

Abstract
One fundamental feature of mutant forms of p53 consists in their accumulation at high levels in tumors. At least in the case of neomorphic p53 mutations, which acquire oncogenic activity, stabilization is a driving force for tumor progression. It is well documented that p53 mutants are resistant to proteasome-dependent degradation compared with wild-type p53, but the exact identity of the pathways that affect mutant p53 stability is still debated. We have recently shown that macroautophagy (autophagy) provides a route for p53 mutant degradation during restriction of glucose. Here we further show that in basal conditions of growth, inhibition of autophagy with chemical inhibitors or by downregulation of the essential autophagic genes ATG1/Ulk1, Beclin-1 or ATG5, results in p53 mutant stabilization. Conversely, overexpression of Beclin-1 or ATG1/Ulk1 leads to p53 mutant depletion. Furthermore, we found that in many cell lines, prolonged inhibition of the proteasome does not stabilize mutant p53 but leads to its autophagic-mediated degradation. Therefore, we conclude that autophagy is a key mechanism for regulating the stability of several p53 mutants. We discuss plausible mechanisms involved in this newly identified degradation pathway as well as the possible role played by autophagy during tumor evolution driven by mutant p53.
Keywords: p53, mutant, mutations, autophagy, proteasome, ubiquitin tumor, cancer
Substantial lines of evidence indicate that the ability of mutant forms of p53 to elude proteolysis is a central step for tumor progression and metastasis in vivo.1-4 While multiple mechanisms for mutant p53-influenced tumorgenesis likely exist,5-9 and may differ in a mutation- and tissue-specific fashion,10 it is clear that in established tumors many p53 mutants escape the physiological mechanisms that control the stability of the wild-type protein, accumulating at abnormally high levels and leading to gain of oncogenic activities. However, a rather surprising finding was that in knock-in mouse models expressing p53R172H—equivalent to the human p53H175R—p53 expression was low in normal tissues as well as in some tumors.4,11 This expression profile observed in mice may mirror the heterogeneous pattern of expression seen in human tumor biopsies as well, which often display a mixed population of cells characterized by high or very low p53 mutant levels within the same sample.12-14 These observations have led to the important conclusion that like wild-type p53, mutant p53 is also inherently unstable, but accumulation arises as a result of additional events that occur in a tissue-or cell-specific fashion during tumor evolution. With these considerations in mind, the identification of the pathways that destabilize mutant p53 and their manipulation may open novel therapeutic avenues to effectively target their pro-oncogenic activities.
The proteasome and macroautophagy (henceforth referred to as autophagy) regulate the half-life of many proteins.15 While the proteasome predominantly targets short-lived ubiquitinated proteins, autophagy degrades long-lived misfolded proteins, intracellular aggregates or various damaged organelles. We previously reported that autophagy provides a route for p53 mutant degradation when cells are cultured in the absence of glucose.16-18 Here, we present additional data showing that autophagy, rather than the proteasome, crucially contributes to regulation of p53 mutant stability in basal conditions of growth. By keeping into account other studies demonstrating substantially opposite effects of autophagy on the stability of wild-type p53,19 along with the observation that autophagic genes are often lost in many human cancers,20,21 we discuss the implications of these findings.
Autophagy Controls Mutant p53 Stability
During the course of our previous work we found that in MDA-231 cells harboring p53R280K, a short-time treatment with the proteasome inhibitor MG132 did not substantially affect mutant p53 levels, while after a longer treatment time MG132 promoted a dramatic p53 loss, in spite of the fact that MG132 effectively prevented degradation of ubiquitinated protein substrates (Fig. 1A, compare lanes 2–4 and 6–8 with lanes 1 and 5, respectively). Similar results were obtained in other cell lines harboring p53 mutations, specifically in T47D (p53L194F), MDA-468 (p53R273H) (Fig. 1B), in TOV (p53H175R, not shown), as well as in the p53 null H1299 cell line ectopically expressing p53G245A or p53R175H (Fig. 1C). To examine the relationship between p53 stability and the proteasome further, we studied the degradation profile of p53R175H in either H1299 or TOV cells or of wild-type p53 in MCF7 cells in conditions in which the activity of the proteasome was pulsed-chased with a transient MG132 treatment, followed by a wash-out. The combination of these experiments revealed a stark inverse correlation between proteasomal activity and p53 mutant levels, such that when the proteasome was inhibited p53 mutant levels dropped (Fig. 1A-C), while during the wash-out phase, p53 mutant accumulated (Fig. 1D and E, compare lanes 2–4 with lane 1 in each panel). An opposite pattern of expression was seen with wild-type p53 (Fig. 1F), consistent with the known function of the proteasome as the major pathway for native p53 degradation.

