

Abstract
Innate instruction of adaptive immunity was proposed more than 20 years ago as a mechanism by which long-lived lymphocyte responses are targeted to appropriate antigens. At the time Charles Janeway proposed this theory, most of the innate immune receptors were unknown and the pivotal role of the dendritic cell in instructing T cell priming was debated. There is now overwhelming evidence that the innate and adaptive branches of the immune system must interact to generate immunity. Much of this work has focused on families of innate immune receptors called pattern recognition receptors (PRRs) on dendritic cells, which translate these inflammatory triggers into productive T cell responses. Nevertheless, we are only beginning to understand how these defense molecules shape the generation of immunity. We review the varied roles of one class of PRRs, the NOD-like receptors (NLRs), in immune responses and propose a new model in which adaptive immunity requires coordinated PRR activation within the dendritic cell.
NLRs: pattern recognition “receptors”
The immune system possesses a repertoire of receptors activated by evolutionarily conserved molecular patterns that are broadly classified into two categories on the basis of their origin: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). As the term suggests, PAMPs refer to molecules that are exogenous in origin and are found particularly on a group of pathogens. These include such “patterns” as cell wall components, flagella, lipoproteins and nucleic acids of bacterial, fungal and viral origin [1]. PAMPs can also be referred to as microbe-associated molecular patterns (MAMPs) to indicate that not all microbes are pathogens, most notably commensal flora, although this distinction depends on the immunocompetence of the host. In contrast to PAMPs, DAMPs are endogenous molecules which, upon stress or damage to the host cell, are released or modified and are capable of initiating an inflammatory response. A few examples of these include DNA-binding proteins such as high-mobility group protein 1 (HMGB1), heat-shock proteins (HSPs), extracellular ATP and uric acid crystals [2].
The innate branch of the immune system detects these molecular patterns in part through numerous germ line-encoded pattern recognition receptors (PRRs). Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), RIG-I-like receptors (RLR), AIM2-like receptors (ALR) and C-type lectin receptors (CLR) are not only central to the rapid innate immune response but also have direct effects on initiating adaptive immunity [3]. Although much attention is paid to the role of the above PRRs in the immune response, a broader repertoire of pattern recognition molecules (PRMs) including complement components [4], scavenger receptors, SIRP-α [5], pentraxins [6] and the KIR family of receptors on natural killer (NK) cells [7] detect host invasion, damage, aging or alteration; these sensors of altered or damaged patterns are outside of the scope of this review but are also crucial in the maintenance of immune homeostasis.
TLRs are the most-well characterized pattern recognition receptors. They are membrane-bound and expressed on both the cell surface and in endosomal compartments; TLRs primarily recognize PAMPs, although examples of DAMPs activating TLRs also exist [8]. The CLRs, which are either membrane-bound or secreted, bind to carbohydrate moieties such as mannose, fucose and β-glucan on pathogens such as Candida albicans, as well as to host-derived glycoproteins and F-actin [9, 10]. The other PRRs scan a topologically different part of the cell – the integrity of cytosolic contents. RLRs are cytoplasmic helicases that sense single- or double-stranded RNA [11], whereas ALRs respond to bacterial or viral-derived cytoplasmic double-stranded DNA [12]. NLRs are also cytoplasmic, and like many PRRs, they respond to a range of both PAMPs and DAMPs.
Some NLRs detect PAMPs in the cytosol, indicating cellular invasion by the pathogen or loss of cellular compartmentalization, such as bacterial muramyl-dipeptide (MDP) recognition by NOD2 or the bacterial secretion system molecule PrgJ by NAIP2 [13]. Other NLRs “sense” cytosolic PAMPs but indirectly through potentially cell-derived signals or adapter molecules (e.g. sensing of acylated lipopeptides by NLRP7 or sensing of flagellin by NLRC4) [14, 15]. Finally, some NLR members are activated following the loss of cell membrane integrity, including the insertion of pores or ion channels through cellular membranes (e.g. NLRP1 activation by Lethal Toxin and NLRP3 activation by influenza M2 protein as well as numerous other pathogen-derived toxins) [16–18], insertion of bacterial secretion systems into the plasma membrane (e.g., NLRC4-Type III secretion systems) [19, 20] or membrane rupture (e.g. NLRP3-insoluble crystals) [21]. Altogether, by recognizing the loss of cellular integrity, NLRs seem to detect a different kind of insult than other PRRs and might be able to act as a “back-up” system if a pathogen has successfully bypassed TLR detection. Accordingly, the cellular inflammatory program following NLR triggering is often distinct from that induced by an activated TLR and can be even quite different between different NLR members.
