mothers and offspring, and by generating a predicted model of the structure of this domain. Structural prediction involved the use of three separate algorithms; Robetta, HHpred/MODELLER and I-TASSER. We used the transmission disequilibrium test (TDT) to investigate familial association in the ALSPAC study cohort (N = 2,292 mother-offspring pairs). Linear regression models were used to examine the association of genotype with mean measurements of adiposity (Body Mass Index (BMI), waist circumference and Dual-energy X-ray absorptiometry (DXA) assessed fat mass), and logistic regression was used to examine the association with odds of obesity. Modelling showed that the R125W mutation occurs in a location of the TBC1D1 PTB domain that is predicted to have a function in a putative protein:protein interaction. We did not detect an association between R125W and BMI (mean per allele difference 0.27 kg/m2 (95% Confidence Interval: 0.00, 0.53) P = 0.05) or obesity (odds ratio 1.01 (95% Confidence Interval: 0.77, 1.31, P = 0.96) in offspring after adjusting for multiple comparisons. Furthermore, there was no evidence to suggest that there was familial association between R125W and obesity (χ2 = 0.06, P = 0.80). Our analysis suggests that R125W in TBC1D1 plays a role in the binding of an effector protein, but we find no evidence that the R125W variant is related to mean BMI or odds of obesity in a general population sample.
Introduction
Obesity has become a major cause of morbidity and mortality in the industrialised world, substantially through the impact on incidence of type 2 diabetes mellitus and coronary heart disease [1]. While environmental change has driven the increase in obesity, genetic contributions may highlight aspects of pathogenesis and novel pathways susceptible to new interventions (drug or nutritional) [2]–[4].
A non-synonymous polymorphism (rs35859249, C to T) in TBC1D1, referred here as the R125W variant or 125W risk allele, has been previously reported to be associated with familial obesity in women [5], [6]. R125 is conserved as an Arg or Gln in mammals and locates within the first phosphotyrosine-binding (PTB) domain of TBC1D1 [5]. Stone et al. [7] initially identified a predisposition locus at 4 p15–14, in which TBC1D1 locates, to severe obesity (Body Mass Index (BMI)>35 kg/m2) in pedigrees of US Caucasian females. They analysed 435 pedigrees of European descent where on average 10 subjects in each family had a BMI≥35 kg/m2. They identified 10 pedigrees from this sample that had a logarithm (base 10) of odds (LOD) score≥1.0 in 4 p15–14 and found strong evidence for obesity linkage at this locus in females. However, there was no prior rationale for sex differences and no gene*sex interaction tests were presented. In a subsequent study, Stone et al. [5] reported linkage of the TBC1D1 125W risk allele with severe familial obesity in females only, using the same 10 aforementioned pedigrees. This familial association of the R125W variant with obesity has since been replicated in a European cohort consisting of 9,714 French individuals, through the over-transmission of the 125W allele into obese offspring [6]. Using the 97th percentile as their threshold for obesity, there was a borderline significant over-transmission of 125W risk allele into obese subjects (P = 0.05). After stratifying by sex they found evidence of a familial association in females (P = 0.008) but did not present any results of a gene*sex interaction test. Neither group that have observed the associations in families of probands with extreme obesity have found replication in population samples, using a sample size of 137 unrelated females (BMI≥35 kg/m2) and 4,634 general population individuals [5], [6].
TBC1D1 is a Rab-GTPase Activating Protein, which links the signals generated by insulin and muscle contraction to the molecular machinery facilitating glucose uptake into muscle cells [8]–[10] where the protein is most highly expressed [11]. Expression of the R125W TBC1D1 mutant in mouse skeletal muscle impaired insulin-stimulated, but not contractile-stimulated, glucose uptake [12].
Recent genetic evidence from the Swiss Jim Lambert (SJL) mouse strain, which express a truncated form of the TBC1D1 protein, suggests that TBC1D1 may have a more direct role in whole-body energy homeostasis [13] as these mice were lean and resistant to high fat diet-induced obesity. Depletion of TBC1D1 in the C2C12 muscle cell line resulted in increased fatty acid uptake and oxidation, consistent with similar observations in glycolytic muscles of SJL mice [13]. Furthermore, sequence variation at TBC1D1 identified it as a major quantitative trait locus in distinguishing growth characteristics between chickens bred for meat-producing or egg-laying [14] additionally supporting a role for TBC1D1 in metabolism and growth.
Given the evidence that the TBC1D1 R125W variant is associated with familial extreme obesity but does not appear to be associated with obesity in general population cohorts [5], [6] together with the physiological relevance of TBC1D1 in regulating glucose and lipid homeostasis, we set out both to (i) model the location of R125 in the first PTB domain of TBC1D1 and (ii) undertake a further study of the familial association of the R125W variant with obesity in a general population birth cohort of 2,292 white British mother-offspring pairs.