Figure 1. Prolonged proteasome inhibition leads to p53 mutant depletion. (A) MDA-231 cells were treated with vehicle control, (DMSO) (lanes 1 and 5) or with 5 μM (lanes 2 and 6); 10 μM (lane 3 and 7) or 20 μM (lane 4 and 8) of MG132 for 3 h (lanes 1–4) or 18 h (lane 5–9). The expression levels of p53 or ubiquitinated proteins and actin are shown. (B) T47D (lanes 1–4) or MDA-468 (lanes 5 and 6) were mock treated (lanes 1 and 5) or treated with increasing concentrations of MG132 for 16 h. The expression levels of LC3, p53 and ubiquitinated proteins are shown. (C) Proteasome block induces p53 depletion in cells ectopically expressing p53. For these experiments, we employed H1299 cells ectopically expressing mutant p53G245 or p53H175R under the control of a tetracycline-regulated promoter.15 Cells were subjected to a short doxycycline treatment for 6 h, then they were washed and received fresh media lacking (lanes 1 and 4) or containing 10 μM (lanes 2 and 5) or 20 μM (lanes 3 and 6) MG132 for additional 12 h. The levels of ubiquitinated proteins, p62, p53 and LC3 are shown. (D–F) H1299 cells expressing p53R175H (D), lanes 1–4), TOV cells (E) or MCF7 cells (F) were treated with 20 μM MG132 for 8 to 12 h (indicated as time 0; lane 1 in all panels). After this time, cells were washed twice with PBS and were re-incubated in regular media for 4 h (lane 2), for 8 h (lane 3) or for 12 h (lane 4). The anti-p53 and anti-actin immunoblots are shown.
Since we have recently shown that autophagy provides a route for mutant p53 disruption during glucose restriction, we asked whether this degradation pathway is also responsible for p53 mutant degradation due to proteasome inhibition. Indeed, there is now substantial evidence that proteasomal inhibition enhances the load of misfolded proteins and triggers autophagy as a compensatory mechanism for their degradation.15,22 First, autophagy was activated in cells treated with MG132, as demonstrated by the decreased levels of p62 and/or the increased conversion of LC3 (Fig. 1B and C). We then examined the effects of MG132 in cells transfected with the plasmids harboring the shRNA for the essential autophagic genes ATG1/Ulk1 (Ulk1) or ATG5, relative to control shRNA transfected cells. As expected, MG132 failed to induce a robust activation of autophagy and LC3 conversion in cells transfected with the shRNA for these genes relative to control cells. More importantly, MG132-induced p53 degradation was entirely rescued in cells where autophagy is defective (Fig. 2A, compare lanes 5 and 6 and 8 and 9 with lanes 2 and 3).

Figure 2. Inhibition of autophagy increases mutant p53 half-life. (A) MDA-231 cells were transfected with control shRNA (lanes 1–3) or with shRNA for ATG1/Ulk1 (indicated as Ulk1, lanes 4–6) or ATG5 (lanes 7–9). Cells were left untreated (lanes 1, 4 and 7) or were treated with 10 μM (lanes 2, 5 and 8) or 20 μM (lanes 3, 6 and 9) MG132 for 12 h. The panels show the immunoblots for Ulk1, ATG5, LC3 and p53 (with two different exposures of the anti-p53 blot outlined by the vertical line). (B) Assessment of p53 expression half-life in the absence (lanes 1–3) or presence of MG132 (20 μM, lanes 4–6) or chloroquine (50 μM, lanes 7–9). A group of cells received CHX for 12 h (lanes 2, 5 and 8) or 24 h (lanes 3, 6 and 9). The levels of ubiquitinated proteins, of p53, p62, LC3 and actin were assessed. Note the pattern of LC3 conversion and the difference in p62 levels in the MG132 and CHQ treated samples, relative to control.
Viewed together, our results outlined the importance of autophagy in regulation of mutant p53 stability. Additional experiments were performed to better clarify the role of autophagy in regulation of mutant p53 stability. First, we studied the half-life of mutant p53 in cells where either autophagy or the proteasome were inhibited. By employing cycloheximide (CHX) chase experiments, we confirmed that mutant p53 degradation was faster in the presence of MG132, consistent with our previous results (Fig. 2B, lanes 4–6 vs. lanes 1–3). In contrast, in cells treated with the autophagic inhibitor chloroquine, p53 stability was increased (Fig. 2B, lanes 7–9 vs. lanes 1–3). We next generated stable cell lines harboring the Ulk1 or control shRNAs. We found that the expression levels of mutant p53 and its half-life, as assessed with CHX chase experiments, were increased in stable MDA-231 clones expressing the Ulk1-shRNA relative to control cells (Fig. 3A and B). Similarly, downregulation of either Beclin-1 or of ATG5 with RNA interference led to p53 mutant accumulation (Fig. 3C and D). To further corroborate these results, we asked whether superimposing the expression of autophagic genes also affects p53 mutant levels. We determined that overexpression of either Beclin-1 or Ulk1 led to downregulation of mutant p53 levels in MDA-231 cells and in H1299 cells ectopically expressing p53H175R, respectively (Fig. 3E and F). Thus, we conclude that autophagy provides a key route for mutant p53 destabilization.

