

Abstract
FBH1 is a member of the UvrD family of DNA helicases and plays a crucial role in the response to DNA replication stress. In particular, upon DNA replication stress, FBH1 promotes double-strand breakage and activation of the DNA-PK and ATM signaling cascades in a helicase-dependent manner. In the present manuscript, we show that FBH1 is often deleted or mutated in melanoma cells, which results in their increased survival in response to replicative stress. Accordingly, FBH1 depletion promotes UV-mediated transformation of human melanocytes. Thus, FBH1 inactivation appears to contribute to oncogenic transformation by allowing survival of cells undergoing replicative stress due to external factors such as UV irradiation.
Keywords: FBH1, F-box protein, helicase, DNA replication stress, melanoma
Introduction
DNA replication stress is one of the most common and dangerous forms of genotoxic stress, which, if not quickly resolved, leads to double-strand breaks (DSBs) and apoptosis to prevent the propagation of DNA lesions. FBH1 is a member of the UvrD familiy of DNA helicases, which in mammalian cells has been shown to be recruited to the DNA replication stress-induced single-stranded DNA (ssDNA) to promote further ssDNA generation.1,2 Significantly, FBH1 helicase activity is necessary for the efficient induction of DSBs and the DSB signaling cascade in response to DNA replication stress.1 FBH1 silencing in the presence of DNA replication stress results in a reduction in the activating phosphorylation events in ATM and DNA-PK (on Ser1981 and Ser2056, respectively).1 As a consequence, ATM and DNA-PK substrates, such as RPA2 (on Ser4 and Ser8), p53 (on Ser15) and H2AX (on Ser139), are also less phosphorylated. In contrast, the ATR-CHK1 cascade is preserved after FBH1 depletion, and downstream signaling in this pathway (e.g., the phosphorylation of RPA2 on Ser33) is not affected.1 Importantly, FBH1 confers sensitivity to catastrophic DNA replication stress, and FBH1 silencing decreases apoptosis in cell treated with hydroxyurea (HU) or gemcitabine.1 Moreover, in response to UV irradiation of cells synchronized in S phase, FBH1 depletion attenuates the DSB signaling cascade and promotes cell survival.1
Results
Since increased survival is a characteristic of cancer cells, we considered the possibility that abnormal FBH1 levels or activity may contribute to oncogenic transformation. The GSK cancer cell line genome profiling database shows that the FBH1 locus is deleted in melanoma and lung cancer cell lines more often than in other types of cell lines (Fig. S1A). Therefore, we used high-density, single nucleotide polymorphism (HD-SNP) genotyping arrays in a separate cohort of 19 melanoma cell lines3 and found FBH1 deletions (including a focal deletion) in 55% of cases (Fig. 1A). Accordingly, gene copy analyses in the NCBI-GEO database4 show hemizygous or homozygous deletions of the locus containing FBH1 in 32% of melanoma cell lines (n = 25), 63% of malignant melanoma cell lines derived from metastatic tumors (n = 60) and 36% of primary melanomas (n = 51) (Supplementary Information, Fig. S1B–E). We also performed an immunoblot analysis of the NCI-60 cell line panel,5 and found that four of the nine melanoma cell lines display low FBH1 protein levels (Fig. 1B). Interestingly, the COSMIC database reports a monoallelic point mutation in FBH1 in the MZ7-MEL melanoma cell line, which we confirmed by sequencing cDNA from these cells (Fig. S2A). Strikingly, this naturally occurring Asp698Asn mutation is the same mutation that we and others previously generated to inactivate FBH1 helicase activity,1,2 suggesting loss of helicase activity in this tumor. Similarly, mutations in the helicase domain (Pro793Ser and Pro844Ser) have been identified in primary melanomas.6

Figure 1. Melanoma cells expressing either low levels of FBH1 or mutated FBH1 display an attenuated DSB signaling response to hydroxyurea. (A) Affymetrix genome-wide human HD-SNP 6.0 array copy number data of the FBH1 locus in 19 melanoma cell lines and in two primary neonatal melanomas is shown. The data are also accessible at the NCBI-GEO (www.ncbi.nlm.nih.gov/geo/) database,4 accession #GSE22305.3 (B) The NCI-60 cell line panel was obtained from the Developmental Therapeutics Program of the National Cancer Institute (http://dtp.cancer.gov/branches/btb/characterizationNCI60.html).5 Whole-cell extracts were subjected to immunoblotting for the indicated proteins. Four melanoma cell lines (SK-MEL-28, SK-MEL-5, UACC-257 and UACC-62), out of the nine in this panel, display low FBH1 protein levels. (C) The indicated melanoma cells were treated with hydroxyurea (HU) for 24 h (time 0) and released for 2 h into fresh medium. After harvesting, cells were fractionated into soluble and chromatin fractions, and lysates were immunoblotted for the indicated proteins. (D) The indicated melanoma cells were treated with hydroxyurea for 72 h, released, cultured for an additional 15–20 d and stained with crystal violet. Images show representative examples.