Results
Homology Model of TBC1D1 PTB1 Domain
To gain insight into the molecular impact of an Arg to Trp substitution we generated homology models of the first PTB domain of human TBC1D1. The best available template for homology modelling TBC1D1 PTB1, the PTB domain of AIDA1 (2M38; DOI:10.2210/pdb2m38/pdb), has a sequence identity of 24% which is a little lower than the required≥30% required for reliable fold identification from a single template. However, the Hidden Markov methods applied in the homology modelling servers used here confirm that the PTB1 domain possesses the standard PTB-domain fold. For example, the HHpred E-values for the top 10 template structures are between 10−36 and 10−33 and the average of the Cα Root-mean-square deviation (RMSD) between models built with these templates and 2M38 is 2.25 Å, i.e. the same fold in each case. Despite this rather low (<25%) primary sequence homology to known PTB domain structures, models of PTB1 generated from three independent servers were very similar with only 1.2Å–2.2 Å RMSD between the C-alphas of their secondary structural elements (Table 1). The models differed more in the prediction of the loop regions (Figure S1), which is unsurprising given these regions are intrinsically more flexible and are likely to exhibit an ensemble of rapidly interconverting conformations in the physiological environment. The homology model generated by Robetta server, based on the structure of human Numb-like protein (PDB ID: 3F0W), was determined, by PROCHECK analysis, to have better geometric quality than the models created by HHpred/Modeller and I- TASSER servers (Table S1 and Method S1). The predicted model (Figure 1A) consists of the core PTB fold, a β sandwich of perpendicular antiparallel β sheets (β1–β4 and β5–β7) flanked by a C-terminal α helix (α2), with an additional α helix between β1 and β2. As shown in Figure 1A, R125 locates within a loop between β6 and β7 with the side chain capable of orienting towards the cleft formed between β5 and α2. Numerous structures solved in the presence of peptides support this groove as a canonical peptide binding site particularly for the NPX(p)Y/F motif which upon binding directs the main chain of the peptide towards the β6/β7 loop of the PTB domain [15]. Given the proximity of R125 to a putative peptide binding cleft, substitution of a positively-charged, hydrophilic arginine to a bulky, neutral, more hydrophobic tryptophan could have significant repercussions on the biological function of this domain.
Table 1. Comparison of homology models generated by Robetta, HHpred/MODELLER and I-TASSER servers.
| Model | RSMD | |
| All C alphas | C alphas of secondary structure* | |
| MODELLER – Robetta | 3.8 Å | 2.0 Å |
| MODELLER – I-TASSER | 3.4 Å | 1.2 Å |
| I-TASSER – Robetta | 4.3 Å | 2.2 Å |
Manually determined C alphas of secondary structure correspond to human TBC1D1 residues 18–56, 61–79, 92–120, 128–136 and 142–162.
Figure 1. Location of R125 in the homology model of human TBC1D1 PTB1.

(A) Homology based model of TBC1D1 PTB domain (residues 13–161) predicted by Robetta server (http://robetta.bakerlab.org/). The R125 residue (red) is orientated towards the cleft formed between β5 and α2 (purple). (B) Solved structure of IRS-1 PTB domain (PDB ID: 1IRS, [47]). The IL-4 phosphopeptide (red) lies within the cleft formed between the β5 and α2 (purple), a type I β turn redirects the peptide such that the phosphorylated Tyr (shown) is orientated towards the β6/β7 loop.
Before examining the associations of R125W with phenotypes in our cohort, we used a range of prediction tools that were recently evaluated [16] to assess the pathogenicity of the variant. The conclusions differed, as the Polyphen and SIFT tools predicted the variant to be harmful, whereas MutPred and SNPsGO reported it to have a neutral effect (Table 2).
Table 2. Evaluation of the predicted pathogenicity of R125W according to a range of prediction tools.
| Prediction Tool | Effect |
| SIFT | DELETERIOUS |
| PolyPhen | DAMAGING |
| PolyPhen2 HumDiv | DAMAGING |
| PolyPhen2 HumVar | DAMAGING |
| PANTHER | UNKNOWN |
| MutPred | NEUTRAL |
| SNPsGO | NEUTRAL |
Study Sample Characteristics
8,228 mothers and 9,193 offspring were successfully genotyped for the R125W variant with a minor allele frequency (MAF) of 9.0% and 8.9% respectively for the 125W allele. The genotype frequencies for both mothers and children were consistent with Hardy-Weinberg equilibrium (P = 0.24 and 0.18 respectively). These MAF in mothers and offspring in our cohort were consistent with those found in previous general population studies [5], [6].
Table 3 shows the study characteristics. All offspring measurements were taken during the 15+year focus clinic (mean age: 15.5 years; range: 14.3–17.6 years). The adiposity phenotypes measured were BMI (mean: 21.4 kg/m2, range 14.1 kg/m2–39.7 kg/m2), waist circumference (mean: 76.5 cm, range 54.3 cm–125.9 cm) and (Dual-energy X-ray absorptiometry) DXA-assessed fat mass 14.1 kg (0.6 kg–70.9 kg). Adiposity measurements differed somewhat between males and females but the magnitude of these differences was small, despite the low p-values, which reflected the large sample size for these gender comparisons. For the mothers, BMI was the only adiposity measurement available; this was calculated from their self-report of prepregnancy height and weight given at mean age 27.7 when they were recruited in early pregnancy (mean BMI = 23.0 kg/m2, range = 12.5 kg/m2–51.6 kg/m2).
