

Abstract
Hemorrhagic shock (HS) promotes the development of systemic inflammatory response syndrome (SIRS) and organ injury by activating and priming the innate immune system for an exaggerated inflammatory response through, as of yet, unclear mechanisms. IL-1β also plays an important role in the development of post-HS SIRS and active IL-1β production is tightly controlled by the inflammasome. Pyrin, a protein of 781 amino-acids with pyrin domain (PYD) at the N-terminal, negatively regulates inflammasome activation through interaction with nucleotide-binding oligomerization domain-like receptor protein (NLRP). Expression of pyrin can be induced by LPS and cytokines, and IL-10 is a known potent inducer of pyrin expression in macrophages. In the present study, we tested the hypothesis that HS downregulates IL-10, and therefore decreases pyrin expression to promote inflammasome activation and subsequent IL-1β processing and secretion in the lungs. Our results show that LPS, while activating Nlrp3 inflammasome in the lungs, also induced pyrin expression, which in turn suppressed inflammasome activation. More importantly, LPS-mediated upregulation of IL-10 enhanced pyrin expression, which serves, particularly in later phases, as a potent negative feedback mechanism regulating inflammasome activation. However, HS-mediated suppression of IL-10 expression in alveolar macrophages (AM) attenuated the upregulation of pyrin in AM and lung endothelial cells, and thereby significantly enhanced inflammasome activation and IL-1β secretion in the lungs. This study demonstrates a novel mechanism by which HS suppresses negative feedback regulation of Nlrp3 inflammasome to enhance IL-1β secretion in response to subsequent LPS challenge, and so primes for inflammation.
Keywords: IL-1β, IL-10, alveolar macrophages, lung vascular endothelial cells
Introduction
Hemorrhagic shock (HS), as a result of major trauma and surgical operation, promotes the development of systemic inflammatory response syndrome (SIRS) and organ injury by activating and priming the innate immune system for an exaggerated inflammatory response through, as of yet, unclear mechanisms. Interleukin-1β (IL-1β) is one of the key pro-inflammatory mediators of inflammation and studies have shown that HS induces IL-1β expression and secretion, which in turn promotes the development of SIRS (1–7). IL–1β not only causes inflammation itself, but more importantly also induces the expression of many other pro-inflammatory cytokines and adhesion molecules, which further exaggerate inflammation (8).
The production of active IL-1β is tightly controlled by the formation and activation of the inflammasome, which is comprised of NOD-like receptors (NLRs), caspase-1 and apoptosis-associated speck-like protein containing a CARD domain (ASC) (9–11). IL-1β is synthesized initially as an inactive precursor molecule (pro- IL-1β p35), which must be cleaved by caspase-1 at amino acid position 116 to produce the actively mature IL-1β (p17) that is then secreted in response to stimulating signals. Caspase-1 is also synthesized as an inactive 45-kDa protein (procaspase-1) that undergoes autocatalytic processing after assembly of the inflammasome in response to an appropriate stimulus (12). However, it is not clear how HS augments inflammasome activation and IL-1β processing in the lungs.
Pyrin, a protein of 781 amino-acids, is an important regulator of the inflammasome (13). Pyrin was primarily recognized as a protein that is encoded by the gene responsible for Familial Mediterranean Fever (FMF), a recessively inherited systemic auto-inflammatory disease (14, 15). The N-terminal of pyrin constitutes a protein domain named pyrin domain (PYD), a member of the death effector-fold domain (16, 17). The PYD found in pyrin is also present in nucleotide-binding oligomerization domain-like receptor protein (NLRP; the nomenclature used in mouse is Nlrp). The NLRP proteins are involved in the formation of the inflammasome that triggers activation of caspase-1 followed by processing and release of active IL-1β (18, 19). It has been reported that pyrin may serve as a negative regulator for caspase-1 activity and therefore inhibit pro-IL-1β processing (13, 20, 21). Pyrin is expressed in neutrophils, monocytes and macrophages, and its expression is tightly regulated by cytokines (22), with IL-10 reported to induce pyrin expression in macrophages (20).
The immunosuppressive cytokine IL-10 has been shown to inhibit activation of cells of the monocyte/macrophage lineage, causing impaired LPS-induced release of various cytokines and chemokines (23, 24). In the in vivo setting, IL-10 administration in models of ischemiareperfusion has been shown to abrogate organ injury, whereas neutralization or deficiency of IL-10 generally aggravates the inflammatory response (25–27). Studies investigating bronchoalveolar lavage (BAL) levels of IL-10 in patients with ARDS showed that although increased IL-10 levels in the lavage fluid were frequently observed (~90% of these patients), the median IL-10 levels in non-survivors were significantly lower than that detected in surviving patients (28). We have previously demonstrated, by using in vivo HS mouse model and ex vivo LPS challenge of alveolar macrophage (AM), that HS suppresses LPS-induced IL-10 expression in AM through inhibiting IL-10 gene transcription, and this effect of HS correlates with augmented lung inflammation (29). Together, these data show that IL-10 is an important contributor to the anti-inflammatory response of the lung during injury, and impaired expression of IL-10 may augment the overall magnitude of inflammation.
In the present study, we tested the hypothesis that HS may act through downregulating IL-10, and therefore decreasing pyrin expression to promote inflammasome activation and resultant IL-1β processing and secretion in the lungs. Our results show that LPS, while activating Nlrp3 inflammasome in the mouse lungs, also induced pyrin expression, which in turn suppressed inflammasome activation. More importantly, LPS-mediated upregulation of IL-10 enhanced pyrin expression, which serves as a potent negative feedback mechanism regulating inflammasome activation at later time points. However, HS-mediated suppression of IL-10 expression in AM attenuated LPS-induced upregulation of pyrin in AM and lung endothelial cells, and thereby significantly enhanced inflammasome activation and IL-1β secretion in the lungs. In aggregate, these findings suggest a novel mechanism by which HS suppresses negative regulation of Nlrp3 inflammasome and enhances IL-1β secretion in the lungs with subsequent LPS challenge, and therefore primes for inflammation.
Materials and Methods
Materials
Recombinant IL-10, LEAF™ purified anti-mouse IL-10 neutralizing antibody, and mouse IL-10 ELISA MAX™ Deluxe kit were purchased from BioLegend (San Diego, CA). Nonimmune rabbit IgG (item I5006) was purchased from Sigma-Aldrich. Polyclonal anti-Pyrin antibody for Western blotting was purchased from Santa Cruz (Santa Cruz, CA). All other chemicals were obtained from Sigma-Aldrich, except where noted.