Figure 3. Autophagy controls mutant p53 expression levels. (A) p53 expression and half-life were studied in stable MDA-231 cells harboring control shRNAs (lanes 1 and 2) or the shRNAs for Ulk1 (lanes 3 and 4) in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 50 μM CHX for 24 h. Note the presence of high molecular weight forms of p53 displaying a migration pattern consistent with ubiquitination in Ulk1-shRNA cells. In (B), the relative expression levels of Ulk1 in cells harboring the Ulk1 or control shRNA are shown. (C) p53, Beclin and actin expression levels in TOV cells transfected with control or Beclin-specific siRNA. Images shown are derived from the same autoradiogram, where lanes between relevant samples were cut. (D) The panel shows p53 expression in control shRNA harboring cells or in two different clones expressing the ATG5 shRNA (indicated as ATG5-1 and 2). (E) MDA-231 cells were transfected with control DNA (pcDNA/TO, 8 μg/plate) or with identical concentration of the cDNA expressing Beclin-1. p53, Beclin-1 and actin levels were assessed 48–72 h after transfection. (F) H1299 cells infected with control adenovirus (lanes 1 and 2) or with p53RH175H-expressing adenovirus (lanes 3 and 4) were transfected with a control vector (lanes 1 and 3) or with the Ulk1 expressing vector (lanes 3 and 4). The expression levels of Ulk1, p53 and tubulin are shown. Note that endogenous Ulk1 is not detectable in H1299 cells at exposure times in which the ectopically expressed is clearly visible.
Opposite Effects of Autophagy on the Stability of Mutant and Wild-Type p53: Fate or Coincidence?
We have demonstrated that inhibition of autophagy leads to mutant p53 accumulation, while its activation via proteosomal block or during glucose restriction, induces degradation which can be blocked by inhibiting the expression of various autophagic genes (this study and ref. 16). Our data showing that mutant p53 interacts with components of the autophagic apparatus, such as Beclin-1 and p62,16 together with results presented here strongly argue that mutant p53 is a direct substrate for autophagic degradation. It is relevant to note that wild-type p53 and autophagy cross-talk in a substantially opposite fashion. A chemical inhibitor of autophagy, spautin-1, or mono-allelic deletion of Beclin-1 both destabilize wild-type p53 by inducing its ubiquitination and proteasome-dependent degradation.19 Thus, autophagy promotes stabilization of wild-type p53 but destabilization of mutant p53. We then reasoned that this differential effect of autophagy on the stability of p53 depending upon its mutational status and oncogenic capacity is unlikely to be a restricted phenomenon. In fact, the analysis of the available literature reveals that mutant p53 is not the only example of an oncogenic protein that can be targeted for degradation via autophagy (summarized in Fig. 4). The members of the Dishevelled (Dvl) family are essential mediators of Wnt/β-catenin signal pathway, the dysregulation of which promotes tumor development in various tissues.23 Induction of autophagy during starvation promotes degradation of all three Dvl family members and also destabilizes β-catenin, thus extinguishing Wnt oncogenic signaling. Similarly, two master regulators of NFκB, namely IκB kinase (IKK) and NFκB-inducing kinase (NIK), which are highly expressed in many cancers, are also direct substrates of autophagy.24,25 Other relevant examples of oncogenic molecules degraded via autophagy include the BCR-ABL26 and PML-RARA27 fusion proteins, the Ret tyrosine kinase28 as well as the viral oncogenes KIT,29 (the v-KIT Hardy-Zuckerman feline sarcoma homolog) and large T antigen of JC virus.30 Thus, the available data are consistent with the idea that autophagy acts as a tumor barrier but add a new twist to this concept, specifically suggesting that degradation of oncogenic proteins, including mutant p53, might be a relevant aspect of the tumor-suppressive activity of autophagy. In the case of Dvl, NIK, BCR-ABL and PML-RARA, it is not entirely clear to what extent basal autophagy contributes to their degradation, while autophagic disruption occurs when autophagy is stimulated above basal levels by stress signals (e.g., starvation) or by drugs (e.g., arsenic trioxide or geldanamycin). Our experiments showing that the manipulation of autophagic genes in the absence of any stress signal is sufficient to modify mutant p53 levels establish that basal autophagy controls mutant p53 degradation, and that this proteolytic pathway is enhanced when autophagy is stimulated by proteasome inhibition or by glucose restriction. An important question is how autophagy recognizes and targets for disruption mutant forms of p53 or other oncogenic proteins. By analogy with proteins involved in the pathogenesis of neurodegenerative disorders, which are relevant substrates for autophagic clearance, below we discuss the nature of these possible discriminatory signals.