We then selected five melanoma cell lines [two with high levels of FBH1 (SK-MEL2 and SK-MEL 147), two carrying only one FBH1 allele and expressing low FBH1 levels (SK-MEL28 and 501-MEL) and the cell line with the Asp698Asn mutation (MZ7-MEL)] and investigated their response to treatment with HU. Significantly, compared with cells expressing high levels of FBH1, MZ7-MEL cells and cells with low FBH1 levels showed impaired activation of ATM and DNA-PK, with less p-RPA2(Ser4/Ser8), γ-H2AX (i.e., H2AX phosphorylated on Ser139) and p-p53(Ser15) (Fig. 1C; Fig. S2B and data not shown). Finally, cell lines with low levels of FBH1 or inactivated FBH1 were less sensitive to HU than cells with high FBH1 expression in a clonogenic survival assay (Fig. 1D).
We then depleted FBH1 in SK-MEL2 cells (which express high FBH1 levels) and found that the DSB signaling response (as determined by analyzing p-ATM, p-DNA-PK, γ-H2AX and p-p53) was strongly attenuated (Fig. 2A). Accordingly, when FBH1 expression was forced in SK-MEL28 cells (which display low FBH1 levels), the DSB signaling response was much more robust than in SK-MEL28 cells infected with an empty virus (Fig. 2B)

Figure 2. Modulation of FBH1 expression changes the DSB signaling response in melanoma cells. (A) SK-MEL2 cells infected with lentiviruses encoding shRNAs targeting either LacZ or FBH1 mRNA were treated with hydroxyurea (HU) for 24 h. After harvesting, cells were fractionated into soluble and chromatin fractions, and the latter were immunoblotted for the indicated proteins. (B) SK-MEL28 cells stably infected with either an empty virus (MSCV) or a virus expressing FBH1 were treated with HU for the indicated times. After harvesting, cells were fractionated into soluble and chromatin fractions, and lysates were immunoblotted as indicated.
Using UV irradiation to induce transformation7 and anchorage-independent growth as readout for the transformed phenotype, we observed that depletion of FBH1 in HMEL468 immortalized primary human melanocytes8 resulted in a dramatic increase in soft agar colony formation (Fig. 3). In addition to the increased number of colonies, FBH1-depleted cells formed considerably larger colonies than control cells (Fig. 3). Thus, FBH1 protects melanocytes from UV-induced transformation, as indicated by the ability of the surviving cells to grow in an anchorage-independent manner.