Although the triggers and function of numerous NLRs remain unknown, some trends have emerged from those NLRs that have been characterized and we have attempted to group these into a functional classification scheme outlined in Table 1. One well-defined role of a number of NLRs, as well as non-NLRs such as AIM2 and IFI16, is the nucleation of a multi-molecular complex termed an “inflammasome” consisting of the adaptor molecule apoptosis-associated speck-like protein containing a card (ASC) and the inflammatory caspase, caspase-1. The defining feature of an active inflammasome is the proteolytic processing and subsequent secretion of interleukin (IL)-1β and IL-18 by active caspase-1.
Table 1. Proposed functional classification of NLRs.
Although four subgroups of NLRs exist based on domain structure groupings [116], a distinct grouping emerges based on functional studies of particular NLRs characterized to-date. All of these functions can influence the outcome of DC activation although many of these proposed effects require more work to confirm their true function specifically within the DC. It should be noted that although some NLR members fall into multiple categories in the table above, in general each NLR appears only once and therefore might be specialized to a particular function.
| Activity | Triggers | NLRs | Outcomes | Proposed DC Effect | Ref |
|---|---|---|---|---|---|
| Transcriptional regulation | Cytokines | CIITA NLRC5 |
MHC Class I & II transcription | Antigen presentation | [73, 109] |
| TLR-like activity | PAMP recognition | NOD1 NOD2 |
Synergistic induction of inflammatory signaling cascade (NF-κB, MAPK) | Activation (Figure 1b) | [46, 49, 110] |
| Inflammasome nucleation | DAMP or PAMP sensing | NLRP1 NLRP2 NLRP3 NLRP6 NLRP7 NLRP12 NLRC4/N AIP2,5 |
IL-1β secretion IL-18 secretion Pyroposis |
Enhanced T cell priming? | [14, 22, 62, 111–113] |
| Signaling modulator | ? | NLRP2 NLRP4 NLRP6 NLRP12 NLRX1 NLRC3 |
Inhibit NF-κB pathways | Modification of TLR induced maturation | [55, 57, 60, 63, 66, 114, 115] |
| Signaling molecule? | ? | NLRP10 NLRP12 |
Migration | Licensing (Figure 1b) | [90, 94, 95] |
Of all inflammasomes, NLRP3 is the best characterized. It is activated upon stimulation of cells with a wide array of chemically diverse DAMPs, including pore forming toxins, products of cell death (e.g., ATP, uric acid), haptens, endogenously and exogenously-derived insoluble crystals (e.g., alum, cholesterol, silica), protein aggregates (e.g., β-amyloid, islet amyloid polypeptide) and adjuvant microparticles [2, 22]. Although a wide range of PAMPs from fungal zymosan to viral RNA have also been suggested to activate the NLRP3 inflammasome [23–25], it has been difficult to clearly delineate whether the PAMP itself or rather the damage caused by the PAMP/organism actually initiates NLRP3 activation [16]. We argue that the overwhelming commonality to NLRP3 triggers is the induction of cell damage [22] and therefore most PAMPs that induce NLRP3 activity will also follow this rule. These kinds of questions will remain unanswered until we have identified ligands that directly bind to and regulate NLRP3. For NLRP3, and indeed most NLRs studied to date with only a couple of exceptions [26–29], there is little evidence that any directly bind to the PAMPs or DAMPs that induce their activation.
Given the striking breadth of chemical and physical characteristics of NLRP3 triggers, it is unlikely that each agonist directly binds NLRP3 in a typical ligand-receptor paradigm. Instead, we and others have proposed that these triggers share in common the disruption of cellular homeostasis or integrity of specific intracellular compartmentalization [22, 30, 31] (Box 1; Figure 1a) - analogous to the release of cytochrome c from the inner mitochondrial membrane to initiate apoptosome formation (Box 2). A number of different mechanisms have been proposed by which NLRP3 activity is directly regulated downstream of DAMP or PAMP stimulation including NLRP3 deubiquitination, cellular potassium efflux, interaction with the phosphorylated protein kinase PKR or phagolysosome destabilization [30, 32–34]; however, multiple studies have recently pointed to mitochondrially-derived signals, ranging from reactive oxygen species (ROS) to mtDNA to ATP, as direct ligands nucleating the NLRP3 inflammasome [35–39]. In combination with the colocalization of NLPR3 and ASC to mitochondrial organelle clusters during inflammasome activation [39] these data suggest that cellular damage is sensed by the mitochondria and desequestration of molecules from this organelle might directly program the cell for inflammation (inflammasome) or death (apoptosome) (Box 1–2).