Mouse model of Hemorrhagic shock and resuscitation
Male C57BL/6 WT mice were purchased from the Jackson Laboratory (Bar Harbor, ME). TLR4 knockout (TLR4−/−) mice and MyD88 knockout (MyD88−/−) mice were bred in Dr. Billiar's lab at the University of Pittsburgh; Nlrp3 knockout (Nlrp3−/−) mice were obtained from Millennium Pharmaceuticals (Cambridge, MA) and bred in Dr. Billiar's lab; all mice used are on a C57BL/6 background. All experimental protocols involving animals were approved by Institutional Animal Care and Use Committee of VA Pittsburgh Healthcare System and University of Pittsburgh. Mice were 12–14 weeks of age at the time of experiments and were maintained on standard rodent chow and water ad libitum. The mice were not fasted before experiments. Animals were anesthetized with 50 mg/kg of ketamine and 5 mg/kg of xylazine via intraperitoneal (i.p.) administration. Femoral arteries were cannulated for monitoring of mean arterial pressure (MAP), blood withdrawal and resuscitation. HS was initiated by blood withdrawal and reduction of the MAP to 40 mmHg within 20 min. Blood was collected into a 1 ml syringe and heparinized to prevent clotting. In order to exclude the effect of heparin on immune processes, equal amounts of heparin (10 U) were injected into sham animals through the cannulated femoral artery during the sham operation. After a hypotensive period of 1 h, animals were resuscitated by transfusion of the shed blood and Ringer's Lactate (RL) in a volume equal to that of shed blood, over a period of 20 min. The catheters were then removed, the femoral artery was ligated, and the incisions were closed. Sham animals underwent the same surgical procedures without hemorrhage and resuscitation. At 2 h after resuscitation, LPS in a dose of 100 μg/kg B.W. was injected intratracheally (i.t.) into the mice (HS-LPS model). The animals remained anesthetized throughout the entire experimental period under the influence by ketamine and xylazine. At various time points after resuscitation (0 to 10 h), either bronchoalveolar lavage (BAL) was performed and BAL fluid was collected or lung tissue was harvested for experimental analysis.
AMϕ isolation
BAL was performed as previously described (30). Normally the BAL fluid contains ~91% of AMϕ, and ~9% of other cells including PMN, lymphocytes, and erythrocytes. The immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) was used to isolate AMϕ from BAL fluid. Magnetic nanoparticle-conjugated antibodies (anti-mouse Gr-1, anti-CD4, anti-CD8, and anti-CD45R/B220 antibodies; BD Biosciences Pharmingen, San Diego, CA) were chosen to label and remove PMN and lymphocytes. The resulting cells consisted of >98% macrophages, and cell viability was >95%.
Mouse lung vascular endothelial cell (MLVEC) isolation and characterization
MLVEC were isolated using a previously described method (31) that was modified in our laboratory as follows. Briefly, mice were anesthetized with 50 mg/kg of ketamine and 5 mg/kg of xylazine i.p. The chest cavity was opened and the right ventricle was cannulated. PBS was infused to remove blood from lungs. Peripheral lung tissue was cut into approximately 1 mm3 dices and prepared and cultured in a 60-mm culture dish in growth medium (MEM D-Val medium containing 2 mM glutamine, 10% FBS, 5% human serum, 50 μg/ml penicillin/streptomycin, 5 μg/ml heparin, 1 μg/ml hydrocortisone, 80 μg/ml endothelial cell growth supplement from bovine brain, 5 μg/ml amphotericin, and 5 μg/ml mycoplasma removal agent) at 37°C with 5% CO2 for 60 h. The adherent cells were continued in culture for 3 days after removal of the tissue dices, followed by purification using biotin-conjugated rat anti-mouse CD31 (PECAM-1) monoclonal antibody and BD IMag streptavidin particles plus-DM, and the immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) following the manufacturer's instructions. The cells were allowed to grow for 3 to 4 days after purification. The cells were characterized by their cobblestone morphology, uptake of Dil-Ac-LDL (Biomedical Technologies Inc., Stoughton, MA), and staining for factor VIII-related antigen (Sigma Chemical Co., St. Louis, MO). MLVEC passaged between 3 and 5 times were used in experiments.
AM-MLVEC co-incubation
AM-MLVEC co-incubation was performed using Transwell™ plates (Corning Incorporated Life Sciences, Acton, MA). AM were collected from BAL fluid from HS or sham mice, treated with LPS for 2 h, and then transferred into the top well of Transwell in a concentration of 5×105 cells/well. MLVEC in the bottom well were prepared as described above. The co-cultures were then incubated for up to 8 h in DMEM containing 10% FCS.
Coimmunoprecipitation and immunoblotting analysis
Mouse lung tissue or MLVEC were homogenized or lysed (~1 × 106 cells/ml) in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 20 mM PMSF). The supernatants were quantified, and 600 μg of total protein for each sample was then immunoprecipitated with anti-ASC antibody (Santa Cruz Biotechnologies, CA). The immunoprecipitated proteins were separated on a 10% SDS-PAGE gel, and were then electroblotted onto PVDF membrane and blocked for 1 h at room temperature with Tris-buffered saline containing 3% non-fat dried milk. Pyrin and Nlrp3 proteins were detected by probing the membranes with anti-pyrin and anti-Nlrp3 antibodies (Santa Cruz Biotechnologies, CA) at 1:500 dilution, respectively, and detected with Clean-Blot IP Detection Reagent (Thermo Scientific, Rockford, IL) following the manufacturer's instructions. Blots were then stripped and reprobed with anti-ASC antibody and again detected with Clean-Blot IP Detection Reagent. Caspase-1 cleavage in the lung tissue and MLVEC was measured by detecting its p10 fragment in Western blot using rabbit polyclonal anti-mouse caspase-1 p10 (Santa Cruz Biotechnologies, CA).
Measurement of IL-1β, pro-IL-1β, and IL-10
IL-1β, Pro-IL-1β, and IL-10 in BAL fluid, lung tissue, cell supernatant, and cell culture media were measured using ELISA Ready-Set-Go kit for mouse IL-1β (eBioscience, San Diego, CA), Mouse IL-1β Pro-form ELISA Ready-Set-Go kit (eBioscience, San Diego, CA), and ELISA kit for mouse IL-10 (BioLegend, San Diego, CA), respectively, following the manufacturers' instructions.
RNA extraction and quantitative real-time PCR
Total RNA was isolated from lung tissue, AM and MLVEC by TRI-REAGENT (Molecular Research Center, Cincinnati, OH) following manufacture's instruction. Real time RT-PCR was done using a single step real time RT-PCR kit (Qiagen, Valencia, CA) in a Bio-Rad iQ5 real time PCR machine (Bio-Rad Laboratories, Hercules, CA) using SYBR Green detection protocol. The following gene specific primers were used for amplifying genes: IL-1β forward GCAACTGTTCCTGAACTCAACT, reverse ATCTTTTGGGGTCCGTCAACT; GAPDH forward TGACCACAGTCCATGCCATC, reverse GACGGACACATTGGGGGTAG. Reverse transcription was performed for 30 minutes at 50°C then reverse transcriptase was inactivated at 95°C for 15 min. Amplification was performed with cycling conditions of 94°C for 15 sec, 57°C for 30 sec, and 72°C for 30 sec for 35 cycles. After amplification protocol was over, PCR product was subjected to melt curve analysis using Bio-Rad iQ5 software. Fold change was calculated using the ΔΔCT method (32) and the value for the GAPDH gene, which was normalized to untreated mouse lung tissue, AM or MLVEC.
Transfection of siRNA in MLVEC
Pyrin siRNA, control siRNA, and transfection kit were purchased from Santa Cruz Biotechnologies. MLVEC (2 × 105 cells) were seeded in a six well tissue culture plate, and incubated at 37°C in a CO2 incubator until the cells were 80% confluent. The cells were then transfected with Pyrin siRNA or control siRNA using the siRNA transfection kit following the manufacturer's instructions. At 24 – 72 h after the transfection, pyrin protein expression in the transfected cells was analyzed by Western blot. Since we observed a confirmed knockdown of pyrin in the MLVEC at 48 h after siRNA transfection, we set this time point as time 0 for the experiments using LPS and or IL-10 treatment.