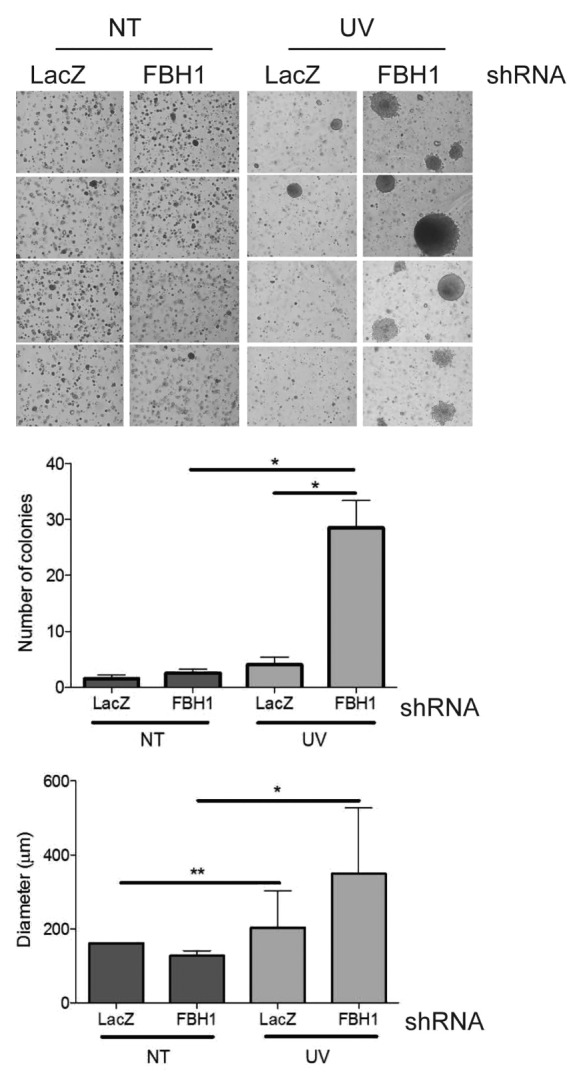
Figure 3. FBH1-depleted immortalized melanocytes are prone to transformation. HMEL468 immortalized, primary human melanocytes infected with lentiviruses encoding shRNAs targeting either LacZ or FBH1 mRNA were assayed for effects on anchorage-independent growth in soft agar. The images on the left show representative examples. The graph in the middle shows the number of colonies in three independent experiments (+/− SD). The graph on the right shows the diameter of colonies in three independent experiments (+/− SD). Single asterisk denotes significant difference (p < 0.05), and double asterisk denotes (p < 0.01) calculated by Student’s t-test.
Discussion
Replication stress occurs nearly every time the cell replicates its DNA, and this form of endogenous DNA damage can be exacerbated by external factors, such as UV irradiation or oncogene expression.9 Fork stalling is consequently among the most common and dangerous forms of genotoxic stress, and failure to resolve this lesion may result in genome instability. Therefore, either the DNA lesion is repaired or apoptosis is induced to eliminate cells with damaged DNA. The molecular mechanisms that induce cell death due to DNA replication stress are not well understood, but it is clear that DSB formation plays a crucial step in the process. We have demonstrated a role for FBH1 in the generation of DSBs and the induction of cell death following DNA replication stress.1
The GSK cancer cell line genome profiling database shows that the FBH1 locus is deleted in both melanoma and lung cancer cell lines more often than in cell lines originating from different tissues (Fig. S1A). Interestingly, skin and lung are the organs of first contact for most environmental agents (such as UV irradiation and smoke) that may affect DNA replication. We found that multiple melanoma cell lines lack proper FBH1 function, and the FBH1 status of melanoma cell lines inversely correlates with their resistance to DNA replication stress (Fig. 1B–D; Fig. S2). We also observed that FBH1 depletion enhances UV-induced transformation of immortalized melanocytes (Fig. 3). UV irradiation induces DNA photoproducts, mostly pyrimidine dimers (i.e., adjacent thymidines and cytosines forming covalent linkages). The bulge created by these pyrimidine dimers is a physical block to DNA replication. Therefore, these lesions, if not repaired, induce fork stalling and replication stress during S phase. The induction of apoptosis following blockage of DNA replication by pyrimidine dimers requires the formation of DSBs.10 Similar to the role of FBH1 in the response to HU-mediated replication stress, FBH1 plays a crucial role in the efficient activation of the DSB signaling cascade and the induction of apoptosis in response to UV-induced DNA replication stress.1 As UV irradiation plays a significant role in melanomagenesis, the elucidation of the molecular mechanisms behind the response to UV irradiation is of paramount clinic relevance. Our studies suggest that FBH1 inactivation may contribute to UV-induced oncogenic transformation by allowing survival of melanocytes that did not properly resolve lesions generated by pyrimidine dimers posing a threat to genome stability.