Text box 1. The canary in the coalmine and NLRP3 inflammasome activation.
It has already been proposed that the NLRP3 inflammasome “monitors the activity of the mitochondrion” [30], but perhaps more specifically, mitochondria monitor the integrity and homeostasis of the cell; in this way, mitochondria act as a “canary in a coal mine” to sense the integrity of the cell and then orchestrate the cellular response to perturbations, whether metabolic changes, inflammation (e.g., inflammasome) or death (e.g., apoptosome). Because mitochondria likely originated from a prokaryotic symbiotic relationship within the eukaryotic cell [117], it is plausible that during evolution mitochondrial molecules with non-eukaryotic motifs would have been preferentially selected to serve as DAMPs to nucleate the inflammasome response. Identification of the direct ligand(s) that binds and activates NLRP3 will be crucial to our molecular understanding of NLR regulation and inflammasome formation.
Figure 1. Two-step models of DC activation for (a) inflammasomes and (b) T cell priming.
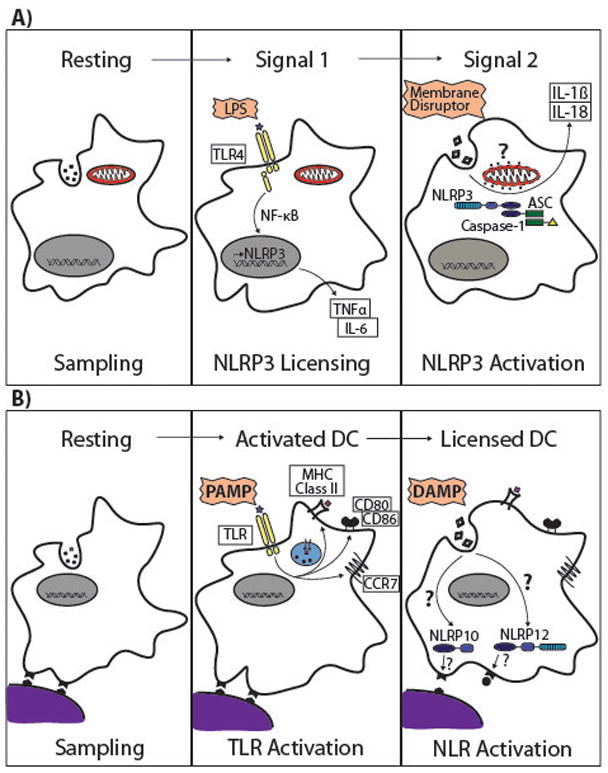
(a) TLR induced (or cytokine induced) NLRP3 and pro-cytokine transcription is a licensing signal for NLR inflammasome activity (“Signal 1”). Even so, TLR signalling is not sufficient to enable inflammasome nucleation and function. In the case of NLRP3, we propose that inflammasome activation requires a second signal that induces cell damage (“Membrane Disruptor”) resulting, through unknown pathways, in the release of a motochondrially-sequestered molecule (“?”). We propose that this endogenous ligand then directly induces NLRP3 inflammasome activation with one of the well-established sequelae of IL-1β and IL-18 secretion. This model is not restricted to inflammasome activation in DCs. LPS, lipopolysaccharide; TLR4, Toll-like receptor 4; ASC, apoptosis-associated speck-like protein containing a CARD. (b) Similar to the two-signal model of T cell activation, we propose a two PRR model of DC activation in which neither PAMPs nor DAMPs alone are sufficient to induce a fully licensed DC for T cell priming. PAMP-induced DC maturation upregulates most of the requisite molecules to effectively prime a naïve CD4+ T cell including costimulatory molecules, MHC class II and CCR7 surface expression (“Activated DC”). It should be noted that this step has been shown to be mediated by more than TLRs and particular NLRs that induce inflammatory signalling cascades can also regulate DC activation. A fully licensed DC requires a second signal initiated by the detection of a damaged host (DAMP). For example, without activation of NLRs such as NLRP10 and NLRP12, the dendritic cell is unable to migrate to draining lymph nodes even if the DC is activated, thus preventing unnecessary lymphocyte priming. PAMP, pathogen associated molecular pattern; DAMP, damage associated molecular pattern.
Text box 2. Apoptosome.