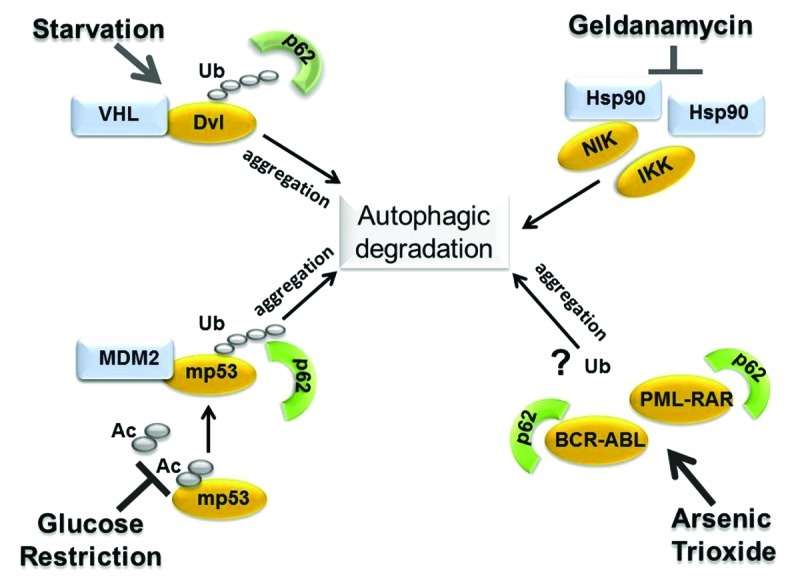
Figure 4. Summary of the available literature depicting relevant examples of oncogenic proteins degraded via autophagy and the molecular changes involved (see text for explanation). Autophagic degradation of members of the Dishevelled (Dvl) family is triggered by starvation and involves Dvl ubiquitination, in turn mediated by VHL, as well as aggregation and the interaction with p62.23 Glucose restriction induces mutant p53 deacetylation,16 increases the interaction with MDM2 as well as ubiquitination and mutant p53 detection in p62-positive aggregates. In the case of NIK and of IkB-kinase, IKK, autophagic degradation is induced by treatment with the Hsp90 inhibitor, geldanamycin, which, in turn, leads to release of these proteins from the interaction with Hsp90.24,25 In this case, ubiquitination is apparently dispensable for autophagy-dependent degradation. In two other studies, treatment with arsenic trioxide induced aggregation and the interaction of BCR-ABL or PML-RARA with p62 followed by autophagic degradation.26,27 Whether degradation requires ubiquitination was not directly addressed in these studies.
Discriminatory Signals for p53 Mutant Autophagic Disruption: Aggregation, Ubiquitination and Protein-Protein Interactions
Proteins targeted for autophagic degradation are typically misfolded proteins that form either macro- or micro-aggregates that are too large to fit in the narrow proteosomal channel and, thus, are unsuitable for proteosomal disruption.31,32 Protein misfolding and aggregation arise as a result of a variety of mechanisms, including mutations, post-translational modifications, excessive synthesis, environmental or intracellular stress. Since many p53 mutations have a misfolded configuration and display a high tendency to aggregate, they possess the characteristics of typical autophagic substrates. Protein aggregates are tagged for autophagic degradation with modalities very similar to those employed by the proteasome, in that they require chaperones, ubiquitin and a variety of ubiquitin-modifying enzymes and are eventually recognized by specific autophagic receptors, such as p62 and NBR1. We have shown previously that during glucose restriction mutant p53 becomes deacetylated and ubiquitinated, colocalizing in p62-positive aggregates, and autophagic degradation requires the activity of the E3 ligase MDM216 (Fig. 4). Similarly, autophagic disruption of Dvl during starvation leads to aggregation and requires VHL-mediated E3 ubiquitination activity.23 Because MDM2 or VHL also target their substrates for ubiquitin-dependent proteasomal disruption, a relevant question is whether discriminatory signals exist that specifically divert a set of substrates, including mutant p53, from the proteasome directing them toward autophagic degradation. While many misfolded denatured proteins are delivered to the lysosomes directly by the Hsc70 complex via chaperone-mediated-autophagy31 (CMA), the p53 protein does not contain the canonical consensus motif “KFERQ” that mediates Hsc70 recognition and CMA-dependent degradation. As in the case of other proteins, an attractive possibility is that at least one discriminatory signal is provided by the topology of ubiquitin chains, in turn dictated by selective protein-protein interactions (depicted in Fig. 5). Ubiquitin possesses seven internal lysines (K6, K11, K27, K29, K33, K48 and K63) each of which can potentially be used for the formation of polyubiquitin chains. While the proteasome predominantly recognizes K48-linked-chains, p62 and NBR1 interact preferentially with K63-linked ubiquitinated substrates.31 Ubiquitination of K63, in turn, can be catalyzed via the action of a unique and specific E2 enzyme, Ubc13,33 as well as by specific combinations of various E2 and E3 ubiquitin-modifying enzymes. It was previously shown that Ubc13 induces K63-dependent ubiquitination of wild-type p53, preventing its proteasomal degradation while modifying its subcellular localization and transcriptional activity.34 It is currently unknown whether mutant p53 is similarly capable of binding to Ubc13 and the topology of ubiquin chains linked to mutant p53 has not been fully elucidated yet. However, several studies have documented that under basal conditions of growth many types of p53 mutants are poorly ubiquitinated, in part due to a negative interference with the E3 ubiquitin ligase MDM2, and ubiquitination requires the activity of CHIP, a chaperone-dependent ubiquitin E3 ligase.35,36 Intriguingly, in addition to promoting proteasomal clearance of many proteins, CHIP can also interact with Ubc13, catalyzing K63-linked ubiquitin chain formation,37 and this activity is likely involved in the degradation of α-synuclein aggregates via autophagy.31 Similarly, while MDM2 predominantly catalyzes K48-linked chain formation, recent studies have shown that in the presence of its family member MDMX and of UbC13, it can also promote K63 chain formation.38,39 Therefore, one could speculate that intracellular or environmental stress signals promote the p53 interaction with specific combinations of E2 and E3 ubiquitin-modifying enzymes, including MDM2 or CHIP, which would edit the architecture of ubiquitinated chains bound to mutant p53 making it capable to be recognized by autophagic receptors. In this scenario, modification of mutant p53 through other post-translational modifications and its subcellular localization are likely to play a key role as well. For example, in the case of wild-type p53, various modifications, including acetylation, prevent ubiquitination. Even though how post-translational modifications influence p53 mutant activity is largely unclear, at least in the case of glucose restriction, deacetylation appears to be a pre-requisite for mutant p53 autophagic degradation,16 strongly arguing that acetylation and ubiquitination are competing signals for autophagic disruption as well.