Table 3. Study Characteristics in the ALSPAC cohort.
| Characteristic | Males | Females | P (no sex difference) | ||
| N | Mean (SD) | N | Mean (SD) | ||
| Age (years) | 2,026 | 15.5 (0.3) | 2,195 | 15.6 (0.3) | 0.03 |
| BMI (kg/m2) | 2,007 | 21.0 (3.3) | 2,158 | 21.7 (3.6) | <0.001 |
| Waist Circumference (cm) | 1,598 | 76.8 (8.6) | 1,823 | 76.2 (8.7) | 0.05 |
| DXA Fat Mass (kg) | 2,003 | 10.0 (6.6) | 2,150 | 17.9 (7.6) | <0.001 |
| Maternal Age (years) | 3,421 | 29.0 (4.8) | 3,263 | 28.8 (4.7) | 0.04 |
| Maternal Pre-pregnancy BMI (kg/m2) | 3,421 | 23.0 (3.8) | 3,263 | 22.9 (3.9) | 0.29 |
Data are presented as mean (standard deviation), BMI – Body Mass Index; DXA - Dual-energy X-ray absorptiometry; BP – Blood Pressure.
Association between R125W and Adiposity Phenotypes in ALSPAC
We found no evidence for an association of the R125W variant with mean BMI at mean age 15.5 years in the ALSPAC offspring (Table 4). The results of the Kolmogorov-Smirnov test for equality of distributions suggested there was no difference between the BMI distributions of the two homozygous genotypes (D = 0.128, P = 0.813) (Figure 2). After adjustment of multiple testing there was no strong statistical evidence that R125W was associated with any of the phenotypes that we examined as continuous traits. We also found no evidence of an association of R125W with odds of obesity at age 15.5 years, or that associations differed by sex for any phenotypes (Table S2 shows sex specific analyses; P for R125W*sex interaction all> = 0.33). Using the pre-pregnancy data from 6,684 mothers in the cohort, we found no strong evidence of association between R125W and mean BMI or odds of obesity (Table 5).
Table 4. Association of TBC1D1 R125W genotypes with adiposity phenotypes in the ALSPAC Cohort.
| No. of Obs | C/C | C/T | T/T | OR/Padd ** | ||
| N (%) | 9,193 | 7,600 (82.67%) | 1,529 (16.63%) | 64 (0.7%) | ||
| Sex (Male/Female) | 9,193 | 3,949/3,651 | 800/729 | 28/36 | ||
| BMI (kg/m2) | 4,165 | 21.4 (3.5) | 21.1 (3.0) | 21.1 (2.8) | 0.05 | |
| Continuous traits | Waist Circumference (cm) | 3,421 | 76.7 (8.8) | 75.8 (7.8) | 74.3 (6.1) | 0.02 |
| DXA Fat Mass (kg) | 4,153 | 14.2 (8.4) | 13.6 (7.2) | 13.7 (7.4) | 0.42 | |
| Odds of Obesity | Lean (BMI<25 kg/m2)* | 3,404 | 2,793 | 588 | 23 | reference |
| Obesity (BMI≥30 kg/m2) | 155 | 137 | 17 | 1 | 0.63 (0.39–1.01), P = 0.06 |
Data are presented as mean (standard deviation), BMI – Body Mass Index; DXA - Dual-energy X-ray absorptiometry; BP – Blood Pressure.
Odd Ratios are calculated in comparison to lean subjects based on the additive model of inheritance. Lean, overweight and obesity cut-offs points are based on IOTF equivalents for offspring aged 15.5.
The Bonferroni corrected p-value equivalent to 0.05 is 0.0125.
Figure 2. Kernel Density Plot to Show BMI Distribution at mean age 15.5 years between Homozygous Genotypes.
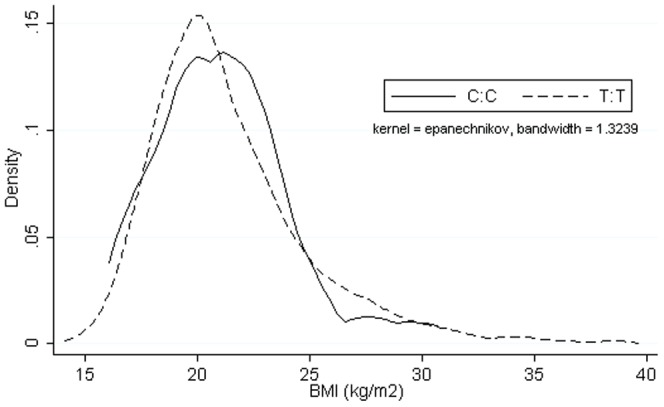
Table 5. TBC1D1 R125W Genotype Frequencies of Mothers in the ALSPAC Cohort according to Overweight and Obese status.