A cytosolic pathway parallel to inflammasome activation exists in the cell to regulate apoptosis. A mitochondrial-sequestered molecule, cytochrome c, is released into the cytosol and recognized by Apaf-1, which can then form a platform for caspase-9 activation called the apoptosome [118]. Subsequent activation of execution caspases ultimately results in apoptosis. Although the cellular outcome is significantly different, the apoptosome and inflammasome have numerous similarities; the “somes” in both are multimolecular platforms that result in activation of caspase enzymes, many of the domains of Apaf-1 are shared and central to NLR function and the upstream triggers of both are possibly released mitochondrial molecules.
This model suggests that NLRP3, and perhaps other NLRs as well, sense a pattern of cell stress rather than the actual agonists, analogous to the Guard Theory of plant immunity, which is based on resistance molecules with significant homology to NLR molecules (i.e., NB-LRR R proteins) [40]. It further suggests that NLRs should induce an immunological response distinct from TLR-induced activation to specify the detection of a different type of immune insult, in this case cell disruption rather than the presence of microbes. Indeed, triggering of NLRs regulates a wide range of cellular programs from the modulation of signalling cascades (e.g., NOD2) to gene expression (e.g., CIITA) to autophagy (e.g., NLRP4) (Box 3) to cellular migration (e.g., NLRP10). As will be discussed below, many of these cellular effects distinguish NLRs from other PRRs and might allow for distal communication of the presence of host damage to the adaptive immune system.
Text box 3. NLRs and autophagy.
Eukaryotic cells use a fundamental process called autophagy to degrade macromolecular structures in the cytoplasm. In DCs, the autophagic machinery has been shown to be crucial in transporting cytoplasmic antigens (such as HSV-2) to MHC Class II, thus regulating CD4+ T cell responses [119]. Both NOD1 and NOD2 were reported to induce autophagy in response to bacterial infections like Shigella flexneri by directly interacting with the autophagic protein ATG16L1 or indirectly through RIP2-kinase activation [120, 121]. Certain NLRs like NLRP4 have also been found to inhibit the initiation and maturation of autophagy by associating with Beclin-1 and class C vacuolar protein-sorting complex [122]. Finally, autophagy has been found to negatively regulate NLRP3 inflammasome activation, and this is believed to be a result of defective mitochondrial clearance by autophagic processes [31].
Beyond IL-1β
The primary cell type that bridges the innate and adaptive immune branches of the immune system is the dendritic cell (DC), glibly called “nature’s dirty little secret” by Charles Janeway because it acts as nature’s adjuvant [41] to translate immunologists’ “dirty little secret” (the use of microbially-based adjuvants) [42] into productive adaptive immune responses. This versatile group of cells can induce particular acute pro-inflammatory cytokine responses but are also the master regulators of naïve T cell fate. During steady-state, immature DCs continuously sample their environment for potential antigens. They express low levels of co-stimulatory molecules on their surface, high levels of major histocompatibility complex (MHC) class II molecules in endosomal compartments and have enhanced endocytic capacities [43]. The complex maturation program that a DC must undergo to productively prime a naïve T cell can be broken down to four discrete steps: i) uptake of antigen that is followed by decreased phagocytic capacity; ii) processing and presentation of antigen with enhanced surface expression of MHC II; iii) upregulation of costimulatory molecules (the licensing signals for T cell priming); and iv) migration to the draining lymph node (LN) where the APC interacts with naïve T cells.
