

Abstract
Redox signaling is emerging as an essential mechanism in the regulation of biological activities of the cell. The HGF/c-Met signaling pathway has been implicated as a key regulator of the cellular redox homeostasis and oxidative stress. We previously demonstrated that genetic deletion of c-met in hepatocytes disrupts redox homeostasis by a mechanism involving NADPH oxidase. Here, we were focused to address the mechanism of NADPH oxidase regulation by HGF/c-Met signaling in primary mouse hepatocytes and its relevance. HGF induced a biphasic mechanism of NADPH oxidase regulation. The first phase employed the rapid increase in production of ROS as signaling effectors to activate the Nrf2-mediated protective response resulting in up-regulation of the antioxidant proteins, such as NAD(P)H quinone oxidoreductase and γ-glutamylcysteine synthetase. The second phase operated under a prolonged HGF exposure, caused a suppression of the NADPH oxidase components, including NOX2, NOX4, p22 and p67, and was able to abrogate the TGFβ-induced ROS production and improve cell viability. In conclusion, HGF/c-Met induces a Nrf2-mediated protective response by a double mechanism driven by NADPH oxidase.
Keywords: HGF, c-Met, NADPH oxidase, hepatocytes, ROS
1. INTRODUCTION
The hepatocyte growth factor (HGF) was originally identified as a potent mitogen for hepatocytes. It exerts multiple functions in a wide range of cell types by the interaction with its receptor c-Met which recruits key signal transduction proteins to control proliferation, survival, morphogenesis, motogenesis and antioxidant response [1].
A large body of data indicates that HGF/c-Met signaling pathway protects against oxidative stress-induced cellular damage. We have shown that HGF upregulates the antioxidant and antiapoptotic proteins, such as Bcl-2, superoxide dismutase (SOD), catalase and Mcl-1 as well as induces glutathione (GSH) synthesis [2, 3]. Although it has been considered that reactive oxygen species (ROS) are only noxious entities for the cell, it is clear today that this effect is dependent on the time of production and concentration, and that basal levels of ROS are needed for the activation of many signaling pathways driving vital physiological functions, including antioxidant defense.
Nrf2 is a transcription factor that regulates the expression of a wide range of antioxidant and cytoprotective genes, particularly in the liver [4]. Under normal conditions, Nrf2 resides in the cytoplasm forming an inactive complex with its inhibitor keap1, an adaptor protein for the E3-based ligase Cullin 3 which mediates the proteosomal degradation of Nrf2. Following a ROS-mediated stimulus, specific cysteine residues in keap1 are oxidized releasing Nrf2 which migrates to the nucleus to initiate the transcription of antioxidant and phase II and III enzymes [4].
In addition to the control of oxidative stress, HGF/c-Met pathway can also regulate the basal cellular redox status. Using conditional mouse genetics, we have demonstrated that the loss of c-Met in liver caused a profound misregulation of genes involved in the antioxidative stress response and GSH metabolism due to the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [2, 5] and predisposed c-Met conditional knockout mice to chemically-induced hepatocarcinogenesis [6].
The NADPH oxidase is a multi-component membrane-bound enzyme that catalyzes the reduction of oxygen to super oxide anion, using NADPH as an electron donor. It was first described in phagocytic cells as a key source of ROS for bacteria killing, but it is also found in many cell types [7], including liver cells [2, 8]. The function of NADPH oxidase in non-phagocytic cells is not completely understood, but there is evidence of involvement in signal transduction events [9]. Overexpression and overactivation of NADPH oxidase have been reported to induce oxidative stress in the liver [5], cardiovascular system [10], and lungs [11], among many other tissues. Furthermore, the sustained overactivation of the NADPH oxidase is highly correlated with the development of hepatic fibrosis [12], liver damage induced by hepatitis C virus infection [13], and cancer [14].
The family of NADPH oxidases consists of several homologous catalytic membrane proteins (NOX1-5, Duox 1–2) and some regulatory proteins (p67, p47, p40, Noxo1, Rac, etc). The latter are located in the cytoplasm under normal conditions, and translocate to the membrane after appropriate stimulus to ensemble the active NADPH oxidase complex. The exception is p22, an ubiquitously expressed subunit required for activation and stabilization of NOX1-4, which resides in membranes and is believed either to assist in the folding of the NOX or participate in the electron transport [7].
In this study we attempted to characterize the mechanisms of NADPH oxidase regulation by the HGF/c-Met pathway in primary mouse hepatocytes. We report that HGF/c-Met controls NADPH oxidase by dual mechanisms. The first is stimulatory, inducing oxidase activity, followed by an inhibitory effect resulting in repression of NADPH oxidase components. We also provide evidence that HGF induces a NADPH oxidase-dependent Nrf2 activation as a part of the survival function elicited by HGF/c-Met signaling pathway in hepatocytes.