Data presentation and statistical analysis
The data are presented as mean ± SEM of the indicated number of experiments. Statistical significance among group means was assessed by ANOVA. Student Neuman- Keuls post-hoc test was performed. Differences were considered significant at p<0.05.
Results
HS augments Nlrp3 inflammasome activation in response to LPS in the lung
To determine whether enhanced activation of inflammasome might contribute to shock-enhanced IL-1β processing, we examined inflammasome activation in the lung by detecting the association of Nlrp3 and ASC, as well as caspase-1 cleavage in animals treated with LPS with or without prior HS. Animals were subjected to HS or sham operation, and then given LPS or saline vehicle (SAL) intratracheally at 2 h after resuscitation. Lung tissue and BAL fluid were then recovered at 2 to 8 h after LPS or SAL. The association of Nlrp3 and ASC was determined by using coimmunoprecipitation and immunoblotting analysis. Lung tissue from sham and shock alone animals demonstrated a very low level of association between Nlrp3 and ASC (Fig. 1A and 1B), as well as an undetectable caspase-1 cleavage (Fig. 1C). Administration of LPS to sham animals induced an increase in the association between Nlrp3 and ASC and cleavage of caspase-1 in the lung by 4 h, which increased further by 8 h (Fig. 1A, 1B, and 1C). Whereas, animals subjected to HS before LPS exhibited at 8 h a noticeable increase in the association between Nlrp3 and ASC and cleavage of caspase-1 as compared with that in the lungs from sham/LPS treated animals at the same time point (Fig. 1A, 1B, and 1C). The increased activation of inflammasome and caspase-1 seen in the lungs from HS/LPS-treated animals resulted in a marked increase in IL-1β in BAL fluid, which represents the secretion of IL-1β from pulmonary cells, as shown in Fig. 1D. The alterations in IL-1β mRNA and pro-IL-1β in the lungs induced by LPS and HS/LPS were similar (Fig. 1E and 1F), suggesting that HS augments IL-1β secretion through enhancing inflammasome activation, rather than IL-1β expression. On the other hand, lung tissue recovered from Nlrp3−/− mice failed to show a significant increase in activation of caspase-1 in the lungs and IL-1β in BAL fluid at 8 h after LPS (Fig. 1G and 1H), which indicated an important role of Nlrp3 inflammasome in controlling lung IL-1β secretion in a setting of HS.
Figure 1. HS primes for augmented Nlrp3 inflammasome activation and IL-1β secretion in the lungs.
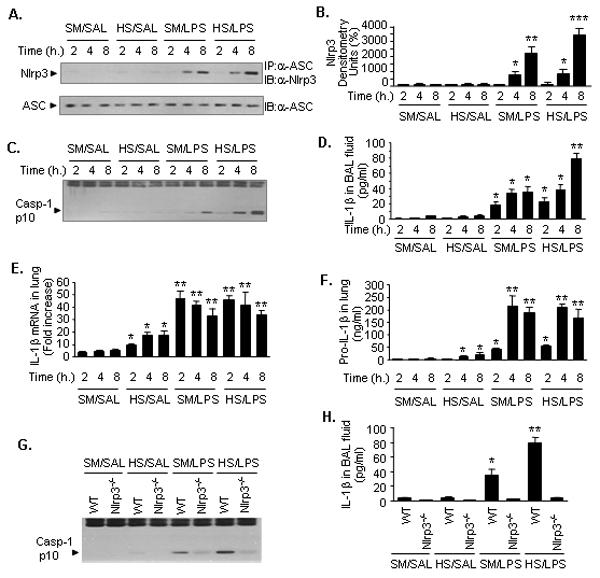
WT (C57BL/6) mice and Nlrp3−/− mice were subjected to HS or sham operation (SM) followed by LPS or saline (SAL) i.t. at 2 h after resuscitation as detailed in the Material and Methods. Lung tissue and BAL fluid were recovered at the time (h) as indicated after LPS or saline. All images are representatives of five independent experiments, and graphs depict the value of mean and SEM, n=5. ** P< 0.01 compared with all other groups; * P< 0.01 compared with the groups labeled with no asterisk. A. The association of Nlrp3 and ASC was detected using immunoprecipitation (IP) with anti-ASC antibody followed by immunoblotting (IB) for Nlrp3 and ASC. B. Densitometry of Nlrp3 shown in A. C. Caspase-1 cleavage product p10 fragments were detected using Western blotting. D. IL-1β in BAL fluid measured by ELISA. E. Pro-IL-1β in the lungs measured by ELISA. F. IL-1β mRNA in the lung tissue measured by quantitative real-time PCR. G. IP for ASC followed by IB for Nlrp3 and ASC in the lung tissue that was collected from WT or Nlrp3−/− mice at 8 h after i.t. LPS or saline. H. IL-1β in BAL fluids that were recovered from WT or Nlrp3−/− mice at 8 h after i.t. LPS or saline.
To determine whether AM contribute to the enhanced inflammasome activation detected in whole lung tissue, BAL cells were recovered at 2 h after HS/resuscitation and enriched for AM by using immunomagnetic separation system. The AM were then incubated in vitro in the presence or absence of LPS (1μg/ml) to evaluate activation of inflammasome and caspase-1, as well as IL-1β release. AM from sham animals treated with LPS exhibited an increased association of Nlrp3 and ASC, and increased caspase-1 cleavage at 4 h and 8 h (Fig. 2A). There was also increased IL-1β secretion in the cell culture medium by 8 h (Fig. 2B). However, in the AM that were isolated from HS mice, LPS induced a markedly augmented association of Nlrp3 and ASC, cleavage of caspase-1, and release of IL-1β (Fig. 2A and 2B). Noticeably again, there was no significant difference in the levels of IL-1β mRNA and pro-IL-1β expression in AM between the groups treated with LPS and HS/LPS, respectively (Fig. 2C and 2D). The results suggested that the release of IL-1β is mainly controlled by the activation of inflammasome while sufficient pro-IL-1β was available. Considered together, these data support the notion of an important role for HS in augmenting the LPS-induced Nlrp3 inflammasome activation in the lung and AM.
Figure 2. HS primes for enhanced Nlrp3 inflammasome activation and IL-1β secretion in the AM.
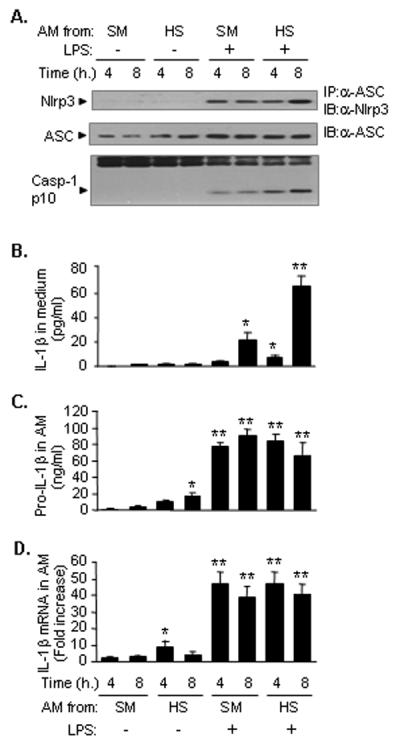
WT (C57BL/6) mice were subjected to HS or sham operation (SM), and at 2 h after resuscitation, AM were recovered from BAL fluid, and incubated in vitro in the presence or absence of LPS (1μg/ml) for 4 and 8 h. Cell lysates underwent immunoprecipitation (IP) for ASC followed by immunoblotting (IB) for Nlrp3 and ASC, and caspase-1 p10 fragments were detected by Western blotting (A), and IL-1β in the cell culture media was measured with ELISA (B), Pro-IL-1β in the AM supernatants was measured with ELISA (C), and IL-1β mRNA in the AM was measured by quantitative real-time PCR (D). The images are representatives of five independent experiments. The graph shows the mean and SEM, n=5. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk.