| BMI Category | No. of Obs | C/C | C/T | T/T | OR/Padd ** | |
| N (%) | - | 6,684 | 5,540 (82.88%) | 1,099 (16.44%) | 45 (0.68%) | |
| Association across Genotypes | BMI (kg/m2) | 6,684 | 23.0 (3.8) | 22.8 (3.9) | 23.1 (3.9) | 0.19 |
| Odds of Obesity | Lean (BMI<25 kg/m2)* | 5,261 | 4,353 (82.74%) | 873 (16.59%) | 35 (0.67%) | reference |
| Obesity (BMI≥30 kg/m2) | 366 | 301 (82.24%) | 64 (17.49%) | 1 (0.27%) | 1.01 (0.77–1.31), P = 0.96 |
Data are presented as mean (standard deviation), BMI – Body Mass Index.
Odd Ratios are calculated in comparison to lean subjects based on the additive model of inheritance.
The Bonferroni corrected p-value equivalent to 0.05 is 0.025.
Familial Association between R125W and Obesity
To investigate the over-transmission of the R125W risk allele from heterozygous mothers to affected (i.e. obese) offspring or from affected mothers to heterozygous offspring we applied the Transmission Disequilibrium Test (TDT). We found no evidence of an association of R125W with familial obesity for any of the different thresholds used to define obesity (Table 6).
Table 6. Results of Transmission Disequilibrium Test Assessing Association between R125W Variant and Familial Obesity.
| Affected Case | BMI Percentile Threshold for Obesity | McNemar's χ2 Statistic | P-value | McNemar's χ2 Statistic (Females) | P-value | McNemar's χ2 Statistic (Males) | P-value |
| Offspring | IOTF | 0.06 | 0.80 | 0.00 | 1.00 | 0.14 | 0.71 |
| 97th | 0.67 | 0.41 | 2.00 | 0.16 | 0.00 | 1.00 | |
| 90th | 2.29 | 0.13 | 1.67 | 0.20 | 0.69 | 0.41 | |
| 75th | 0.05 | 0.83 | 0.349 | 0.56 | 0.95 | 0.33 | |
| 50th | 0.26 | 0.61 | 0.24 | 0.62 | 0.05 | 0.83 | |
| Mothers | IOTF | 0.05 | 0.83 | 0.40 | 0.53 | 0.82 | 0.37 |
| 97th | 0.00 | 1.00 | 0.14 | 0.71 | 0.20 | 0.66 | |
| 90th | 0.47 | 0.49 | 0.00 | 1.00 | 0.80 | 0.37 | |
| 75th | 1.09 | 0.30 | 1.20 | 0.27 | 0.18 | 0.67 | |
| 50th | 1.03 | 0.31 | 0.11 | 0.75 | 1.15 | 0.28 |
Discussion
Our homology modelling locates R125 within a region of a PTB domain predicted to mediate protein-protein interactions; as such a tryptophan substitution would be anticipated to have deleterious functional consequences. We found no strong evidence that the TBC1D1 R125W variant was associated with adiposity phenotypes in a general population. Furthermore, we did not find any evidence for association with familial obesity.
The R125W variant of TBC1D1 lies within the first of the two PTB domains of the protein. No structure of the TBC1D1 PTB domain is currently available to provide clues as to the role of R125 in the function of the protein. However, there are several structures of other PTB domains with sufficient amino acid sequence homology to allow homology modelling of this region of TBC1D1. We employed three different modelling algorithms to obtain a predicted structure. All methods used (i) highlighted R125 as being located in a loop formed between two β-sheets within the PTB domain structure (β6 and β7; Figure 1A) and (ii) oriented the positively charged guanidinium side-chain of R125 towards the base of a groove formed between an α-helix (α2) and a β-sheet (β5). This groove is highly reminiscent of the phosphopeptide binding groove found in the Shc and Insulin Receptor-1 (IRS1) PTB domains, where the phosphotyrosine on the peptide interacts with the guanidinium side-chains of arginine residues found at the base (compare Figure 1A with the structure of the IRS1 PTB domain in Figure 1B).
Our modelling suggests that R125 in TBC1D1 plays a role in the binding of an effector protein, possibly via a mechanism that involves phosphorylation although it should be noted that several PTB domains can bind peptide ligands in a phosphorylation-independent manner [17]. In most mammals this residue is conserved as an arginine, which is positively charged, however in some (including the rat) it exists as a glutamine (Q) that is polar but uncharged. The consequences of an R125Q substitution on the binding of a peptide ligand are difficult to predict and could be subtle, however substitution of either amino acid for a tryptophan, which is a hydrophobic aromatic residue, would be expected to be highly deleterious to function either by replacing an important electrostatic interaction by a hydrophobic one, or by significantly altering the conformational preference of this loop region.