2. MATERIALS AND METHODS
2.1. Materials
Collagenase type I was purchased from Worthington, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH) from Invitrogen (catalog number c-6827), and recombinant HGF was from PeproTech (Rocky Hill, NJ). All others chemicals were purchased from Sigma-Aldrich (San Louis, MO).
2.2. Mice, Hepatocyte Isolation, and Culture
C57Bl/6 and MetKO mice were maintained in pathogen-free housing and cared for in accordance with the Universidad Autonoma Metropolitana and NIH Guide for the Care and Use of Laboratory Animals. Hepatocytes were isolated from 8–10-week-old male mice by a two-step collagenase perfusion, as described [15]. The viability was >90% as assessed by trypan blue exclusion. Hepatocytes were seeded at 2.13 × 105 cells per cm2 in 10-cm dishes (Nalge, Nunc) in the Ham’s F-12/Dulbecco’s modified Eagle’s basal hepatocyte growth medium supplemented with 10% fetal bovine serum. After a 4 h attachment, the medium was replaced to a serum-free basal hepatocyte growth medium. The following day, cells were treated with 50 ng/ml HGF or with inhibitors of NADPH oxidase (DPI, 10 μM) or PKC (Chelerytrine, 10 μM).
2.3. Western blot analysis
Western blot analysis was performed as previously described [2] using antibodies listed in Supplementary Table 1.
2.4. Immunoprecipitation
Cells were treated and then harvested and subjected to immunoprecipitation (IP), as previously reported [16], SDS-PAGE, using anti-Keap1 or anti-p47 antibodies (antibody dilution 1:50 in both cases).
2.5. Electrophoretic mobility shift assay
Activation of Nrf2 was examined by electrophoretic mobility shift assay (EMSA) using a consensus oligonucleotide, 5′-TTTTCTGCTGACTCAAGGTCCG-3′ as previously we reported [17]. Probe was labeled by T4 polynucleotide kinase with [γ-32P] ATP (3000 Ci/mmol). Nuclear and cytosolic extracts were prepared and EMSA assay was performed as we previously described [17].
2.6. Analysis of NADPH Oxidase Activity
Specific NADPH oxidase activity was determined by monitoring the rate of consumption of NADPH inhibition by addition of 10 μM DPI 30 min before the assay as described in a number of papers, including ours [2, 8, 16]. Results were expressed as picomol of oxidized NADPH per min per mg of protein.
2.7. Quantitative real-time RT - PCR
Total RNA was extracted with Qiazol reagent (Qiagen 79306) from primary hepatocytes untreated and treated with HGF for 1, 2 and 6 h. Isolated RNA was treated with DNase (RQ1 RNase-Free DNase) to remove traces of genomic DNA contamination. One step RT-PCR amplification was carried out with PCR cycler Rotor-Gene Q system (Qiagen, Australia) Quantitative real–time RT–PCR was carried out by using the Rotor-Gene SYBR Green RT-PCR Kit (Qiagen) with 300nM of sense and antisense primer and 250 ng/ul of RNA to final volume of 10 μl reaction. qRT-PCR conditions were as follows: 10 min at 55°C and 5 min at 95°C, followed by 40 cycles of PCR for 5 s at 95°C for denaturatio n, 10 s at 60°C for annealing and extension. Results are expressed relative to the housekeeping gene actin and relative quantification was performed by using REST-08 Software (Relative Expression Software Tool 2008). After 40 amplification cycles, a melting analysis curve was carried out to verify the correct product by its specific melting temperature. Primers used in this analysis are shown in Supplementary Table 2.
2.8. Reactive oxygen species determination
Reactive oxygen species (ROS) content was determined using 2′-7′-dichlorodihydrofluorescein diacetate (DCFH), a cell-permeable non-fluorescent probe that is de-esterified intracellularly and converted to the highly fluorescent 2′-7′-dichlorofluorescein upon oxidation by reactive oxygen species, particularly peroxides [2]. Cells were seeded in 96-well plates (5×104 cells/well) and subjected to different experimental conditions after overnight stabilization in culture. Cells were incubated with 5 μM DCFH for 30 min before harvesting. DCFH derived fluorescence was monitored using a DTX 880 multimodal detector (Beckman Coulter) with excitation wavelength of 480 and emission wavelength of 520.
2.9. Protein oxidation
Carbonyls groups, as a result of amino acid oxidation, were determined by the Oxyblot kit (Merck Millipore, Billerica, MA) following manufacturer’s instructions.
2.10 Cell viability
Cell viability was assessed by crystal violet-staining assay as described [2].