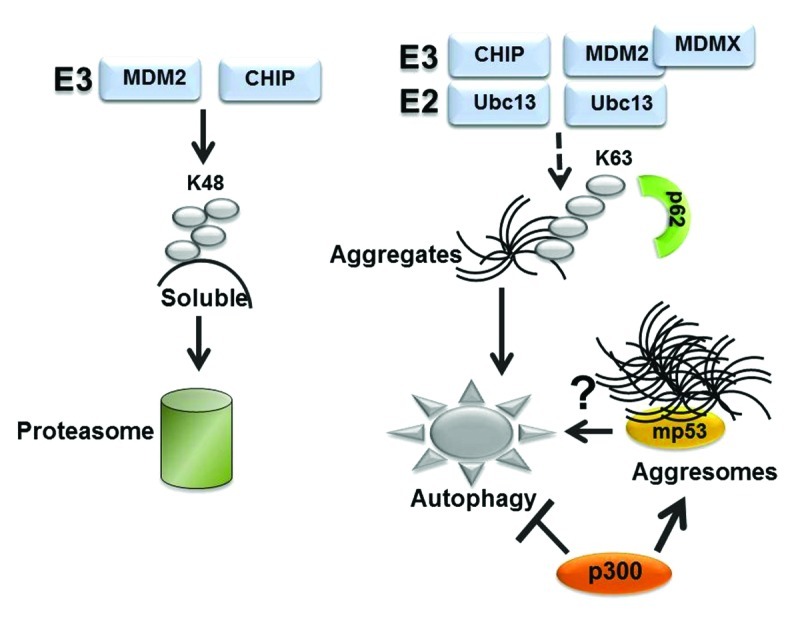
Figure 5. Hypothetical role played by the ubiquitin code in proteasomal or autophagic degradation (see also text for explanation). While substrates modified by K48-linked ubiquitin chains are recognized by the proteasome, K63 chain formation is involved in a variety of other functions, including in the autophagic clearance of micro- or macro-aggregates. CHIP can catalyze K63 ubiquitination when in combination with Ubc13. In the case of MDM2 K63 chain formation required MDMX.37 We speculate that K63-linked ubiquitin chains may play a role in autophagic disruption of mutant p53. Other studies have shown that mutant p53 localize in aggresomes, wherein misfolded proteins are either stored or cleared by autophagy.31 The observation that p300 inhibits autophagy44 and is necessary for aggresome formation43 raises the possibility that autophagic degradation of mutant p53 within aggresomes is regulated by the interaction with p300.
An equally important issue pertains to the role played by mutant p53 aggregation in autophagic degradation. It is amply documented that a variety of aggregated misfolded proteins are degraded via autophagy, and the evidence demonstrating the high tendency of various p53 mutations to aggregate in vitro and in vivo is, at this point, overwhelming.40,41 Recently, a direct correlation has been established between the ability of mutant p53 to form cytoplasmic aggregates, with tumor aggressiveness and the acquisition of oncogenic functions.41 This phenomenon has been attributed to mutant p53-mediated sequestration of various tumor suppressors, including wild-type p53, p63 and p73 in intracellular macro-aggregates called aggresomes. Aggresomes are large cytoplasmic inclusion bodies that act as disposal devices through which abnormally folded proteins that are not destroyed by other proteolytic pathways are secluded and eventually degraded via autophagy.32 Although the presence of mutant forms of p53 in these inclusion bodies is at first glance supportive of the idea that autophagy provides a key route for their degradation, the fate of mutant p53 within aggresomes needs to be further elucidated. For example, in the case of the aggregation-prone protein synphilin-1, while small and disperse cellular aggregates are susceptible to degradation by basal autophagy, when synphilin-1 is secluded into aggresomes, it can be protected from autophagic degradation unless autophagy is super-induced above basal levels by stress signals that in turn modify its ubiquitination code and its movement within these organelles.42 Importantly, we have recently shown that the acetyltransferase p300 is necessary for aggresome formation, and we found that the knockdown of p300 promotes p53 mutant degradation independently of the proteasome (ref. 43 and our unpublished observations). Since p300 can also inhibit autophagy,44 it will be important to determine whether the association of mutant forms of p53 with p300 within aggresomes interferes with their autophagic degradation.