No peptides or proteins have yet been identified that bind to the N-terminal TBC1D1 PTB domain, though it has been suggested that the PTB domains of the related protein, AS160 (TBC1D4), could be involved in dimerisation of the protein [18]. These possibilities, and the consequences of the substitutions, require further experimental investigation in the case of TBC1D1. Our finding of no association between R125W and BMI, other markers of adiposity or odds of obesity, is consistent with two previous general population studies [5], [6]. The reason for the lack of association in general populations when associations have been found with familial obesity (though not in our study) is unclear. The association between the 125W allele and familial obesity was originally identified using linkage analysis amongst pedigrees primarily consisting of families from Utah [7]. The R125W variant was enriched in the individuals in 10 of these families (P = 0.000007) when compared with 846 population controls. Ethnicity and ancestry are unlikely to explain the difference between family studies and results from general population cohorts as the latter are conducted in European origin individuals and the Utah cohort has an ancestry similar with that of Europeans [19].
Another possible explanation for the difference is that we did not preselect our case and control cohorts. Stone et al. [5] selected their case families from either the Health Family Tree Program [20] or families consisting of patients who had previously undergone a gastric bypass [21]. They manually selected pedigrees for their linkage analysis that had at least two affected sisters (BMI≥35 kg/m2) and also carried R125W, while the control subjects were unrelated and taken from different populations [22]. Meyre et al. [6] selected their pedigrees based on at least one person having a BMI>35 kg/m2 and a first degree relative with BMI>30 kg/m2 (435 pedigrees) or pedigrees having at least one obese child (674 pedigrees). In contrast, our study was population based and included all mothers and offspring for whom we had genotype and phenotype data and did not have any inclusion or exclusion criteria based on BMI. As such, our results cannot be interpreted as evidence against the previous findings concerning TBC1D1 [5], [6].
Moreover, if the R125W effect is restricted to, or conditional on, very high BMI, then we would not have had the statistical power to detect it. Initially we only had power of 21.21% using the International Obesity Task Force (IOTF) cut off points analysing the transmission of the 125W risk allele from mothers to affected offspring. However, we were able to control/minimise this affect in our subsequent analyses by using different thresholds for obesity to increase the number of study participants being analysed. In doing so, we achieved power of 41.31% using an obesity cut off of 90%. We also analysed the transmission of the 125W risk allele from affected mothers to offspring to more extensively evaluate the hypothesis.
A further limitation of our study is that the maternal pre-pregnancy BMI was self-reported and therefore is susceptible to recall error. Since the women will not have been aware of their genotype this would be non-differential for the association examined here and would therefore have the statistical expectation of biasing results towards the null. However, the consistent null associations in offspring across a range of directly assessed adiposity measurements could not be explained by this recall error.
Conclusion
Our protein modelling studies support a plausible role of this amino acid in the function of the N-terminal TBC1D1 PTB domain. One initial report and one replication study have suggested that R125W is associated with familial obesity in females, though biologically it remains obscure why there should be a sex difference. Our analyses find no general population effect, nor trend in our smaller number of high BMI families. Biochemical studies of the mutant protein and further studies of families exhibiting extreme obesity may yield further insight.
Materials and Methods
Homology Modelling
Secondary structure prediction of human TBC1D1 (GenBank accession number NP_055988.2) with Jpred3 (www.compbio.dundee.ac.uk/www-jpred/) [23], together with domain coordinates from pfam (http://pfam.sanger.ac.uk/) [24], identified residues 13–161 to contain secondary structure elements consistent with a PTB domain topology. Tertiary structure of this region was predicted by Robetta (http://robetta.bakerlab.org/) [25], HHpred/MODELLER (http://toolkit.tuebingen.mpg.de/modeller) [26], [27] and I-TASSER servers (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) [28], [29] using default parameters. Root-mean-square deviation (RMSD) values were calculated from superimposition of modelled structures with InsightII (Accelrys, San Diego, CA). Geometric quality of the models was assessed, based on the Ramachandran plot of residues and main and side chain parameters, using PROCHECK [30]. USCF Chimera (version 1.7; [31]) was used for the structure visualisation.
Cohort Description
The Avon Longitudinal Study of Parents and Children (ALSPAC) is a population-based cohort study investigating genetic and environmental factors that affect the health and development of children. The study methods are described in detail elsewhere [32], [33] (http://www.bristol.ac.uk/alspac). Briefly, 14,541 pregnant women residents in the former region of Avon, UK, with an expected delivery date between 1st April 1991 and 31st December 1992, were eligible to take part in ALSPAC. There were 14,062 live born children, 13,988 of whom were alive at 1 year.
Ethical approval was obtained from the ALSPAC Law and Ethics Committee and the Southmead, Frenchay, United Bristol Healthcare NHS Trust (UBHT) and Weston Research Ethics Committees. Written informed consent was obtained from parents for all measurements made. In total, genotyping was attempted on DNA from 9,020 mothers and 10,920 offspring. All genotyping was performed by KBioscience (Herts, UK).