Our results have further clinical relevance since they suggest that the assessment of FBH1 levels and/or functional status could aid in the identification of melanoma patients that would benefit from treatment with HU or other drugs11 with a similar mechanism of action.
Materials and Methods
Cell lines and cell treatments
HEK-293T cells were maintained in DMEM supplemented with 10% bovine serum (Invitrogen), and U2OS cells were maintained in DMEM supplemented with 10% fetal bovine serum (Invitrogen). HMEL468 cells are PMEL/hTERT/CDK4(R24C)/p53DD/BRAFV600E immortalized primary human melanocytes and were cultured as described.8 Hydroxyurea were from Sigma and used at concentrations of 2 mM. UV was irradiated using XL-1000 UV crosslinker (Spectronics Corporation).
Biochemical methods
Extract preparation and immunoblotting have previously been described.12 Whole-cell lysates were generated by lysing 1 volume of cells with three volumes of SDS lysis buffer [50 mM TRIS pH 6.8, 2%SDS, 10% glycerol, 1 mM Na3VO4, 1 mM NaF and protease inhibitor mix (Roche)]. For chromatin fractionation, cells were extracted with CSK buffer13 for 5 min on ice and centrifuged for 3 min at 1,300 g. The insoluble pellets were digested with Turbo nuclease to generate the chromatin fraction.
Plasmids, siRNA and shRNA
FBH1 mutants were generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene). Both wild type and mutants were subcloned into the pMSCV retroviral vector. All cDNAs were completely sequenced. ON-Target siRNAs to FBH1 were purchased from Dharmacon. Generation of lentivirus encoding shRNAs targeting human FBH1 was performed as previously described.14 The target sequence used to knockdown human FBH1 is 5′-GCAATAGGATTCACTACAA-3′.
Antibodies
Mouse monoclonal antibodies were from Calbiochem (RPA2), Sigma (anti-FLAG M2), Abcam (phospho-S2056 DNA-PK), Cell Signaling Technology (phospho-S1981 ATM) and Millipore (γ-H2AX). Rabbit polyclonal antibodies were from Invitrogen (CUL1, SKP1), and Bethyl Laboratories [phospho-RPA2 (S4S8) and phopho-RPA2 (S33)]. The generation of an anti-FBH1 antibody was previously described.1
Transient transfections and retrovirus-mediated gene transfer
For retrovirus production, GP-293 (Clontech) packaging cells were transfected with FuGENE-6 reagent according to the manufacturer’s instructions. Forty-eight hours after transfection, the virus-containing medium was collected and supplemented with 8 mg/ml polybrene (Sigma). Cells were infected by replacing the cell culture medium with the viral supernatant for 6 h. siRNA duplexes were transfected into subconfluent U2OS cells using HiPerfect reagent (QIAGEN) according to the manufacturer’s instructions.
Clonogenic assay
Cells were transfected with siRNAs to FBH1 or LacZ, and after 48 h, they were, replated and incubated with HU for 48 h. Cells were then released into fresh media for another 7–10 d to allow colony formation. The colonies were washed with PBS and stained with Crystal Violet (Sigma).