Is There a Role for Autophagy within the Context of p53-Mutant Driven Oncogenesis?
Mice where the p53H172R alleles equivalent to the human hot spot p53H175R replace the endogenous wild-type gene display a heterogeneous pattern of p53 accumulation in normal tissues as well as in various tumors.4,12-14 These observations have suggested that p53 mutant accumulation arises as a result of secondary events that occur in a cell-, tissue- or tumor-specific manner. Therefore, a relevant question is whether inactivation of autophagy is one of such secondary event that contributes to p53-mutant stabilization during tumor evolution. Significantly, mono-allelic deletion of Beclin-1 is found with a frequency nearly similar to that annotated for mutations of the p53 gene, being detectable in 75% ovarian cancers, 50% breast cancers and 40% prostate cancers.20,21,45,46 Similarly, mutations or deletions or frame shift mutations of many autophagic genes have been reported in a variety of human tumors. Additionally, we and others have recently demonstrated that at least some types of p53 mutations inhibit the extent of autophagic flux.16,47 Together, these data suggest that loss of autophagy in p53 mutant tumors might occur indirectly (i.e., due to genomic alterations) or via mutant p53-dependent inhibition of autophagy. Again, opposite to the mutant proteins, wild-type p53 stimulates autophagy especially in response to DNA damage.48
Several studies have shown that loss of autophagy promotes genomic instability and aneuploidy while also providing an inflammatory microenvironment that promotes tumorigenesis.49 In keeping with the finding that autophagy stabilizes wild-type p53, as depicted in Figure 6, the available data provide for a speculative, yet plausible, scenario, whereby wild-type p53 and autophagy work together to preserve genomic stability, whereas autophagic inhibition could bring a double bonus in a p53 mutant setting, first by allowing stabilization, second by creating a genetically unstable environment that might cooperate with mutant p53 during oncogenesis. In fact, it is well recognized that p53 mutations lead to genomic instability to an extent much greater than that seen in p53-null cells, likely due to gain of oncogenic functions.50 In light of these considerations, it will be very important to cross mice expressing p53 mutations of the type found in human tumors, for example p53H172R mice, with mice where the dosage of autophagic genes is reduced. It is reasonable to expect that loss of autophagy will promote p53 mutant accumulation and accelerate tumor progression.
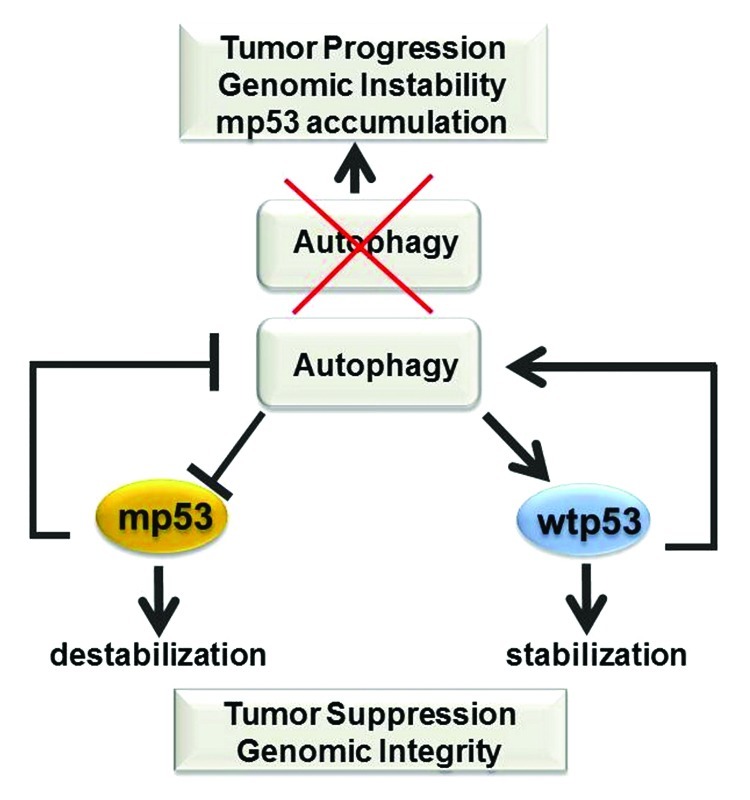
Figure 6. Significance of the cross-talk between wild-type and mutant p53 with autophagy. The figure depicts a summary of some of the available data highlighting a possible antagonistic feedback loop between mutant p53 and autophagy in contrast to cooperative effects described between wild-type p53 and autophagy, and their consequences on tumorigenesis (see also text for explanations).