Data Collection
All measurements were taken by a trained research team during the 15+year focus clinic (mean age: 15.5 years; range: 14.3–17.6 years). Height was measured to the nearest 0.1 cm using a Leicester Height Measure (Holtain Crosswell, Dyfed) and weight was measured to the nearest 0.1 kg using Tanita electronic scales. Waist circumference was measured to the nearest 1 mm at the mid-point between the lower ribs and the pelvic bone with a flexible tape. DXA determined fat mass was assessed at the clinic using a Lunar Prodigy scanner (Madison, WI, USA) with pediatric scanning software.
At the time of pregnancy ALSPAC mothers were asked in a mailed questionnaire to retrospectively report their pre-pregnancy weight and their height; these were used to calculate maternal pre-pregnancy BMI.
Genotyping
Genotyping of the TBC1D1 variant R125W was undertaken in 9,020 mothers and 10,920 offspring. All genotyping was performed by KBiosciences (Herts, UK) using their own system of fluorescence based competitive allele-specific PCR (KASPar). Details of assay design are available from the KBiosciences website (http://www.kbioscience.co.uk).
9,193 offspring were successfully genotyped for the R125W variant. Study participants with a reported (maternal) non-white ethnic origin (N = 218) were excluded from analyses. A further 72 offspring were removed from analyses as they were either the second born child or born in a multiple pregnancy. Of the remaining 8,903 offspring, 4,165 had been successfully measured for height and weight at the 15+year focus clinic. 3,421 had their waist circumference measurement taken and 4,153 had a DXA assessed fat mass measurement recorded.
8,228 mothers were successfully genotyped for the R125W variant and 10,569 mothers with a reported white ethnic origin had filled out a questionnaire concerning their pre-pregnancy BMI. There was an overlap of 6,748 mothers who were included in both of these groups. A further 64 subjects were removed before any analysis took place due to missing data of their offspring at the pregnancy, leaving our sample size at 6,684.
From the 6,684 mothers that were included in our BMI analysis, 2,292 of them had an offspring amongst the 4,165 subjects taken from the offspring analysis. This meant we had 2,292 mother-offspring pairs available to investigate for familial association.
Statistical Analysis
We used Pearson's χ2-test amongst all offspring and mothers separately to examine if the genotype distributions were consistent with Hardy-Weinberg equilibrium [34]. Direct associations between the R125W variant and obesity-related phenotypes in the offspring were assessed by linear regression using an additive model of inheritance as this was previously described as the most suitable for R125W [5]. Logistic regression was used to examine the association of R125W with odds of obesity, again using an additive model of inheritance. Multiple comparisons were adjusted for using the Bonferroni correction [35], with a p-value threshold of 0.0125 being the equivalent of the conventional 0.05. The Kolmogorov-Smirnov test for equality of distributions [36] was used to compare the BMI distributions between the two homozygous genotypes. We used the maternal data to assess the association between the R125W variant and adult (pre-pregnancy) BMI and overweight/obesity using linear and logistic models, respectively. Any possible interaction between genotype and sex in the offspring analyses was assessed using the likelihood ratio test to compare two regression models, one which was simply adjusted for sex and another which also included an interaction term for genotype*sex. Furthermore, we analysed males and females separately to verify whether associations appeared similar in males and females.
We applied the transmission disequilibrium test (TDT) [37] to investigate familial association between the R125W polymorphism and obesity. Initially, we used the International Obesity Task Force (IOTF) cut off points for obesity [38] (BMI = 28.6 kg/m2 in females and 29.29 kg/m2 in males at age 15.5) and analysed transmissions between mother and affected child. We repeated this analysis looking at mother-son and mother-daughter pairs separately to accommodate for the previous reports associating the R125W variant with familial obesity in females. It has been suggested that the TDT suffers from a lower statistical power [39], [40] compared with case-control studies of unrelated individuals; however, in our subsequent analysis we used different thresholds for obesity to increase the number of study participants being analysed and control/minimise this affect. We extended this work by classifying the mothers as the affected case and then repeated our analysis, initially using BMI cut off point of 30 kg/m2.
Stata 12.0 software (StataCorp, College Station, TX) was used for general statistical analyses.
Handling of Heterozygote Transmissions
There is literature concerning the issue of missing parental data when testing for family based association [41]–[43]. However, techniques involved in these circumstances may incorporate bias into the study [44]. When using the TDT, including with genotype data for both parents, there may be ambiguity concerning which alleles are transmitted from parents who are heterozygous to offspring who are also heterozygous [45], [46]. Since paternal genotype data was not available, we used an approach based on conditional probability, which takes into account the MAF of the R125W variant, to calculate the probability that the risk allele has been transmitted to the affected offspring. The approach is detailed in the supplementary material (Method S2).