HS causes impaired up-regulation of pyrin in the lung
Pyrin has been suggested as an inhibitor of caspase-1 activation and subsequent processing of IL-1β (13, 20). To address whether pyrin is involved in the regulation of inflammasome activation in the lung in a setting of HS, we first detected the level of pyrin expression in the lung and AM following HS and/or LPS. Using the animal model of HS/LPS treatments, we found that antecedent HS significantly impaired the expression of pyrin protein in the lungs in response to LPS, as shown in Fig. 3A. This observation was recapitulated in the AM that were isolated from either sham or HS mice and treated with LPS ex vivo. As shown in Fig. 3B, LPS upregulated the expression of pyrin protein in AM from sham animals, whereas HS markedly attenuated the LPS-induced expression of pyrin.
Figure 3. HS causes impaired upregulation of pyrin in the lung.
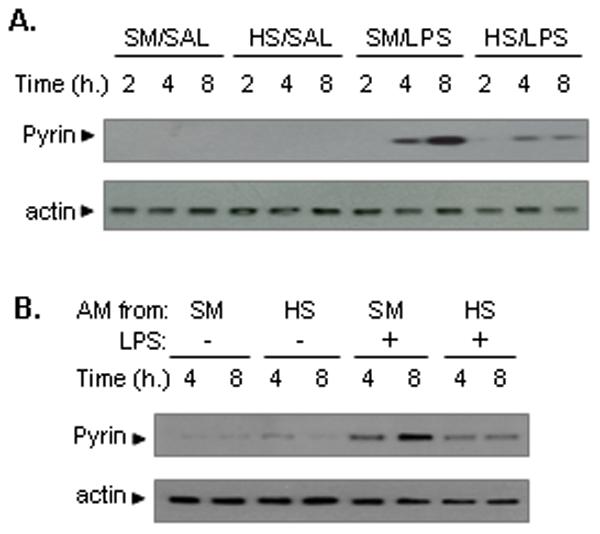
A. WT mice were subjected to HS or sham operation (SM) followed by LPS or saline (SAL) i.t. at 2 h after resuscitation. Lung tissues were recovered after 2 to 8 h. Pyrin expression in the lung tissues was detected by Western blotting. The images are representatives of three independent experiments. B. WT mice were subjected to HS or sham operation (SM), and at 2 h after resuscitation, AM were recovered from BAL fluid, and incubated ex vivo in the presence or absence of LPS (1μg/ml) for 4 and 8 h. Pyrin expression in AM was detected by Western blotting. The images are representatives of four independent experiments.
HS-suppressed IL-10 expression is responsible for impaired pyrin induction
Studies have shown that pyrin expression can be modulated by several cytokines (21). IL-10 has been shown to induce pyrin expression in a manner that is dependent on Jak3, Stat 6 and NF-κB, since the genetic deficiency of these genes diminished IL-10-induced pyrin expression (20). We have previously reported that HS impairs LPS-induced upregulation of IL-10, which in turn exaggerates lung inflammation (29). In this study, we further confirmed that LPS induced the increase expression of IL-10 in the lung acting through TLR4 and MyD88 signaling pathways, since IL-10 expression in TLR4−/− or MyD88−/− mouse lung tissue after LPS challenge were low and not altered by HS, whereas HS attenuated the LPS-induced IL-10 lung expression in WT mice (Fig. 4A).
Figure 4. HS-suppressed IL-10 expression is responsible for impaired pyrin induction in the lung.
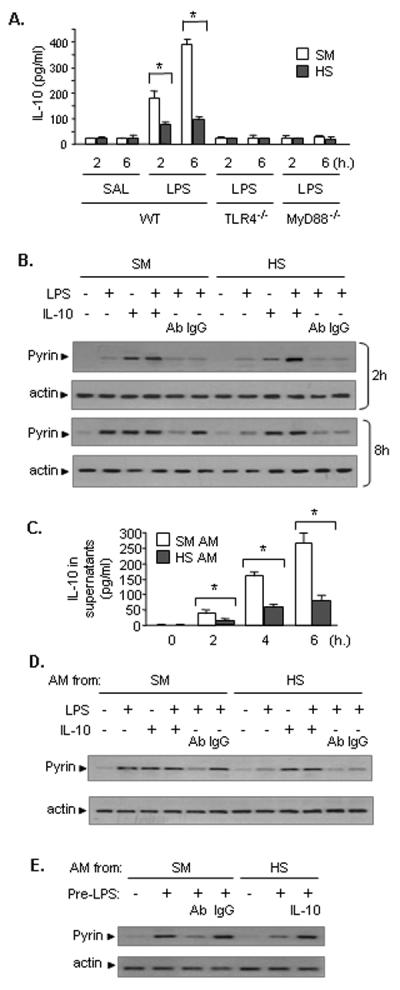
A. HS acting through TLR4 and MyD88 signaling pathway suppresses IL-10 expression in the lung in response to LPS. WT mice, TLR4−/− mice, and MyD88−/− mice were subjected to HS or sham operation (SM) followed by LPS or saline (SAL) i.t. at 2 h after resuscitation. Lung tissues were recovered 2 to 6 h after LPS (or saline) i.t. IL-10 levels in the lung digest homogenates (100 mg of tissue) were quantified by ELISA. The graph shows the mean and SEM, n=5. * p < 0.01. B. IL-10 augments LPS-induced pyrin expression in the lung. WT mice were subjected to HS or sham operation (SM) followed by LPS or saline (SAL) together with i.t. recombinant IL-10 (10 μg/kg B.W.) or neutralizing antibody (Ab) against IL-10 or non-specific IgG control antibody (2 mg/kg B.W.) at 2 h after resuscitation. Lung tissues were recovered at 2 and 8 h after i.t. LPS (or saline). Pyrin expression in the lung tissue digest homogenates was detected by Western blotting. The images are representatives of four independent experiments. C. Ex vivo experiments show that antecedent HS impairs LPS-induced IL-10 secretion from AM. WT (C57BL/6) mice were subjected to HS or sham operation (SM), and at 2 h after resuscitation, AM were recovered from BAL fluid, and incubated in vitro in the presence or absence of LPS (1μg/ml) for up to 6 h. IL-10 in the cell culture media was measured with ELISA. The graph shows the mean and SEM, n=5. * p < 0.01. D. Ex vivo experiments demonstrate important role of IL-10 in enhancing LPS-induced pyrin expression in AM. AM from SM or HS animals, as described in part C (above), were treated with LPS (1 μg/ml) and/or recombinant IL-10 (2 ng/ml), neutralizing Ab to IL-10 (2 μg/ml), or control non-specific IgG antibody for 8 h, and pyrin expression in the AM was detected by Western blotting. The images are representatives of four independent experiments. E. IL-10 derived from AM plays a critical role in enhancing pyrin expression in response to LPS in lung EC. AM from either sham or HS animals, as described in part C (above), at a concentration of 5 × 105 cell/well were pre-treated with LPS for 2 h and then were transferred to top well of a Transwell® plate, in which the bottom well was layered with MLVEC. The co-cultures were incubated for 6 h in the presence or absence of either neutralizing Ab against IL-10 or recombinant IL-10. Non-specific IgG was also added to some wells as control. Pyrin expression in the MLVEC lysates was detected by Western blotting. The images are representatives of four independent experiments.