2.11. Protein content determination
Protein concentration was determined using a bicinchoninic acid (BCA) kit (Pierce Thermo Scientific, Rockford, ILl), according to the manufacturer’s instructions.
2.12. Statistical analysis
The data are presented as mean ± S.E. for at least three independent experiments. Comparisons between groups were performed using the ANOVA test followed by the Tukey variance analysis. Differences were considered significant at p≤ 0.05.
3. RESULTS
3.1 HGF generates transient increase in NADPH oxidase-mediated ROS production
Assessment of the NADPH activity showed an increase as early as 1 min after HGF exposure, which reached a peak value by 30 min (Figure 1A). The enzymatic activity decreased within the next 1–6 h, and fell below the basal activity by 24 h after HGF treatment. To verify whether this effect was related to ROS production, we measured peroxides content by using DCFH. ROS production was significantly increased (1.6-fold) already 1 min after HGF treatment and remained significantly elevated up to 30 min (Figure 1B). ROS production decreased by 60 min and by 360 min was far below the basal levels. Pretreatment with diphenylene iodonium (DPI, 10μM), an extensively used inhibitor of NADPH oxidase, for 30 min before HGF exposure, completely abrogated the early time HGF-mediated increase in ROS. No effect was observed in the late decrease in ROS content (Figure 1B), supporting the fact that HGF-induced ROS generation is driven by NADPH oxidase activity as previously we reported [2].
Figure 1.

HGF stimulates the NADPH oxidase activity. (A) Primary mouse hepatocytes were treated with 50 ng/ml HGF for indicated times and NADPH oxidase activity was assayed as depicted under Materials and Methods. Each column represents the mean ± SE of at least four independent experiments carried out by triplicate. (B) HGF increases production of NADPH oxidase-derived peroxides. Hepatocytes were pretreated, or not with DPI 10 μM for 30 min before stimulation with 50 ng/ml HGF. De-esterified DCFH fluorescence was monitored at the indicated times after 30 min incubation with DCFH (5 μM). Each point represents the mean ± SEM of at least four independent experiments carried out by triplicate. * P ≤ 0.05 vs not treated cells (NT); # P ≤ 0.05 vs 30 min of HGF treatment.
3.2. HGF induces a NADPH oxidase-mediated Nrf2 activation and the expression of cytoprotective genes
To address the significance of HGF-mediated mild ROS generation, we hypothesized that it could activate survival pathways, such as Nrf2. Supporting the HGF-mediated Nrf2 activation, DNA binding activity of Nrf2 started to increase 30 min after HGF exposure and peaked at 60 min (Figure 2A). This result was corroborated by the nuclear translocation of Nrf2 shown by Western blotting (Figure 2B).
Figure 2.
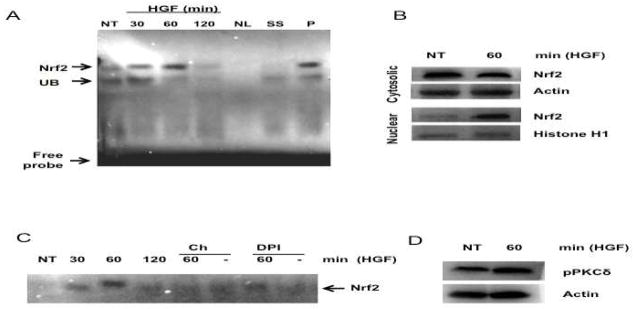
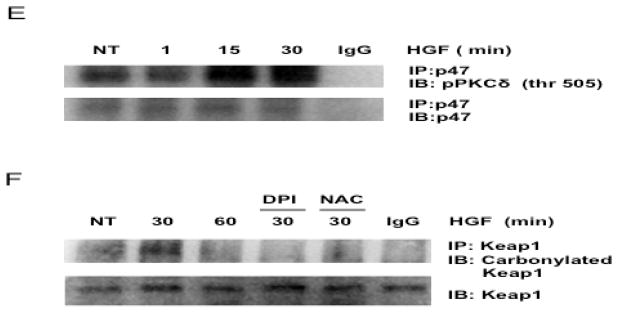
HGF activates Nrf2. (A) Electrophoretic mobility shift assay (EMSA) of Nrf2. Nuclear extracts were prepared from primary hepatocytes treated with 50 ng/ml HGF for indicated times and subjected to EMSA analysis. NT, not treated cells; NC, negative control without protein; SS, super shift using anti-Nrf2 antibody; P, positive control (80 μM tert-butylhydroquinone for 12 h); UB, unspecific band. (B) Nuclear translocation of Nrf2. Cytosolic and nuclear proteins were prepared from cells treated with HGF for 60 min and subjected to Western blot analysis using anti-Nrf2. Actin and Histone H1 were used as loading controls for cytosolic and nuclear proteins respectively. (C) EMSA of Nrf2. Cells were pretreated with 10 μM diphenylene iodonium (DPI) or 10 μM chelerythrine (Ch) for 30 min and stimulated with 50 ng/ml HGF for 30, 60, and, 120 min and subjected to EMSA analysis. (D) PKCδ activation. Total protein was prepared from cells treated with HGF for 60 min and subjected to Western blot analysis using anti-pPKCδ (thr 505). Actin was used as loading control. (E) p47 coimmunoprecipitates with PKCδ. Cells were treated with HGF for 15 and 30 min, cell lysate were prepared and immunoprecipitated with anti-p47 antibody, PKCδ was detected with anti-pPKCδ (thr 505). (F) Keap1 oxidation. Keap1 was immunoprecipitated and resulted protein was assayed with the oxyblot kit. N-acetyl Cysteine (NAC, 25 mM) and DPI (10 μM) were used as unspecific antioxidant and NADPH oxidase inhibitor respectively. Representative images of at least three independent experiments are shown.