In conclusion, the finding that autophagy regulates the stability of some mutant forms of p53 opens a variety of important mechanistic questions that could have key implications for both the understanding of the pathogenesis of p53 mutant tumors as well enlighten novel avenues for possible therapeutic interventions, for example aimed at activating autophagy in tumors harboring p53 mutations. However, given the complexity of the p53 mutational spectrum, it will be, at the same time, challenging to establish whether and how the type of mutation or the cellular context influence the cross-talk with autophagy.
Materials and Methods
Cells, antibodies and reagents
The cell lines employed in this study were obtained from the tissue culture core facility at LCCC. Cells were grown in Dulbecco' s modified Eagle's medium (DMEM, 25 mM glucose, with glutamine and pyruvate from Invitrogen) and supplemented with 10% fetal calf serum (FCS). The antibodies were as follows: p53 (FL393 Santa Cruz Biotechnology, Inc.); p53 Ab-1, Ab-3 and Ab-6 (Calbiochem); actin (I19; Santa Cruz Biotechnology, Inc.); LC3 (MBL #PM036; LC3 Novus Biol. #NB100-2220); ubiquitin (monoclonal mix made of P4D1, SCBT and 13-1600, Zymed); ATG-5 (Cell Signaling, #D169); ULK1 (Santa Cruz, SC-33182). The stable MDA-231 cell lines harboring the control, or Ulk1, or ATG5 shRNAs were generated by transfecting the plasmids harboring the specific constructs (see below) and were then selected with puromycin treatment.
Treatment conditions and immunoblots
Cell extracts were prepared in RIPA buffer supplemented with protease inhibitor cocktail (Roche) and 5 mM Betamercaptoethanol, 10 mM N-ethylmalemide (NEM) as well as phosphorylation inhibitors. For the assessment of p53 levels in MG132-treated cells, cells were plated at 30–50% confluency the day before treatment, and MG132 was added either overnight or for the indicated time points, at the concentrations specified in the legend of each figure. Cells were typically scraped in the culturing media, washed in PBS, re-suspended in 10 volumes (relatively to the packed pellet) of RIPA buffer and incubated on ice for 20 min after vortexing 2–3 times. Cell lysates were cleared by centrifugation at 4°C for 15 min. In some of these experiments, the pellets were re-extracted, and p53 levels were analyzed in both the first and second extraction. Pellets were typically completely extracted with RIPA bufer, but the DNA was never sheared. To enable p53 detection in a quantitative range, 0.5–2 μg of cell extracts were analyzed in immunoblot.
Sequence of siRNAs or shRNAs
The shRNA plasmids for ATG5 were purchased from Genecopia (#HSH022804-mU6), while Ulk1 was from Origene (#TF308491). Four shRNA per target were first screened based on their ability to downregulate the expression levels of proteins of interest. Typically a combination of two shRNA was used to achieve stable knockdown. The sequences of shRNA were as follows: for the ATG5: shRNA1: TCCTTGGAACATCACAGTA; shRNA2: CACTGTCCATCTAAGGATG. For Ulk1: 1: shRNA1: CTTCCAGGAAATGGCTAATTCTGTCTACC. The siRNA for Beclin-1 was from Ambion (#195717).
Acknowledgments
This work was supported by NIH-R01 CA102746 grants to M.L.A. and by Award Number P30CA051008 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24128
References
- 1.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–86. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer. 2009;9:831–41. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- 3.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 4.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, et al. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2011;10:1679–89. doi: 10.4161/cc.10.10.15642. [DOI] [PubMed] [Google Scholar]
- 6.Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget. 2011;2:654–6. doi: 10.18632/oncotarget.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quante T, Otto B, Brázdová M, Kejnovská I, Deppert W, Tolstonog GV. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Cell Cycle. 2012;11:3290–303. doi: 10.4161/cc.21646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valenti F, Fausti F, Biagioni F, Shay T, Fontemaggi G, Domany E, et al. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle. 2011;10:4330–40. doi: 10.4161/cc.10.24.18682. [DOI] [PubMed] [Google Scholar]
- 9.Haupt S, Haupt Y. Mutant p53 subverts PLK2 function in a novel, reinforced loop of corruption. Cell Cycle. 2012;11:217–8. doi: 10.4161/cc.11.2.18977. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Tarapore RS, Jarmel MH, Tetreault MP, Katz JP. p53 mutation alters the effect of the esophageal tumor suppressor KLF5 on keratinocyte proliferation. Cell Cycle. 2012;11:4033–9. doi: 10.4161/cc.22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Ebina M, Martínez A, Birrer MJ, Ilona Linnoila R. In situ detection of unexpected patterns of mutant p53 gene expression in non-small cell lung cancers. Oncogene. 2001;20:2579–86. doi: 10.1038/sj.onc.1204351. [DOI] [PubMed] [Google Scholar]
- 13.Ebina M, Steinberg SM, Mulshine JL, Linnoila RI. Relationship of p53 overexpression and up-regulation of proliferating cell nuclear antigen with the clinical course of non-small cell lung cancer. Cancer Res. 1994;54:2496–503. [PubMed] [Google Scholar]