In order to elucidate whether IL-10 expression impaired by HS contributes to reduced pyrin induction, recombinant IL-10 (10 μg/kg B.W.) or neutralizing antibody against IL-10 (2 mg/kg B.W.) was administered i.t. along with LPS or saline to sham or HS animals at 2 h after resuscitation, and pyrin protein expression in the lungs was then detected at 2 and 8 h afterwards. As shown in Fig. 4B, at the 2 h time point, exogenous IL-10 induced a greater increase in pulmonary pyrin in both sham and HS mice as compared to that in the groups treated with LPS alone, and the combination of LPS and IL-10 induced an even higher expression of pyrin in the lungs. At 8 h after LPS, exogenous IL-10 significantly enhanced the LPS-induced pyrin expression in HS animals, and this was significantly attenuated in both sham and HS mice by neutralizing antibody against IL-10 (Fig. 4B). These findings suggest that the failed upregulation of IL-10 contributes to the suppressed pyrin expression following HS/LPS, especially at the later time point.
To test the role of AM in regulating pyrin expression in the lungs, AM collected from sham or HS mice were treated ex vivo with LPS, and IL-10 level in the cell culture media was measured after 0 to 6 h using ELISA. As shown in Fig. 4C, LPS induced an increase in IL-10 from the AM of sham animals. However, antecedent HS decreased the LPS-induced IL-10 release from AM (Fig. 4C). The alterations in pyrin expression in the AM at 8 h after LPS treatment are shown in Fig. 4D. LPS, IL-10, or a combination of LPS and IL-10 upregulated pyrin expression in the AM that were recovered from sham animals, and neutralizing antibody against IL-10 significantly attenuated the LPS-induced pyrin expression. In contrast, in the AM those were collected from HS animals, the LPS-induced pyrin expression was suppressed, but was able to be restored by exogenous IL-10 (Fig. 4D). Noteworthy, IL-10 alone was sufficient to induce a high level of expression of pyrin in the AM from HS animals (Fig. 4D). Taken together, these results suggest that AM through the release of IL-10 enhanced LPS-induced pyrin expression in an autocrine manner.
The role of AM-derived IL-10 in enhancing pyrin expression in lung endothelial cells, a major cell population in the lung, was also evaluated. AM from either sham or HS animals were treated with LPS for 2 h and then transferred to the top well of a Transwell® plate, in which the bottom well was layered with mouse lung vascular endothelial cells (MLVEC). The co-cultures were then incubated for 6 h in the presence or absence of either neutralizing antibody against IL-10, or recombinant IL-10. The changes in pyrin expression in the MLVEC were detected as shown in Fig. 4E. LPS-treated AM that were isolated from sham animals were able to induce pyrin expression in MLVEC, and this induction was attenuated by IL-10 antibody. AM that were recovered from HS mice and treated with LPS failed to induce a significant expression of pyrin in MLVEC. However, addition of recombinant IL-10 restored the pyrin expression in the MLVEC to similar levels as the MLVEC cocultured with sham AM (Fig. 4E). These results further suggest an important role of AM-derived IL-10 in inducing pyrin expression in the lungs.
Impaired pyrin induction contributes to HS-augmented inflammasome activation
Next, we tested the hypothesis that HS impaired IL-10 expression and therefore decreased pyrin expression in the lungs is responsible for augmented inflammasome activation and IL-1β secretion following HS/LPS. First, the role of IL-10-pyrin axis in regulating inflammasome activation in the lungs was determined. Mice were subjected to HS followed by intratracheal injection of LPS and neutralizing antibody against IL-10 or recombinant IL-10. The interaction of pyrin and Nlrp3 inflammasome components was determined by coimmunoprecipitation using anti-ASC antibody. With blocking of IL-10 by neutralizing antibody, the binding of pyrin to ASC was decreased, which was allied with an increase in Nlrp3-ASC association in the lung and IL-1β level in the BAL fluid from sham/LPS animals (Fig. 5A and 5B); whereas, in HS/LPS-treated mice, exogenous recombinant IL-10 increased pyrin expression in the immunoprecipitates and weakened the association of Nlrp3 and ASC, which resulted in a decrease in IL-1β in the BAL fluid (Fig. 5A and 5B). In this experimental setting, changes in IL-10 did not significantly cause alteration in pro-IL-1β level in the lungs (Fig. 5C), although IL-1β mRNA expression in the lungs was increased while IL-10 was either blocked by neutralizing antibody or diminished by HS (Fig. 5D). These results demonstrate an important role of IL-10 and pyrin in regulating IL-1β secretion - predominantly through modulating activation of Nlrp3 inflammasome, but not IL-1β expression.
Figure 5. HS-suppressed IL-10 expression is responsible for augmented activation of inflammasome and IL-1β secretion in the lung.
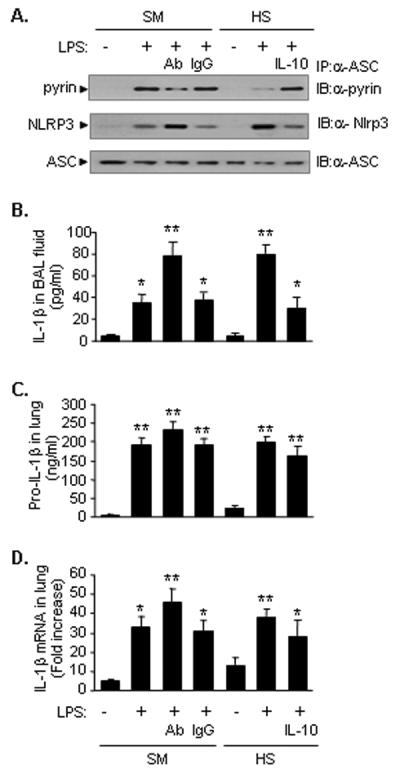
IL-10 augments LPS-induced pyrin expression in the lung. WT mice were subjected to HS or sham operation (SM) followed by LPS or saline (SAL) together with recombinant IL-10 or neutralizing antibody against IL-10 (Ab) or control non-specific IgG antibody (IgG) i.t. at 2 h after resuscitation. BAL fluid and lung tissue were recovered at 8 h after LPS (or saline) i.t. A. The association of pyrin, Nlrp3 and ASC was detected using immunoprecipitation (IP) with anti-ASC antibody followed by immunoblotting (IB) for pyrin, Nlrp3 and ASC, respectively. The images are representatives of five independent experiments. B. IL-1β in BAL fluid was measured with ELISA. C. Pro-IL-1β in the lung tissue was measured with ELISA. D. IL-1β mRNA in the lung tissue was measured by quantitative real-time PCR. The graph shows the mean and SEM from five mice. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk.