Next we determined if HGF activates Nrf2 by the PKC-dependent canonical pathway and tested the involvement of NADPH oxidase in this process. Pretreatment with chelerythrine chloride (Ch), a well-known inhibitor of PKC [18], and DPI abolished the Nrf2 activation induced by HGF (Figure 2C) suggesting that HGF/c-Met signaling pathway use PKC and NADPH oxidase for Nrf2 activation. Since PKC delta (PKCδ) has been reported to be involved in Nrf2 activation in hepatic cell lines [19], we determined the levels of phosphorylated PKCδ (Thr 505) in total protein from hepatocytes treated with HGF for 30 min, indeed, HGF increased the phosphorylation of PKCδ (Figure 2D), further more, p47 coimmunoprecipitates with pPKCδ (Figure 2E), corroborating the PKCδ participation in HGF-induced NADPH oxidase activation. In order to verify if NADPH oxidase-derived ROS are oxidizing Keap1 as a major signal to activate Nrf2, we immunoprecipitated Keap1 using anti-Keap1 antibody (1:50 dilution), IP protein was assayed to detect protein carbonyl modifications, figure 2E shows the oxyblot result observing that HGF induces the increase of Keap1 carbonylation at 30 min of HGF treatment, this effect was prevented with both, the NADPH oxidase inhibitor DPI, and the antioxidant N-acetyl-cysteine(NAC, 25 mM).
To address further the HGF effect on Nrf2 activation, we assayed by Western blot the protein content of NAD(P)H quinone oxidoreductase (NQO1) and γ-glutamyl cysteine synthetase (γ-GCS), two of the main Nrf2 target genes, which reached peak values by 12 h after HGF treatment (Figure 3A). HGF-mediated induction of γ-GCS and NQO1 was abrogated by 30 min pretreatment with DPI (10 μM) supporting participation of the NADPH oxidase (Figure 3B). This result was consistent with our previous data, which showed a maximal antioxidant response in hepatocytes at 12 h after HGF treatment [2, 3].
Figure 3.

HGF induces the expression of Nrf2 target genes. Total proteins were prepared from cells treated with 50 ng/ml HGF for 1, 3, 6, and 12 h and subjected to Western blot analysis using indicated antibodies. In parallel experiments, cells were pretreated with 10 μM diphenylene iodonium (DPI) for 30 min before HGF exposure for 12 h. (A) time-dependent increment in NQO1 and γ-GCS. Representative Western blot and densitometry analysis of protein content relative to actin used as loading control. Results are shown as the means ± SEM of 3 experiments. (B) Inhibition of NADPH oxidase abrogates HGF-induced expression of Nrf2 target genes. Representative Western blot and densitometry of protein levels relative to actin used as internal control. Each bar represents the mean ± SEM of at least three independent experiments carried out in triplicate. * P ≤ 0.05 vs not treated cells (NT); # P ≤ 0.05 vs HGF treated cells (HGF). (C) Effect of HGF pretreatment (12 h) on cell viability in cultures treated with antimycin A (AA, 15 μM) for 0, 6, 12, and 24 h. In parallel experiments, cells were pretreated with DPI for 30 min before addition of HGF. Each point represents the mean ± SEM of at least four independent experiments carried out in triplicate. * P ≤ 0.05 vs not treated cells; # P ≤ vs HGF treated cells (12h) and AA (24h); & P ≤ 0.05 vs cells treated with AA (24h).
To confirm the protective effect of HGF, hepatocytes were exposed to HGF for 12 h and then treated with 15 μM Antimycin A (AA), an inhibitor of respiratory chain, leading to elevated amounts of ROS [20]. HGF protected cells from the cytotoxic effect of AA and increased cell viability from 29% to 82% at 24 h of AA treatment, whereas pretreatment with NADPH oxidase inhibitor abrogated the protective effect elicited by HGF, demonstrating a requirement for NADPH oxidase in HGF/c-Met-mediated hepatoprotection (Figure 3C).