Supporting Information
Superimposed homology models of human TBC1D1 PTB1. Superimposed homology models of TBC1D1 PTB1 (residues 13–161) from Robetta (blue), HHpred/MODELLER (green) and I-TASSER (orange) servers with the R125 side chain displayed as spheres. Shown in yellow is the PTB domain of AIDA1 (2M38; DOI:10.2210/pdb2m38/pdb) which has the highest (24%) amino acid sequence identity to the TBC1D1 PTB1 domain. UCSF Chimera (version 1.7) was used to coordinate superimposition of structures utilising the default parameters of the matchmaker function.
(TIF)
Ramachandran Plot analysis, define d by PROCHECK, of the homology models generated by Robetta, HHpred/MODELLER and I-TASSER servers.
(DOCX)
Average Phenotype measurements according to TBC1D1 R125W genotypes stratified by Gender in Offspring in ALSPAC cohort.
(DOCX)
Assessing the stereochemical quality of the individual homology models.
(DOCX)
Conditional probability for Heterozygotes.
(DOCX)
Acknowledgments
We are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, manager, receptionists and nurses. This publication is the work of the authors and Tom Richardson will serve as guarantor for the contents of this paper.
Funding Statement
This work was supported by the British Heart Foundation (Ref: PG/10/008). The UK Medical Research Council and the Wellcome Trust (Grant ref: 092731) and the University of Bristol provide core support for ALSPAC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. TR is a UK MRC PhD student.
References
- 1. Wormser D, Kaptoge S, Di Angelantonio E, Wood AM, Pennells L, et al. (2011) Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: collaborative analysis of 58 prospective studies. Lancet 377(9771): 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stunkard AJ, Foch TT, Hrubec Z (1986) A twin study of human obesity. JAMA 256: 51–54. [PubMed] [Google Scholar]
- 3. Stunkard AJ, Harris JR, Pedersen NL, McClearn GE (1990) The body-mass index of twins who have been reared apart. N Engl J Med 322: 1483–1487. [DOI] [PubMed] [Google Scholar]
- 4. Loos RJ, Bouchard C (2003) Obesity — is it a genetic disorder? J Intern Med 254: 401–425. [DOI] [PubMed] [Google Scholar]
- 5. Stone S, Abkevich V, Russell DL, Riley R, Timms K, et al. (2006) TBC1D1 is a candidate for a severe obesity gene and evidence for gene/gene interaction in obesity predisposition. Hum Mol Genet 15: 2709–2720. [DOI] [PubMed] [Google Scholar]
- 6. Meyre D, Farge M, Lecoeur C, Proenca C, Durand E, et al. (2008) R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum Mol Genet 17: 1798–1802. [DOI] [PubMed] [Google Scholar]
- 7. Stone S, Abkevich V, Hunt SC, Gutin A, Russell DL, et al. (2002) A major predisposition locus for severe obesity at 4p15–p14. Am J Hum Genet 70: 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, et al. (2008) Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J 409: 449–459. [DOI] [PubMed] [Google Scholar]
- 9. Roach WG, Chavez JA, Miinea CP, Lienhard GE (2007) Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein TBC1D1. Biochem J 403: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor EB, An D, Kramer HF, Yu H, Fujii NL, et al. (2008) Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem 283: 9787–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pehmoller C, Treebak JT, Birk JB, Chen S, Mackintosh C, et al. (2009) Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 297: E665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. An D, Toyoda T, Taylor EB, Yu H, Fujii N, et al. (2010) TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes 59: 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, et al. (2008) TBC1D1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet 40: 1354–1359. [DOI] [PubMed] [Google Scholar]
- 14. Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, et al. (2010) Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464: 587–591. [DOI] [PubMed] [Google Scholar]
- 15. DiNitto JP, Lambright DG (2006) Membrane and juxtamembrane targeting by PH and PTB domains. Biochim Biophys Act 1761: 850–867. [DOI] [PubMed] [Google Scholar]
- 16. Thusberg J, Olatubosun A, Vihinen M, et al. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat 32: 358–368. [DOI] [PubMed] [Google Scholar]
- 17. Forman-Kay JD, Pawson T (1999) Diversity in protein recognition by PTB domains. Curr Opin Struct Biol 9: 690–695. [DOI] [PubMed] [Google Scholar]
- 18. Koumanov F, Richardson JD, Murrow BA, Holman GD (2011) PTB-domain constructs of AS160 inhibit insulin-stimulated GLUT4 vesicle fusion with plasma membrane. J Biol Chem 286: 16574–16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith EM, Wang X, Littrell J, Eckert J, Cole R, et al. (2006) Comparison of linkage disequilibrium patterns between the HapMap CEPH samples and a family-based cohort of Northern European descent. Genomics 88: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Williams RR, Hunt SC, Barlow GK, Chamberlain RM, Weinberg AD, et al. (1988) Health family trees: a tool for finding and helping young members of coronary and cancer prone pedigrees in Texas and Utah. Am J Public Health 78: 1283–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams TD, Avelar E, Cloward T, Crosby RD, Farney RJ, et al. (2005) Design and rationale of the Utah obesity study. A study to assess morbidity following gastric bypass surgery. Contemp Clin Trials 26: 534–551. [DOI] [PubMed] [Google Scholar]