TLR activation regulates multiple aspects of DC maturation including enhanced antigen presentation, decreased phagocytic and macropinocytic capacity, and upregulation of the LN-homing chemokine receptor CCR7 [3, 44]. Further MyD88-dependent activation of nuclear factor kappa B (NF-κB) and mitogen-activated kinase (MAPK) signalling pathways following TLR engagement leads to upregulation of co-stimulatory molecules such as CD80 and CD86 as well as production of pro-inflammatory cytokines such as IL-6 and tumor necrosis factor alpha (TNFα, which are fundamental steps in the maturation of DCs [1, 3] (Figure 1b). Studies of both human and murine DCs have demonstrated that the NLRs NOD1 and NOD2 can act in synergy with TLRs to induce enhanced cytokine production from DCs, which in turn impacts T cell differentiation fate [45, 46]. In fact NOD1 and NOD2 induce intracellular signalling cascades similar to the ones initiated downstream of TLR stimulation in response to intracellular PAMPs such as MDP, in particular NF-kB, through the adaptor molecule RIP2 kinase and possibly in conjunction with other NLRs such as NLRP10 [47–49]. Administration of NOD1 and NOD2 ligands in vivo leads to the recruitment and activation of “inflammatory” CD11bhi DCs, although this was not shown to necessarily be a DC-intrinsic effect of NOD1 or NOD2 [50]. Using mixed bone marrow chimeras, Magalhaes et al subsequently demonstrated that although NOD1 & NOD2 activity in stromal cells was sufficient for inducing Th2 immunity in vivo, DC-intrinsic NOD signalling was required to induce optimal and robust T cell responses and therefore it was proposed that the DC responds to NOD ligands and is critical for antigen-presentation but also requires “trans-activation” by stimulated non-hematopoietic cells [51]. Further, NOD1 and NOD2 ligands have been shown to enhance splenic DC cross-priming of CD8+ T cells [52] although more recent work using in vitro generated DCs demonstrated an inhibitory role of NOD1 and NOD2 ligands on DC cross-presentation [53]. Thus, certain NLRs clearly impact DC-intrinsic signalling cascades that overlap with TLR-induced pathways as well as inciting stimuli from the stroma, both of which tailor the DC-induced adaptive immune response.
Although particular NLRs complement TLR-dependent DC activation, there is an emerging trend of NLRs acting instead as negative regulators of immune signalling cascades such as those critical to DC maturation (Figure 2, Table 1) [54]. It was recently demonstrated that NLRC3 inhibited TLR-dependent NF-κB activation by preventing poly-ubiquitination of TRAF6 [55]. Similarly, NLRP12 was found to inhibit TLR-induced activation of both the canonical NF-κB pathway in monocytes [56] by limiting levels of hyperphosphorylated IRAK1 [57] and the non-canonical NF-κB pathway by associating with HSP90 and inducing proteolysis of NF-κB inducing kinase (NIK) [58, 59]. Other members of the pyrin-containing subfamily of NLRs also negatively impact NF-κB activity through inhibition of IKKα phosphorylation in the case of NLRP4 [60] or through as yet unidentified mechanisms in the case of NLRP2 [61]. In addition to its inflammasome-mediated function in colonic epithelium [62], NLRP6 has also been shown to negatively regulate NF-κB and ERK pathways in hematopoietic cells [63]. Although the role of NLRX1 as a negative regulator of NF-κB activation has been debated [64], separate studies have shown that it impaired RIG-I and TLR-mediated activation of NF-κB by interacting with downstream signalling molecules like the adaptor protein MAVS [65] and IKKβ respectively [66].
Figure 2. Beyond IL-1β.
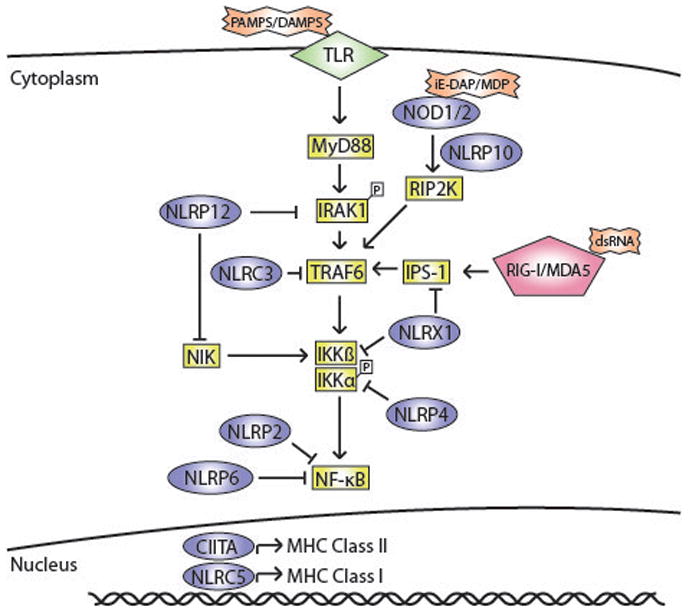
Apart from inflammasomes, NLRs play different roles in modulating DC function. Certain NLRs regulate signalling cascades initiated by other pattern recognition receptors like TLRs and RIG-I/MDA-5. NOD1/2 activation synergistically amplifies TLR-mediated activation of NF-κB thus leading to enhanced DC maturation. Other NLRs negatively regulate NF-κB activation by TLRs and RIG-I/MDA-5. NLRP12, NLRX1 and NLRP4 have been shown to bind and inhibit the activity of specific proteins involved in inflammatory signalling cascades. NLRP2 and NLRP6 have also been shown to impair NF-κB activation but the exact mechanism is yet to be elucidated. Additionally, NLRs like CIITA and NLRC5 can control dendritic cell maturation by transcriptional regulation of MHC Class I and Class II respectively.