3.3 HGF transcriptionally downregulates NADPH oxidase subunits and confers protection against TGFβ toxicity
In order to figure out the decrease, under basal values, on NADPH oxidase activity at 24 h (Figure 1A) we hypothesized a transcriptional effect, on NADPH oxidase components, elicited by HGF. We proceeded to determine the expression of NOX2, NOX4, p22, p67, and p47 using quantitative real-time RT-PCR. The result shows that HGF decreased the abundance of NOX2 mRNA at 3 and 6 h, NOX4 at 6 h, p22 as early as 1 h and p67 at 6 h (Figure 4A–D). The expression levels of p47 and p40 did not change (data not shown). These effects of HGF on the expression of NADPH oxidase components were confirmed by Western blotting (Figure 4E).
Figure 4.

HGF treatment decreases the expression and protein content of NADPH oxidase subunits. qRT-PCR of NOX2 (A); NOX4 (B); p22 (C) and p67 (D). Each column represents the mean ± SEM of three independent experiments. * p≤0.05 vs not treated cells (NT). (E) Whole cell lysates were prepared from cell cultures treated with 50 ng/ml HGF at indicated times and probed by Western blotting using antibodies against NOX4, NOX2, p67 and p22. Actin was used as a loading control.
Finally, to identify the physiological relevance of the HGF-induced NADPH oxidase subunits repression, we first assayed ROS generation in hepatocytes treated with TGFβ (5ng/ml), a growth factor that uses NADPH oxidase to induce cytotoxic effects on hepatocytes [8]. TGFβ increased ROS levels at 1 h (2.8-fold) and 3 h (2.7-fold) after treatment (Figure 5A). ROS production returned to the basal values at the later times of incubation. HGF pretreatment for 12 h abrogated the TGFβ-induced ROS increment. Consistent with previous result, HGF reversed the deleterious effect of TGFβ and increased cell viability from 58% to 94% (Figure 5 B).
Figure 5.

HGF suppresses TGFβ-mediated ROS generation and its cytotoxic effect. (A) ROS generation by TGFβ treatment. Cells were pretreated with 50 ng/ml HGF for 12 h and then exposed to 5 ng/ml TGFβ for 0.5, 1, 3, 6, and, 12 h, de-esterified DCFH fluorescence was recorded after 30 min incubation with DCFH (5 μM). Data are expressed as fold change relative to the untreated cells. Data represents the means ± SEM of at least three independent experiments carried out in triplicate. * P ≤ 0.05 vs untreated cells (0 h); # P ≤ 0.05 vs 1 h treatment with TGFβ, & P ≤ 0.05 vs 3 h treatment with TGFβ. (B) Cell viability in cultures pretreated with HGF for 12 h and then exposed to 5 ng/ml TGFβ for 24 h. Data represent the mean ± SEM of at least three independent experiments. * P ≤ 0.05 vs untreated cells (NT); # P ≤ vs TGFβ treated cells.
4. DISCUSSION
We have previously reported that genetic deletion of c-Met in hepatocytes generates oxidative stress resulting in the overexpression of numerous ROS-responsive genes, many of them driven by Nrf2 [5]. We have further demonstrated that this redox imbalance was due to the over activation of the NADPH oxidase [2]. These data suggested that c-Met/HGF exerts a regulatory control on the NADPH oxidase system, which is lost in c-Met knock-out mice leading the ROS overproduction, oxidative stress and sensitization to apoptosis [2].
Here we provide evidence that HGF/c-Met-driven regulation of the NADPH oxidase keeps this ROS-generating system under control, and thus represents an essential part of the protective mechanism elicited by HGF in normal cells.
Our data show that HGF/c-Met exerts a dual mechanism of NAPDH oxidase regulation: the first one is increasing, and the second one is decreasing the NADPH oxidase activity and ROS production (Figure 1). The NADPH oxidases are membrane-associated, multiunit enzyme complexes that catalyze the reduction of the oxygen to superoxide anion, using NADPH as electron donor. Although the role of these enzymes is best studied in phagocytic cells, there are still many unknown functions in non-phagocytic cells. The main reported function is signal transduction [9]. In contrast to many cytokines, hormones and growth factors (e.g. TNF-α, PDGF, TGFβ, angiotensin II, insulin) causing cytotoxic effects and cell death [8, 21] via NADPH oxidase-mediated ROS production [9], HGF induced only a mild and transient increment in ROS production which did not trigger cellular damage or impairment in cell viability (Figure 3C). Moreover, exposure to HGF protected cells by a NADPH oxidase-associated mechanism (Figure 5B).