In order to reveal the connection between IL-10, pyrin and Nlrp3 inflammasome, we knocked down pyrin in MLVEC using siRNA techniques. At 48 h after transfection of MLVEC with pyrin siRNA, the expression of pyrin protein in response to LPS or IL-10 was significantly decreased in the MLVEC (Fig. 6A). Knockdown of pyrin in MLVEC dramatically increased the LPS-induced Nlrp3-ASC association as determined by coimmunoprecipitation and immunoblotting (Fig. 6B), caspase-1 cleavage (Fig. 6C), and IL-1β release (Fig. 6D) as compared to that in the MLVEC transfected with nonspecific control siRNA. IL-10 attenuated the LPS-induced IL-1β mRNA and pro-IL-1β expression in the MLVEC (Fig. 6E and 6F). However, IL-1β release appeared directly dependent on the activation of inflammasome and caspase-1, rather than the level of pro-IL-1β. Of note, exogenous IL-10 does not decrease inflammasome activation and IL-1β release in the MLVEC with pyrin knockdown, indicating that the IL-10-attenuated inflammasome activation is mediated by pyrin. The changes in pyrin in the immunoprecipitates shown in Fig. 6B suggested that pyrin is not required for the association of Nlrp3 and ASC and subsequent activation of caspase-1; however, the binding of pyrin to the inflammasome complex caused a decrease in the association of Nlrp3 and ASC, caspase-1 activation, and IL-1β secretion.
Figure 6. IL-10-induced Pyrin is an important regulator in inflammasome feedback regulation.
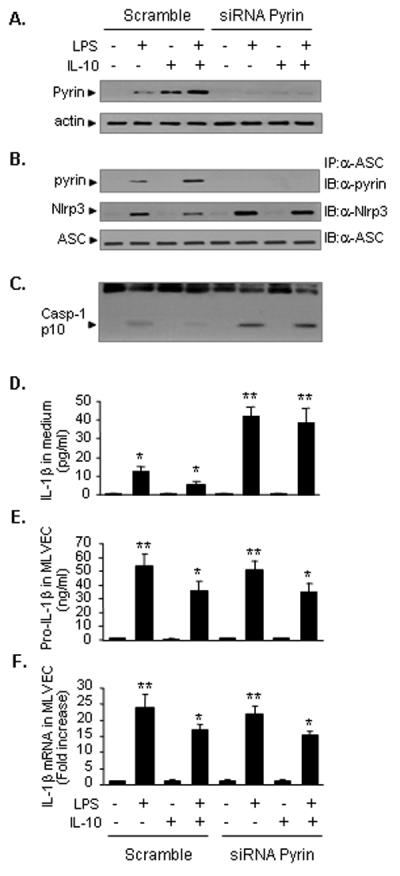
Pyrin in MLVEC was knocked down using siRNA techniques. At 48 h after transfection of pyrin siRNA into MLVEC, the MLVEC were treated with LPS and/or recombinant IL-10 for 8 h. The pyrin protein could not be detected in the cells in response to LPS and/or IL-10 (A). Knockdown of pyrin in MLVEC enhanced LPS-induced Nlrp3-ASC association as detected using immunoprecipitation (IP) with anti-ASC antibody followed by immunoblotting (IB) for pyrin, Nlrp3 and ASC (B), caspase-1 cleavage (C), and IL-1β release in the cell culture media (D), as compared to that in the MLVEC treated with non-specific control siRNA. Exogenous IL-10 attenuated LPS-induced pro IL-1β and IL-1β mRNA expression in MLVEC (E and F). The images are representatives of three independent studies. The graph shows the mean and SEM from three independent experiments. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk.
Discussion
The major cause of late death in HS and trauma patients relates to the development of progressive systemic inflammation and organ failure. The mechanism by which HS augments and exaggerates the inflammatory response remains unclear. IL-1β critically contributes to the development of acute lung injury (33, 34), and SIRS following hemorrhage and trauma (2–7), and elucidating the mechanism underlying HS-primed IL-1β release may result in a new therapeutic strategy for post-hemorrhage SIRS. The present study shows that while LPS induces Nlrp3 inflammasome activation, it also induces pyrin expression in AM and lung EC. Increased pyrin expression inhibits Nlrp3 inflammasome activation and serves as a self-regulatory mechanism. More importantly, LPS induces IL-10 expression in the AM, which in turn augments pyrin expression in AM and EC, and enhances the negative regulation of the inflammasome at later time points. However, HS through the suppression of LPS-induced IL-10 expression prevents this increase in pyrin expression and impairs the negative regulatory mechanism of inflammasome activation, and thus augments IL-1β secretion in the lung (Figure 7).
Figure 7. Model of HS-primed activation of Nlrp3 inflammasome in the lung.
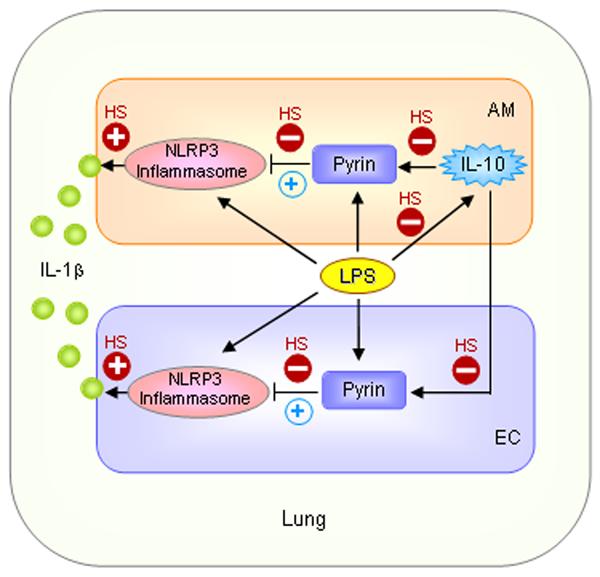
LPS induces Nlrp3 inflammasome activation and pyrin expression in AM and lung EC. Increased pyrin expression inhibits Nlrp3 inflammasome activation and serves as a self-regulatory mechanism. More importantly, LPS induces IL-10 expression in the AM, which in turn augments pyrin expression in AM and EC, and enhances the negative regulation of inflammasome at later time points (blue circled +). Thus, LPS-induced IL-10 is postulated to be a critical feedback mechanism to regulate IL-1β secretion in the lung. However, HS through suppression of LPS-induced IL-10 expression prevents a further increase in pyrin expression and impairs the negative regulatory mechanism of inflammasome activation, and thus, augments IL-1β secretion in the lung.
Pyrin has been shown to inhibit caspase-1 activation and subsequent processing of IL-1β (20). Targeted disruption of the C-terminal portion of pyrin in mice causes increased endotoxin sensitivity and caspase-1 activation, and the macrophages stimulated with LPS produced more IL-1β (20). These data are consistent with the notion that full-length pyrin, but not its truncated version consisting of only the PYD domain, acts as a negative regulator of caspase-1 (35). Pyrin, through its B30.2 domain (also called SPRY domain and RFP) and PYD domains, interacts with inflammasome components caspase-1, ASC, NLRP1, NLRP2 and NLRP3, as well as pro-IL-1β and thereby serves as an inhibitory protein for the inflammasome (13). A recent study, however, reported that a gain-of-function pyrin mutation could cause NLRP3-independent inflammasome activation and autoinflammatory disease (36). That study suggested that pyrin may influence different pathways to lead to a variety of outcomes.