- 22. Smith SC, Goodman GN, Edwards CB (1995) Roux-en-Y gastric bypass: a 7-year retrospective review of 3,855 patients. Obes Surg 5: 314–318. [DOI] [PubMed] [Google Scholar]
- 23. Cole C, Barber JD, Barton GJ (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36: W197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Finn RD, Mistry J, Tate J, Coggill P, Heger A, et al. (2010) The Pfam protein families database. Nucleic Acids Res 38: D211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32: W526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23: 318–326. [DOI] [PubMed] [Google Scholar]
- 27. Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21: 951–960. [DOI] [PubMed] [Google Scholar]
- 28. Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y (2007) Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 69: 108–117. [DOI] [PubMed] [Google Scholar]
- 30. Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067. [DOI] [PubMed] [Google Scholar]
- 31. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612. [DOI] [PubMed] [Google Scholar]
- 32. Golding J, Pembrey M, Jones R (2001) ALSPAC–the Avon Longitudinal Study of Parents and Children. I. Study methodology. Paediatr Perinat Epidemiol 15: 74–87. [DOI] [PubMed] [Google Scholar]
- 33.Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, et al. (2012) Cohort Profile: The Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. International Journal of Epidemiology, doi:10.1093/ije/dys066. [DOI] [PMC free article] [PubMed]
- 34. Rodriguez S, Gaunt TR, Day IN (2009) Hardy-Weinberg Equilibrium Testing of Biological Ascertainment for Mendelian Randomization Studies. Am J Epidemiol 169: 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonferroni CE (1936) Teoria statistica delle classi e calcolo delle probabilit `a. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze 8: 3–62. [Google Scholar]
- 36. Lilliefors HW (1967) On the Kolmogorov-Smirnov Test for Normality with Mean and Variance Unknown. Journal of the American Statistical Association 62: 399–402. [Google Scholar]
- 37. Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52: 506–16. [PMC free article] [PubMed] [Google Scholar]
- 38. Cole TJ, Bellizzi MC, Flegal KM, Dietz WH (2000) Establishing a standard definition for child overweight and obesity worldwide: international survey. BMJ 320: 1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morton NE, Collins A (1998) Tests and estimates of allelic association in complex inheritance. Proc Natl Acad Sci 95: 11389–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Risch N, Teng J (1998) The relative power of family-based and case–control designs for linkage disequilibrium studies of complex human disease. I. DNA pooling. Genome Res 8: 1273–1288. [DOI] [PubMed] [Google Scholar]
- 41. Sun F, Flanders WD, Yang Q, Khoury MJ (1999) Transmission disequilibrium test (TDT) when only one parent is available: the 1-TDT. Am J Epidemiol 150: 97–104. [DOI] [PubMed] [Google Scholar]
- 42. Sebastiani P, Abad MM, Alpargu G, Ramoni MF (2004) Robust transmission/disequilibrium test for incomplete family genotypes. Genetics 168: 2329–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gordon D, Haynes C, Johnnidis C, Patel SB, Bowcock AM, et al. (2004) A transmission disequilibrium test for general pedigrees that is robust to the presence of random genotyping errors and any number of untyped parents. Eur J Hum Genet 12: 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Curtis DR, Sham PC (1995) A note on the application of the transmission disequilibrium test when a parent is missing. Am J Hum Genet 56: 811–812. [PMC free article] [PubMed] [Google Scholar]
- 45. Dudbridge F, Koeleman BC, Todd JA, Clayton DG (2000) Unbiased application of the transmission/disequilibrium test to multilocus haplotypes. Am J Hum Genet 66: 2009–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balding DJ, Bishop MJ, Cannings C (2007) Handbook of statistical genetics Volume 1: , p1271. [Google Scholar]
- 47. Zhou MM, Huang B, Olejniczak ET, Meadows RP, Shuker SB, et al. (1996) Structural basis for IL-4 receptor phosphopeptide recognition by the IRS-1 PTB domain. Nat Struct Biol 3: 388–393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Superimposed homology models of human TBC1D1 PTB1. Superimposed homology models of TBC1D1 PTB1 (residues 13–161) from Robetta (blue), HHpred/MODELLER (green) and I-TASSER (orange) servers with the R125 side chain displayed as spheres. Shown in yellow is the PTB domain of AIDA1 (2M38; DOI:10.2210/pdb2m38/pdb) which has the highest (24%) amino acid sequence identity to the TBC1D1 PTB1 domain. UCSF Chimera (version 1.7) was used to coordinate superimposition of structures utilising the default parameters of the matchmaker function.
(TIF)
Ramachandran Plot analysis, define d by PROCHECK, of the homology models generated by Robetta, HHpred/MODELLER and I-TASSER servers.
(DOCX)
Average Phenotype measurements according to TBC1D1 R125W genotypes stratified by Gender in Offspring in ALSPAC cohort.
(DOCX)
Assessing the stereochemical quality of the individual homology models.
(DOCX)
Conditional probability for Heterozygotes.
(DOCX)