The regulatory role of NLRs on TLR-induced signalling cascades is interesting given that TLR signalling also induces the transcription of particular NLRs such as NLRP3 [67](Figure 1)a. We hypothesize that a TLR-NLR feedback loop might exist in which pathogen-induced TLR signalling primes a cell for NLR responsiveness. However, only upon host damage (i.e., release of DAMPs) would NLRs be activated and in turn inhibit the response to on-going TLR ligation. Presumably this feedback loop limits collateral damage to the host but, as will be discussed next, it might also initiate the transition from the early innate immune response to the later adaptive lymphocyte-based immune response, which depends on a fully licenced DC.
Unique functions of NLRs in dendritic cells
In addition to modulating TLR-induced stimulated pathways, NLRs control distinct and unique pathways in DCs that are crucial for their ability to prime a naive T cell. One obvious example of NLR regulation of the antigen presentation function of DCs is the transcription of MHC molecules (Figure 2 and Table 1). CIITA acts as a transcription factor to regulate the expression of MHC class II, as well as several accessory molecules critical in the MHC class II antigen-presentation pathway such as the invariant chain, HLA-DM and HLA-DO [68]. DCs express a unique variant of CIITA that enhances its activity in regulating MHC class II expression [69]. Furthermore, B cells, macrophages and DCs use distinct promoters to regulate CIITA expression, indicating that a cell-specific regulation of MHC class II by CIITA could determine when and how antigen is presented to CD4+ T cells [70]. Studies by three separate groups demonstrated that NLRC5 regulates expression of MHC class I and related molecules such as β2-microglobulin and Tap-1 in various cell types [71, 72], although its role in regulating MHC class I within DCs has been questioned [73]. Beyond regulation of gene transcription, multiple NLRs have been implicated in controlling T cell-dependent adaptive immune responses, presumably through modulation of DC function.
The NLRP3 inflammasome has been found to be essential for immune processes ranging from anti-tumour cytotoxic T cell responses, T cell mediated immune responses to contact allergens and the adjuvant function of aluminium hydroxide during vaccination [74–77]. Although initially proposed to regulate DC maturation and migration, in part through the secretion of IL-1β, the role of NLRP3 in DCs or the adaptive immune response is not currently clear [76, 78–81]. It has been reported that DC migration to LNs is impaired following vaccination with alum in NLRP3-deficient mice [82]; however, the same group subsequently showed alum-induced inflammatory DC recruitment and allergic responses in a NLRP3-independent but uric acid-dependent manner [83]. In murine models of multiple sclerosis, CD4+ T cells primed with NLRP3- or ASC-deficient splenic DCs were unable to migrate into the central nervous system (CNS) to cause disease [84], and NLRP3-deficient mice had reduced paralysis secondary to a defect in Th1/Th17 induction [85]. In contrast, another study found no role for NLRP3 in the activation of autoimmune T cells using the same model systems [86]. Finally, the ion channel M2 of the influenza virus was shown to activate the NLRP3 inflammasome in bone marrow-derived DCs [16], and a functional inflammasome was required for DC and monocyte recruitment during infection [80], but it was also demonstrated that T cell and antibody responses to influenza did not require NLRP3 [80, 87]. Therefore, the role of NLRP3 in DC function or the regulation of adaptive immunity is far from clear, and the wide variation in immune responses found by different groups using the same genetically modified mice represents a major challenge to our understanding of NLRP3 function. In contrast to NLRP3, the study of several other NLRs has yielded some clearer themes related to the function of dendritic cells as APCs and NLR activity.
NLRs and dendritic cell migration
Dendritic cell capture and presentation of antigen is not enough to prime a T cell; the activated DC must be in the appropriate place to interact with an antigen-specific T cell, the lymph node (Box 4). DC maturation in peripheral tissues is accompanied by a coordinated change in expression of multiple chemokine receptors (e.g., reduced CCR1, CCR2, CCR5 and CXCR1 with concomitant up regulation of CCR7 and CXCR4) [88]; of these receptors, CCR7 is the most crucial determinant of DC homing to the LN following peripheral activation, yet it is not sufficient to ensure DC exit from peripheral tissues [44, 89]. In fact relatively little is known about the molecular regulation of DC release from inflamed tissues prior to chemokine-directed movement. Recent data from a number of groups including our own suggest that certain NLRs, rather than TLRs, might regulate key aspects of this poorly characterized biological process.