In the current study, by using a gene knockout approach we showed that the Nlrp3 inflammasome is part of the major machinery in the lung and AM that induces IL-1β processing in response to HS and LPS. A direct role for pyrin in suppressing Nlrp3 inflammasome activation in the lung cells was shown by two lines of evidence. First, the amount of pyrin presented in the immunoprecipitate of Nlrp3 inflammasome complex directly affects the Nlrp3-ASC association. Second, decreasing pyrin expression by pyrin knockdown with siRNA significantly increased LPS-induced Nlrp3 inflammasome activation, caspase-1 cleavage, and IL-1β secretion in the lung EC. These findings suggest an important role of pyrin in the negative regulation of Nlrp3 inflammasome activation in the lungs in response to LPS, although the exact mechanism by which pyrin inhibits Nlrp3 inflammasome activation remains unclear.
Pyrin expression in lung EC and its role in regulating inflammasome activation are new findings in this study. Lung EC are the major cell populations that compose a considerable part of the lung. Results from our previous (1) and current studies show that EC are able to secret IL-1β in response to HMGB1 and LPS, suggesting that EC are also an important source of IL-1β and conversely, are a target of IL-1β, which causes them to release a range of inflammatory molecules in response to IL-1β stimulation (6, 37, 38). Thus, EC are postulated to be an amplifier of inflammation through the sensing and releasing of IL-1β, and therefore, EC are a good model for understanding the mechanism of regulation of IL-1β secretion in the lung.
IL-10 plays a key role in limiting inflammation and maintaining immunological homeostasis (39)–(40, 41). IL-10 has been shown to inhibit alveolar macrophage production of pro-inflammatory mediators involved in ARDS (42). In a clinical study involving patients of ARDS of different aetiologies (sepsis, multiple trauma, and perforated bowel), it was shown that patients who had ARDS had lower circulating and bronchoalveolar lavage levels of IL-10 than those who were supposed to be at risk, but did not develop the disease (43). Deficient IL-10 responses were shown to contribute to the septic death of burned patients (44), whereas, administration of IL-10 has a protective effect in animal models of ARDS of different aetiologies, such as sepsis and acute pancreatitis (45–47). In the current study, we revealed a new mechanism by which IL-10, through the induction of pyrin expression, suppresses inflammasome activation. We demonstrated in the in vivo and in vitro studies that LPS-induced IL-10 expression contributed to the upregulation of pyrin in AM and lung EC. In the in vivo HS-LPS-treated mouse model, neutralizing antibody against IL-10 attenuated LPS-induced pyrin expression in the lungs, whereas, exogenous IL-10 restored HS-suppressed pyrin expression in response to LPS. The AM-derived IL-10 is therefore important to pyrin induction in both AM and EC, presumably through autocrine and paracrine pathways respectively. Furthermore, we demonstrated that IL-10 suppressed Nlrp3 inflammasome activation in the lungs in HS-LPS-treated mice, whereas, neutralizing antibody against IL-10 enhanced LPS-induced inflammasome activation in the lungs (Fig. 5). These observations are consistent with the effect of IL-10 on inducing pyrin expression. Notably, the IL-10 modulation of IL-1β expression is not a direct regulatory factor controlling IL-1β secretion. As illustrated in the results, although IL-10 decreased the expression of pro-IL-1β in lung EC and IL-1β mRNA in lung and EC, these changes did not lead to a corresponding alteration in IL-1β secretion. The regulation of IL-1β secretion involves multiple steps including regulation of pro- IL-1β expression, inflammasome assembly and activation, caspase-1 activation, IL-1β maturation, and IL-1β secretion. Each step may regulate IL-1β secretion. In our experimental setting, we observed that recombinant IL-10 suppressed LPS-induced expression of IL-1β mRNA and pro-IL-1β in MLVEC; however, pyrin knockdown increased IL-1β secretion. This result suggests that pyrin-regulated inflammasome activation also plays an important role in regulating IL-1β secretion as long as sufficient por-IL-1β is available.
The findings from the current study also demonstrated that LPS through TLR4 and MyD88-dependent signaling induced IL-10 expression in AM, which was, however, significantly suppressed by antecedent HS. LPS-induced IL-10 expression is regulated post-transcriptionally by the RNA-binding protein tristetraprolin (TTP), which destabilizes IL-10 mRNA in activated macrophages (48). Studies investigating IL-10 regulation show that LPS-stimulated TTP-deficient macrophages overproduced IL-10, contained an increased amount of activated STAT3, and showed reduced expression of inflammatory cytokines (48). However, the mechanism by which HS modulates IL-10 expression remains unclear. We have previously reported that HS plus LPS markedly reduced the transcription rate of IL-10 mRNA as compared to LPS alone but did not affect IL-10 mRNA stability. Reduced IL-10 transcription was not caused solely by impaired nuclear translocation of STAT3 and Sp1/Sp3 transcription factors because LPS-induced nuclear translocation of these factors was augmented by antecedent HS (29). Thus, the HS suppressed IL-10 expression in AM is perhaps through inhibition of IL-10 gene transcription rather than destabilization of IL-10 mRNA.
In summary, this study demonstrates a novel mechanism by which HS, through suppression of negative regulation of Nlrp3 inflammasome activation following LPS challenge, enhances IL-1β secretion in the lungs, and therefore primes for inflammation. This study sheds light on the regulatory role of IL-10 and pyrin in the development of post-HS inflammation. Restoring IL-10 and/or pyrin expression may represent a therapeutic strategy to ameliorate the effects of augmented post-HS inflammation.
Abbreviations used in this paper
- ALI
Acute lung injury
- AM
alveolar macrophages
- ARDS
acute respiratory distress syndrome
- ASC
apoptosis-associated speck-like protein containing a CARD domain
- FMF
Familial Mediterranean Fever
- HS
hemorrhagic shock
- NLRs
NOD-like receptors
- NLRP
nucleotide-binding oligomerization domain-like receptor protein
- MLVEC
mouse lung vascular endothelial cell
- SIRS
Systemic inflammatory response syndrome
Footnotes
This work was supported by the National Institutes of Health Grant R01-HL-079669 (J.F. and M.A.W.), National Institutes of Health Center Grant P50-GM-53789 (T.R.B. and J.F.), a VA Merit Award (J.F.), and the 12th five-year key project grant of PLA BWS12J027 (X.S.).