- 14.Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol. 2011;223:116–26. doi: 10.1002/path.2784. [DOI] [PubMed] [Google Scholar]
- 15.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–50. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11:4436–46. doi: 10.4161/cc.22778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon SH, Prives C. Mutant p53 succumbs to starvation. Cell Cycle. 2013;12 doi: 10.4161/cc.23911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagory EL, Giaccia AJ. A low-carb diet kills tumor cells with a mutant p53 tumor suppressor gene: The Atkins diet suppresses tumor growth. Cell Cycle. 2013;12 doi: 10.4161/cc.23948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu EY, Ryan KM. Autophagy and cancer--issues we need to digest. J Cell Sci. 2012;125:2349–58. doi: 10.1242/jcs.093708. [DOI] [PubMed] [Google Scholar]
- 22.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 23.Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol. 2010;12:781–90. doi: 10.1038/ncb2082. [DOI] [PubMed] [Google Scholar]
- 24.Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007;17:520–30. doi: 10.1038/cr.2007.47. [DOI] [PubMed] [Google Scholar]
- 25.Yan P, Qing G, Qu Z, Wu CC, Rabson A, Xiao G. Targeting autophagic regulation of NFkappaB in HTLV-I transformed cells by geldanamycin: implications for therapeutic interventions. Autophagy. 2007;3:600–3. doi: 10.4161/auto.4761. [DOI] [PubMed] [Google Scholar]
- 26.Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120:3555–62. doi: 10.1182/blood-2012-01-402578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116:2324–31. doi: 10.1182/blood-2010-01-261040. [DOI] [PubMed] [Google Scholar]
- 28.Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2012;13:733–40. doi: 10.1038/embor.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li CF, Chen LT. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy. 2013;9:220–33. doi: 10.4161/auto.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sariyer IK, Merabova N, Patel PK, Knezevic T, Rosati A, Turco MC, et al. Bag3-induced autophagy is associated with degradation of JCV oncoprotein, T-Ag. PLoS ONE. 2012;7:e45000. doi: 10.1371/journal.pone.0045000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 2010;1:779–86. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–53. doi: 10.1016/S0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 34.Laine A, Topisirovic I, Zhai D, Reed JC, Borden KL, Ronai Z. Regulation of p53 localization and activity by Ubc13. Mol Cell Biol. 2006;26:8901–13. doi: 10.1128/MCB.01156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9:577–88. doi: 10.1158/1541-7786.MCR-10-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284–95. doi: 10.1128/MCB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C, et al. Chaperoned ubiquitylation--crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell. 2005;20:525–38. doi: 10.1016/j.molcel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 38.David Y, Ternette N, Edelmann MJ, Ziv T, Gayer B, Sertchook R, et al. E3 ligases determine ubiquitination site and conjugate type by enforcing specificity on E2 enzymes. J Biol Chem. 2011;286:44104–15. doi: 10.1074/jbc.M111.234559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sehat B, Andersson S, Girnita L, Larsson O. Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis. Cancer Res. 2008;68:5669–77. doi: 10.1158/0008-5472.CAN-07-6364. [DOI] [PubMed] [Google Scholar]
- 40.Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc Natl Acad Sci USA. 2012;109:13584–9. doi: 10.1073/pnas.1211550109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–95. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 42.Wong E, Bejarano E, Rakshit M, Lee K, Hanson HH, Zaarur N, et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nat Commun. 2012;3:1240. doi: 10.1038/ncomms2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirilyuk A, Shimoji M, Catania J, Sahu G, Pattabiraman N, Giordano A, et al. An intrinsically disordered region of the acetyltransferase p300 with similarity to prion-like domains plays a role in aggregation. PLoS ONE. 2012;7:e48243. doi: 10.1371/journal.pone.0048243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284:6322–8. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell SE, Hickey GI, Lowry WS, White P, Atkinson RJ. Allele loss from chromosome 17 in ovarian cancer. Oncogene. 1990;5:1581–3. [PubMed] [Google Scholar]
- 46.Futreal PA, Söderkvist P, Marks JR, Iglehart JD, Cochran C, Barrett JC, et al. Detection of frequent allelic loss on proximal chromosome 17q in sporadic breast carcinoma using microsatellite length polymorphisms. Cancer Res. 1992;52:2624–7. [PubMed] [Google Scholar]
- 47.Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7:3056–61. doi: 10.4161/cc.7.19.6751. [DOI] [PubMed] [Google Scholar]
- 48.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 49.White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212–7. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem. 2012;113:433–9. doi: 10.1002/jcb.23400. [DOI] [PMC free article] [PubMed] [Google Scholar]