Text box 4. From infection to immunity.
Naïve T cells remain relatively quiescent during their surveillance of secondary lymphoid organs unless they recognize cognate antigen in the context of MHC on the surface of a matured APC (predominantly dendritic cells) in the lymph node. DCs are a class of professional APCs that contribute to the generation of immunity as well as to the maintenance of peripheral tolerance. DCs can be broadly classified into plasmacytoid DC (pDC) and conventional DC (cDC) subsets, and the latter can be further segregated into lymphoid-resident DCs and tissue-resident DCs. DC subsets are extremely diverse in function varying in their anatomic location, expression of markers, gene expression profiles, cytokine production, PRR expression, antigen uptake, processing and presentation capabilities, migratory capacities and in their ability to elicit distinct T cell programs [123]. During steady state, immature DCs continuously sample their environment for potential antigens. They express low levels of co-stimulatory molecules on their surface, high levels of MHC class II molecules in endosomal compartments and have enhanced endocytic capacities. Upon exposure to a pathogen, DCs undergo a maturation process which includes a substantial decrease in phagocytic capacity, increased surface expression of MHC class II (signal 1 for T cell priming) and co-stimulatory molecules (signal 2 for T cell priming) along with a concomitant production of cytokines as well as enhanced migration to draining lymph nodes. In the LNs, mature DCs present these antigens along with costimulatory molecules to naïve T cells thus initiating the first step in adaptive immunity i.e., T cell priming [3].
We recently discovered that NLRP10, a unique NLR that lacks an LRR and does not appear to form an inflammasome, plays an unexpected but important role in DC emigration [90]. NLRP10-deficient mice had impaired CD4+ helper T cell priming as a result of reduced DC migration to draining LNs during immunization with a wide variety of adjuvants at multiple sites [90]. NLRP10 deficiency solely within the DC is sufficient to inhibit migration out of inflamed tissues without affecting the expression and function of CCR7; however, the mechanism by which tissue emigration is impaired remains to be determined (Figure 1b). Further, caspase-1-deficient DCs fail to appropriately migrate during influenza infection and therefore CD8+ T cell priming is impaired [87]. Caspase-11, another inflammasome-related caspase, directly interacts with regulators of cofilin to modulate actin depolymerization and its loss results in migration defects in vivo and in vitro of multiple cell lineages [91]. Ippagunta and colleagues demonstrated that the inflammasome adaptor molecule ASC controlled F-actin polymerization in dendritic cells by regulating the levels of DOCK2, a guanine nucleotide exchange factor that modulates Rac, thereby influencing DC antigen uptake and movement [92]. However, in a recent addendum, the investigators found that the ASC-deficient mice employed in their investigations had defective expression of DOCK2 for unclear reasons, and that this defect was not consistently seen in ASC-deficient mice from other facilities [93]. Therefore, the role of ASC in DC migration remains unclear. Despite this, from the study of particular NLRs or related molecules important for inflammasome function such as caspases, there appears to be a role for NLRs in controlling various aspects of cell movement. Obviously, DC migration is a fundamental step in the initiation of T cell priming and it is interesting that this one step in the DC maturation program might require a second level of regulation perhaps through NLR activity.
Arthur et al. demonstrated that NLRP12-deficient mice had decreased migration of DCs and neutrophils to CCR7 chemokine gradients accompanied by reduced T cell driven inflammation in a model of skin contact hypersensitivity [94] (Figure 1b). However, in a subsequent study using different models of allergic airway disease, the same group showed that there was no difference in allergen-specific CD4+ T cell responses between NLRP12-deficient and wild-type (WT) mice, suggesting that only a subset of DCs is affected by the loss of NLRP12 [95]. Indeed, Langerhans cell movement from the skin appears to be defective in NLRP12-deficient mice [94], whereas NLRP10-deficient mice have a targeted defect in a DC subset that specifically primes naïve CD4+ T cells, the tissue migratory CD11b+ DCs [90]. Perhaps different DC subsets differentially respond to immune insults through the use of distinct NLRs, thereby tailoring the type of lymphocyte-driven adaptive immune response to the inciting agent (Box 4). Although the molecular pathways regulated by NLRs and inflammasomes in DCs are not well defined, an interesting functional theme for these molecules is emerging; the shared domain structures within the NLR family are used in disparate processes many of which control aspects of dendritic cell function, including MHC transcription and modification of TLR-induced maturation and migration (Table 1). Therefore, the original paradigm that TLR stimulation induces a DC maturation program sufficient for T cell priming seems to be incomplete.