The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J. Hemorrhagic Shock Activation of NLRP3 Inflammasome in Lung Endothelial Cells. J Immunol. 2011;187:4809–4817. doi: 10.4049/jimmunol.1102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roumen RM, Hendriks T, van der Ven-Jongekrijg J, Nieuwenhuijzen GA, Sauerwein RW, van der Meer JW, Goris RJ. Cytokine patterns in patients after major vascular surgery, hemorrhagic shock, and severe blunt trauma. Relation with subsequent adult respiratory distress syndrome and multiple organ failure. Ann Surg. 1993;218:769–776. doi: 10.1097/00000658-199312000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roumen RM, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, Hendriks T, Goris RJ. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Crit Care Med. 1995;23:474–480. doi: 10.1097/00003246-199503000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Zhu XL, Zellweger R, Zhu XH, Ayala A, Chaudry IH. Cytokine gene expression in splenic macrophages and Kupffer cells following haemorrhage. Cytokine. 1995;7:8–14. doi: 10.1006/cyto.1995.1002. [DOI] [PubMed] [Google Scholar]
- 5.Shenkar R, Abraham E. Effects of hemorrhage on cytokine gene transcription. Lymphokine Cytokine Res. 1993;12:237–247. [PubMed] [Google Scholar]
- 6.Shenkar R, Coulson WF, Abraham E. Hemorrhage and resuscitation induce alterations in cytokine expression and the development of acute lung injury. Am J Respir Cell Mol Biol. 1994;10:290–297. doi: 10.1165/ajrcmb.10.3.8117448. [DOI] [PubMed] [Google Scholar]
- 7.Abraham E, Richmond NJ, Chang YH. Effects of hemorrhage on interleukin-1 production. Circ Shock. 1988;25:33–40. [PubMed] [Google Scholar]
- 8.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 9.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 10.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 11.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA, Lien E. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 13.Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, Grutter C, Grutter M, Tschopp J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14:1457–1466. doi: 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- 14.Consortium TFF. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 15.Consortium TIF. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 16.Martinon F, Hofmann K, Tschopp J. The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr Biol. 2001;11:R118–120. doi: 10.1016/s0960-9822(01)00056-2. [DOI] [PubMed] [Google Scholar]
- 17.Staub E, Dahl E, Rosenthal A. The DAPIN family: a novel domain links apoptotic and interferon response proteins. Trends Biochem Sci. 2001;26:83–85. doi: 10.1016/s0968-0004(00)01717-5. [DOI] [PubMed] [Google Scholar]
- 18.Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 19.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 20.Chae JJ, Komarow HD, Cheng J, Wood G, Raben N, Liu PP, Kastner DL. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- 21.Grandemange S, Aksentijevich I, Jeru I, Gul A, Touitou I. The regulation of MEFV expression and its role in health and familial Mediterranean fever. Genes Immun. 2011;12:497–503. doi: 10.1038/gene.2011.53. [DOI] [PubMed] [Google Scholar]
- 22.Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, Kingma DW, Horwitz ME, Mansfield E, Holland SM, O'Shea JJ, Rosenberg HF, Malech HL, Kastner DL. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–3231. [PubMed] [Google Scholar]
- 23.Wang P, Wu P, Anthes JC, Siegel MI, Egan RW, Billah MM. Interleukin-10 inhibits interleukin-8 production in human neutrophils. Blood. 1994;83:2678–2683. [PubMed] [Google Scholar]
- 24.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. IL-10 inhibits transcription of cytokine genes in human peripheral blood mononuclear cells. J Immunol. 1994;153:811–816. [PubMed] [Google Scholar]
- 25.Kahlke V, Dohm C, Mees T, Brotzmann K, Schreiber S, Schroder J. Early interleukin-10 treatment improves survival and enhances immune function only in males after hemorrhage and subsequent sepsis. Shock. 2002;18:24–28. doi: 10.1097/00024382-200207000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Kotake Y, Moore DR, Vasquez-Walden A, Tabatabaie T, Sang H. Antioxidant amplifies antibiotic protection in the cecal ligation and puncture model of microbial sepsis through interleukin-10 production. Shock. 2003;19:252–256. doi: 10.1097/00024382-200303000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Welborn MB, 3rd, Moldawer LL, Seeger JM, Minter RM, Huber TS. Role of endogenous interleukin-10 in local and distant organ injury after visceral ischemiareperfusion. Shock. 2003;20:35–40. doi: 10.1097/01.SHK.0000071062.67193.b6. [DOI] [PubMed] [Google Scholar]
- 28.Donnelly SC, Strieter RM, Reid PT, Kunkel SL, Burdick MD, Armstrong I, Mackenzie A, Haslett C. The association between mortality rates and decreased concentrations of interleukin-10 and interleukin-1 receptor antagonist in the lung fluids of patients with the adult respiratory distress syndrome. Ann Intern Med. 1996;125:191–196. doi: 10.7326/0003-4819-125-3-199608010-00005. [DOI] [PubMed] [Google Scholar]
- 29.Khadaroo RG, Fan J, Powers KA, Fann B, Kapus A, Rotstein OD. Impaired induction of IL-10 expression in the lung following hemorrhagic shock. Shock. 2004;22:333–339. doi: 10.1097/01.shk.0000136095.96306.08. [DOI] [PubMed] [Google Scholar]
- 30.Fan J, Marshall JC, Jimenez M, Shek PN, Zagorski J, Rotstein OD. Hemorrhagic shock primes for increased expression of cytokine-induced neutrophil chemoattractant in the lung: role in pulmonary inflammation following lipopolysaccharide. J Immunol. 1998;161:440–447. [PubMed] [Google Scholar]
- 31.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 35.Franchi L, Nunez G. A new twist on the PYRIN Mediterranean coast. Immunity. 2011;34:695–697. doi: 10.1016/j.immuni.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, Katz SI, Kastner DL. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30:1702–1714. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- 38.Rao DA, Tracey KJ, Pober JS. IL-1alpha and IL-1beta are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol. 2007;179:6536–6546. doi: 10.4049/jimmunol.179.10.6536. [DOI] [PubMed] [Google Scholar]
- 39.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 40.Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008;226:205–218. doi: 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo CJ, Fu M, Cryer HG. Interleukin 10 inhibits alveolar macrophage production of inflammatory mediators involved in adult respiratory distress syndrome. J Surg Res. 1998;79:179–184. doi: 10.1006/jsre.1998.5418. [DOI] [PubMed] [Google Scholar]
- 43.Armstrong L, Millar AB. Relative production of tumour necrosis factor alpha and interleukin 10 in adult respiratory distress syndrome. Thorax. 1997;52:442–446. doi: 10.1136/thx.52.5.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh FL, Shen HD, Fang RH. Deficient transforming growth factor beta and interleukin-10 responses contribute to the septic death of burned patients. Burns. 2002;28:631–637. doi: 10.1016/s0305-4179(02)00113-4. [DOI] [PubMed] [Google Scholar]
- 45.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–1208. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kusske AM, Rongione AJ, Ashley SW, McFadden DW, Reber HA. Interleukin-10 prevents death in lethal necrotizing pancreatitis in mice. Surgery. 1996;120:284–288. doi: 10.1016/s0039-6060(96)80299-6. [DOI] [PubMed] [Google Scholar]
- 47.Inoue G. Effect of interleukin-10 (IL-10) on experimental LPS-induced acute lung injury. J Infect Chemother. 2000;6:51–60. doi: 10.1007/s101560050050. [DOI] [PubMed] [Google Scholar]
- 48.Gaba A, Grivennikov SI, Do MV, Stumpo DJ, Blackshear PJ, Karin M. Cutting edge: IL-10-mediated tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production. J Immunol. 2012;189:2089–2093. doi: 10.4049/jimmunol.1201126. [DOI] [PMC free article] [PubMed] [Google Scholar]