Previously we reported that c-Met deleted hepatocytes showed a significant overexpression of Nrf2 target genes, such as Aldh1a1, Gclc, Gsta1 among many others [5], indicating that c-Met/HGF regulates Nrf2 activation for cellular protection and raising a question whether HGF controls Nrf2 by a NADPH oxidase-mediated mechanism.
To address this question, we first confirmed that HGF induced the DNA binding of Nrf2 to its antioxidant response element (ARE) consensus sequence at 0.5–1 h after treatment (Figure 2A). The c-Met-mediated activation of Nrf2 was corroborated by a nuclear translocation of Nrf2 as measured by Western blotting of cytosolic and nuclear protein fractions (Figure 2B).
Furthermore, we provide evidence that HGF-mediated Nrf2 activation is regulated by PKC. Many reports have pointed out that PKC activity is required for an efficient Nrf2 activation in hepatocytes [19, 22, 23] and other cells types [18, 19, 24]. Niture and coworkers (2009) have demonstrated that PKCδ is the major isoform, which phosphorylates Nrf2 in serine 40 leading to stabilization, nuclear localization, and gene expression of cytoprotective genes in HepG2 cells [19]. HGF is known to mediate the formation of 1,2-diacylglycerol and calcium release into the cytosol, the essential cofactors for PKC activation [25, 26]. We found that PKCδ was activated by HGF (Figure 2D), and we showd that the kinease interacts with p47 subunit of the NADPH oxidase (Figure 2E); chelerythrine, a well characterized inhibitor of PKC and known to prevent Nrf2 activation [18], blocked the HGF-mediated Nrf2 DNA binding in hepatocytes (Figure 2C). These data clearly show that HGF activates Nrf2 by a mechanism dependent of PKCδ and NADPH oxidase.
Another condition required for Nrf2 activation is the oxidative modification of specific cysteines on Keap1. Cysteine 273 and 288 have been proposed as redox sensors to release Nrf2 from the complex [27] besides to cysteine 151 oxidation required for Nrf2 activation in HepG2 cells. Our results demonstrate that NADPH oxidase can induce Keap1 oxidation (Figure 2F), even more; NADPH oxidase inhibition with DPI or the antioxidant NAC treatment abrogate the HGF-induced Nrf2 DNA binding. The essential role of NADPH oxidase activity in the activation of Nrf2 and the expression of its target genes was further corroborated by the findings in HepG2 cells overexpressing Keap1. Treatment of these cells with indole analogues increased NADPH oxidase activity, Keap1 oxidation and the expression of γ-GCS, these effects were suppressed by DPI, [28]. Together, our data suggest that HGF/c-Met generates required conditions for Nrf2 activation, which are mediated by NADPH oxidase activity including the PKC-mediated Nrf2 phosphorylation and oxidative signal for cysteine oxidation on Keap1. The Nrf2-driven responses increase the expression of enzymes involved in the transformation, detoxification and transport of xenobiotics; as well as antioxidant defense-related proteins [4], including the prototypical Nrf2-target gene NQO1 and the rate-limiting enzyme on GSH synthesis, γ-GCS [29]. Our data show that HGF induced expression of both NQO1 and γ-GCS in a time-dependent manner. Pretreatment with DPI abrogated the HGF effect, confirming that HGF-mediated stimulation of the NADPH oxidase provides oxidative signal for the Nrf2 activation and expression of protecting proteins (Figure 3A–B). The survival outcome was corroborated by HGF induced NADPH oxidase-dependent protection (Figure 3C) against the toxicity elicited by AA, which decreased the viability in a time-dependent manner whereas HGF pretreatment improved the cell viability, and DPI pretreatment abrogated the HGF effect. Taken together, these data demonstrate that the HGF/c-Met signaling pathway supports a Nrf2-mediated cell survival by a process mediated by the NADPH oxidase. Inhibition of NADPH oxidase activity abrogates Nrf2 activation, expression of survival proteins and sensitizes hepatocytes to death under an oxidative stress challenge.
The second phase of the NADPH oxidase regulation by HGF/c-Met pathway takes place at a later time point (24h) (Figure 1). Long-term treatment with HGF induced a decrease in the expression of several components of the NADPH oxidase (Figure 4), specifically p22, p67, NOX2 and NOX4.