Growth in soft agar
Anchorage-independent growth was assayed by the ability of cells to grow in soft agar. Bottom agar (0.7% agarose) was prepared in medium containing serum in a 6-cm tissue culture dish. HMEL468 cells (0.2 × 106; untreated or treated with 10 J/m2 UVC) were suspended in medium containing serum and 0.35% agar and plated on top of the bottom layer. Cells were incubated at 37°C and colonies quantified after 3 weeks. Photographs were taken with a phase-contrast microscope (Zeiss).
Mutation analysis
Genomic DNA from the MZ7 cell line was prepared as previously described.15 Exons containing potential mutations in the FBH1 gene were amplified using PCR and sequenced. The reference sequence of FBH1 was obtained from the UCSC Human Genome database (mRNA accession NM_032807.3). PCR primers, located ≥ 50 bp upstream or downstream of the target exon boundaries, were designed in the Primer 3 program (http://frodo.wi.mit.edu/primer3/).
Copy number determination by GEO database data analysis
The CEL (SNP) and TXT (CGH) files used for this study were obtained from the National Center for Biotechnology Information, Gene Expression Omnibus (www.ncbi.nlm.nih.gov/projects/geo). Raw data were processed, analyzed and visualized using Genespring GX 11.5. software (SNP; Agilent) or Agilent Genomic Workbench (CGH; Agilent).
Supplementary Material
Acknowledgments
The authors thank M. Aladjem, G. Draetta, T. Heffernan, J. Marszalek and D. Rucando for reagents; J. Borowiec, V. Costanzo, D. Durocher, M. Foiani, H. Klein, J. Lukas and J.R. Skaar for critical reading of the manuscript and the Developmental Therapeutics Program of the National Cancer Institute for providing the NCI-60 cell line panel. M.P. and Y.T.J. are grateful to T.M. Thor and S.O. Hong, respectively, for continuous support. This work was funded by grants from the National Institutes of Health (R01-GM057587, R37-CA076584 and R21-CA161108) to M.P., a fellowship from the Korean National Research Foundation (number KRF-2007-357-C00082) to Y.T.J. and a Ministerio de Educacion y Ciencia fellowship and a National Cancer Center fellowship to M.V.G. M.P. is an Investigator with the Howard Hughes Medical Institute.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24165
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24165
References
- 1.Jeong YT, Rossi M, Cermak L, Saraf A, Florens L, Washburn MP, et al. FBH1 promotes DNA double-strand breakage and apoptosis in response to DNA replication stress. J Cell Biol. 2013;200:141–9. doi: 10.1083/jcb.201209002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fugger K, Mistrik M, Danielsen JR, Dinant C, Falck J, Bartek J, et al. Human Fbh1 helicase contributes to genome maintenance via pro- and anti-recombinase activities. J Cell Biol. 2009;186:655–63. doi: 10.1083/jcb.200812138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rose AE, Poliseno L, Wang J, Clark M, Pearlman A, Wang G, et al. Integrative genomics identifies molecular alterations that challenge the linear model of melanoma progression. Cancer Res. 2011;71:2561–71. doi: 10.1158/0008-5472.CAN-10-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, et al. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 6.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutherland BM, Cimino JS, Delihas N, Shih AG, Oliver RP. Ultraviolet light-induced transformation of human cells to anchorage-independent growth. Cancer Res. 1980;40:1934–9. [PubMed] [Google Scholar]
- 8.Scott KL, Nogueira C, Heffernan TP, van Doorn R, Dhakal S, Hanna JA, et al. Proinvasion metastasis drivers in early-stage melanoma are oncogenes. Cancer Cell. 2011;20:92–103. doi: 10.1016/j.ccr.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batista LF, Kaina B, Meneghini R, Menck CF. How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis. Mutat Res. 2009;681:197–208. doi: 10.1016/j.mrrev.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–31. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 12.D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–42. doi: 10.1038/nature09140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–34. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, et al. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–4. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 15.Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012;481:90–3. doi: 10.1038/nature10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.