According to our findings, there is evidence that harmonized expression or repression of the NADPH oxidase components occurs in other tissues; angiotensin II induces the coordinated expression of both, NOX1 and p22 [7]. In human lung endothelial cells subjected to hyperoxia, a coordinated increase in the expression of NOX1, NOX2, NOX4, p22 mRNA were observed, but only p22, NOX2 and NOX4 were confirmed by Western blot [30]. Here we show that HGF decreases the mRNA and protein levels of NOX2, NOX4, p22 and p67 in a harmonized manner (Figure 4) suggesting a coordinated negative regulation similar to that described in patients with chronic granulomatous disease (CGD), NOX2-deficient patients lack the detectable p22 protein in phagocytes, whereas p22-deficient CGD patients do not express NOX2 protein [31]. It has been reported that NOX2 and p22 are stable only as a heterodimer while monomers are degraded by proteosome 26S [32]. The repression effect brought about by HGF occurs at mRNA, suggesting that HGF could activate signaling messengers, which target activators or repressors of the transcription of NADPH oxidase proteins. This finding may provide an explanation on how HGF antagonizes the effects of TGFβ, TNF-α or FasL by inducing the transcriptional repression of NADPH oxidase components.
5. CONCLUSIONS
In conclusion, we provide evidence that HGF regulate NADPH oxidase in the hepatocytes by a dual mechanism. HGF treatment leads to NADPH oxidase activation and ROS generation, which in turn results in PKC-mediated Nrf2 activation and expression of survival proteins (Figure 6A). In the absent of HGF treatment some NADPH oxidase-dependent cytotoxic factors, such as TGFβ can induce cell death by activating the oxidase-mediated ROS production (Figure 6B).
Figure 6.


Proposed mechanisms of HGF/c-Met-induced NADPH oxidase regulation. (A) post-translational regulation. HGF treatment induces the release of p22 from c-Met-p22 interaction leading to NADPH oxidase assembling and activation. In addition HGF/c-Met activates PKCδ which can phosphorylate Nrf2 and enable NAPDH oxidase activity. Once activated, Nrf2 translocates to the nucleus to drive the expression of survival genes such as NAD(P)H quinone oxidoreductase (NQO1) or γ-glutamylcysteine synthetase (γ-GCS). Transcriptional regulation. (B) In the absent of HGF/c-Met stimulus, NADPH oxidase is functional and can be used by cytotoxic factors such as transforming growth factor beta (TGFβ) to induce cell death. (C) The sustained HGF stimulus leads to the transcriptional repression of NADPH oxidase subunits; this effect could abrogate the cytotoxic action of TGFβ resulting in a survival response.
The second point of HGF-mediated NADPH oxidase regulation is a transcriptional repression of the expression of p22, NOX2, NOX4 and p67. We propose that this outcome constitutes the central mechanism by which HGF/c-Met signaling pathway counteracts the cytotoxic effects of growth factors such as TGFβ (Figure 6C). We are currently studying the molecular mechanism of NADPH oxidase subunits repression.
Supplementary Material
Acknowledgments
This work was partially funded by grants from CONACYT #131707; FUNDHEPA “Antonio Ariza Cañadilla” grant, Universidad Autonoma Metropolitana–Iztapalapa and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
We thank CONACYT for financial support to Denise Clavijo-Cornejo (PhD scholarship holder #233306) and Luis E. Gomez-Quiroz (Estancias Sabaticas al Extranjero #144805). We thank Santamaria-Olmedo, G and Mata-Villegas, A for help with qRT-PCR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Quiroz LE, Factor VM, Kaposi-Novak P, Coulouarn C, Conner EA, Thorgeirsson SS. Hepatocyte-specific c-Met deletion disrupts redox homeostasis and sensitizes to Fas-mediated apoptosis. J Biol Chem. 2008;283:14581–14589. doi: 10.1074/jbc.M707733200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valdes-Arzate A, Luna A, Bucio L, Licona C, Clemens DL, Souza V, Hernandez E, Kershenobich D, Gutierrez-Ruiz MC, Gomez-Quiroz LE. Hepatocyte growth factor protects hepatocytes against oxidative injury induced by ethanol metabolism. Free Radic Biol Med. 2009;47:424–430. doi: 10.1016/j.freeradbiomed.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Klaassen CD, Reisman SA. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol. 2010;244:57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaposi-Novak P, Lee JS, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006;116:1582–1595. doi: 10.1172/JCI27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, Thorgeirsson SS. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67:9844–9851. doi: 10.1158/0008-5472.CAN-07-1905. [DOI] [PubMed] [Google Scholar]
- 7.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 8.Carmona-Cuenca I, Herrera B, Ventura JJ, Roncero C, Fernandez M, Fabregat I. EGF blocks NADPH oxidase activation by TGF-beta in fetal rat hepatocytes, impairing oxidative stress, and cell death. J Cell Physiol. 2006;207:322–330. doi: 10.1002/jcp.20568. [DOI] [PubMed] [Google Scholar]
- 9.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandes DC, Wosniak J, Jr, Pescatore LA, Bertoline MA, Liberman M, Laurindo FR, Santos CX. Analysis of DHE-derived oxidation products by HPLC in the assessment of superoxide production and NADPH oxidase activity in vascular systems. Am J Physiol Cell Physiol. 2007;292:C413–422. doi: 10.1152/ajpcell.00188.2006. [DOI] [PubMed] [Google Scholar]
- 11.Griffith B, Pendyala S, Hecker L, Lee PJ, Natarajan V, Thannickal VJ. NOX enzymes and pulmonary disease. Antioxid Redox Signal. 2009;11:2505–2516. doi: 10.1089/ars.2009.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Minicis S, Seki E, Paik YH, Osterreicher CH, Kodama Y, Kluwe J, Torozzi L, Miyai K, Benedetti A, Schwabe RF, Brenner DA. Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology. 2010;52:1420–1430. doi: 10.1002/hep.23804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudreau HE, Emerson SU, Korzeniowska A, Jendrysik MA, Leto TL. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: a new contributor to HCV-induced oxidative stress. J Virol. 2009;83:12934–12946. doi: 10.1128/JVI.01059-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamata T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009;100:1382–1388. doi: 10.1111/j.1349-7006.2009.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Souza V, del Escobar MC, Bucio L, Hernandez E, Gomez-Quiroz LE, Gutierrez Ruiz MC. NADPH oxidase and ERK1/2 are involved in cadmium induced-STAT3 activation in HepG2 cells. Toxicol Lett. 2009;187:180–186. doi: 10.1016/j.toxlet.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 17.Luna-Lopez A, Triana-Martinez F, Lopez-Diazguerrero NE, Ventura-Gallegos JL, Gutierrez-Ruiz MC, Damian-Matsumura P, Zentella A, Gomez-Quiroz LE, Konigsberg M. Bcl-2 sustains hormetic response by inducing Nrf-2 nuclear translocation in L929 mouse fibroblasts. Free Radic Biol Med. 2010;49:1192–1204. doi: 10.1016/j.freeradbiomed.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Espada S, Rojo AI, Salinas M, Cuadrado A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J Neurochem. 2009;110:1107–1119. doi: 10.1111/j.1471-4159.2009.06208.x. [DOI] [PubMed] [Google Scholar]
- 19.Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452–4464. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Ransac S, Mazat JP. How does antimycin inhibit the bc1 complex? A part-time twin. Biochim Biophys Acta. 2010;1797:1849–1857. doi: 10.1016/j.bbabio.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, Rojanasakul Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 2008;180:3072–3080. doi: 10.4049/jimmunol.180.5.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SG, Kim SO. PKC downstream of Pl3-kinase regulates peroxynitrite formation for Nrf2-mediated GSTA2 induction. Arch Pharm Res. 2004;27:757–762. doi: 10.1007/BF02980145. [DOI] [PubMed] [Google Scholar]
- 23.McNally SJ, Harrison EM, Ross JA, Garden OJ, Wigmore SJ. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int J Mol Med. 2007;19:165–172. [PubMed] [Google Scholar]
- 24.Zhang H, Forman HJ. Acrolein induces heme oxygenase-1 through PKC-delta and PI3K in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2008;38:483–490. doi: 10.1165/rcmb.2007-0260OC. [DOI] [PubMed] [Google Scholar]
- 25.Osada S, Nakashima S, Saji S, Nakamura T, Nozawa Y. Hepatocyte growth factor (HGF) mediates the sustained formation of 1,2-diacylglycerol via phosphatidylcholine-phospholipase C in cultured rat hepatocytes. FEBS Lett. 1992;297:271–274. doi: 10.1016/0014-5793(92)80554-t. [DOI] [PubMed] [Google Scholar]
- 26.Osada S, Saji S, Nakamura T, Nozawa Y. Cytosolic calcium oscillations induced by hepatocyte growth factor (HGF) in single fura-2-loaded cultured hepatocytes: effects of extracellular calcium and protein kinase C. Biochim Biophys Acta. 1992;1135:229–232. doi: 10.1016/0167-4889(92)90142-x. [DOI] [PubMed] [Google Scholar]
- 27.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, Starnes JH, Kelton KR, Summar SR, Sasi S, Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 29.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501:116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pendyala S, Gorshkova IA, Usatyuk PV, He D, Pennathur A, Lambeth JD, Thannickal VJ, Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal. 2009;11:747–764. doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parkos C, Dinauer M, Jesaitis A, Orkin S, Curnutte J. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood. 1989;73:1416–1420. [PubMed] [Google Scholar]
- 32.DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and Maturation of Flavocytochromeb 558 Include Incorporation of Heme as a Prerequisite for Heterodimer Assembly. Journal of Biological Chemistry. 2000;275:13986–13993. doi: 10.1074/jbc.275.18.13986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.