PRR cooperation model
Determination of the migration fate of mature DCs from sites of inflammation is clearly a step that must be carefully regulated to avoid responses to innocuous antigens such as self-molecules or allergens. One major function of the NLRs might be to orchestrate DC movement and thereby regulate one critical aspect of productive innate-adaptive interactions that the TLRs do not [87, 90, 91, 95]. Although TLR activation has been shown to be sufficient for induction of adaptive immunity, the requirement for NLRs in these studies were not addressed [96–98]. It is well-accepted that expression of essential surface molecules for T cell priming are readily upregulated by TLR stimulation including MHC, CCR7 and costimulatory molecules; however, this DC maturation program might be incomplete in that a mature DC in the tissue would not be able to prime a naïve T cell in the LN (Figure 1b). For particular NLRs such as NLRP3, TLR stimulation is also necessary to prime the cell for responses to NLR ligands but again is not sufficient to induce NLR activity (Figure 1a). Therefore TLR stimulation would prime the DC for responsiveness both in the induction of a DC maturation program as well as situating an NLR to respond to damage. By fully licensing a DC for T cell priming the subsequent detection of DAMPs by NLRs would thereby fundamentally shift the nature of the immune response from primarily innate in nature to one that is adaptive. This “division of labor” between the NLRs and TLRs might act as a dual level of regulation to ensure that the powerful adaptive branch of the immune system is only activated upon foreign (e.g. PAMPs) and pathogenic (e.g., release of DAMPs) insults (Figure 1b).
There are numerous “two-hit” or “two-signal” models in biology, from cancer development to inflammasome triggering (Figure 1a) to T cell activation [22, 99, 100]. Here we propose a two-hit model of innate instruction of adaptive immunity, in which TLR activation primes a DC to mature (like releasing one’s foot from the brake of a car), whereas NLR activation licenses the DC to take those signals to awaiting naïve T cells in the lymph node (like putting one’s foot on the gas pedal of said car) [101]. Both steps are required, just as in other models, to ensure specificity of the response, particularly if the response is metabolically costly and potentially dangerous to the host [102]. For example, aspiration of a small amount of Gram-positive mouth flora into the sterile lower airways in the absence of significant tissue damage should induce a rapid but localized innate immune response that limits bacterial spread. Conversely, DAMP activation of particular NLR family members through crush injuries or burns should not induce a T cell response, as the target could be a self-molecule. However, streptococcal lobular pneumonia with significant bacterial burden as well as host tissue damage might require more than a phagocytic and cytokine-induced innate inflammatory response in the lung. Under these circumstances, DCs can employ all the requisite machinery of antigen processing and migration to induce a distal T cell response in the lymph nodes. Elegant studies by Garaude et al. recently provided support for this theory using flagellin-expressing tumor cells; they demonstrated that although TLR5-mediated recognition of flagellin by macrophages was crucial for rapid clearance of the tumor cells, both TLR5 and NLRC4 signalling in the DC led to activation and efficient antigen presentation to tumor-specific CD4+ and CD8+ T cells [103]. As highlighted earlier, NLR and TLR cooperation during an immune response has been well described; in the case of NOD1 and NOD2, which induce overlapping signalling cascades with many TLRs, synergistic immune cell activation appears to fine-tune or augment the innate immune response [104, 105]. By contrast, our PRR cooperation model relies on the sensing of two distinct classes of immune activators, PAMPs versus DAMPs, generating a fundamental shift in the type of immune response from innate to adaptive.
The PRR cooperation model is not novel but instead is a merging of two “old” theories about the regulation of immunity, the Janeway Pattern Recognition Theory [42] and the Matzinger Danger Theory [106]. Just as we began with a description of PAMPs and DAMPs, these two theories fundamentally rely on the detection of PAMPs (C. Janeway) versus DAMPs (P. Matzinger) and diverge in their interpretation of the importance of self versus non-self discrimination in the generation of adaptive immune responses. As has been previously described, both models explain pieces of the observed rules of immunity [107], but in combination with other existing models such as the Missing Self [108] and Guard Hypothesis [40], we believe a holistic description of the rules governing immunity can be described. This is however not a failsafe mechanism; PRRs detect an incredible range of molecules, and it is therefore plausible that a two-hit PRR requirement could be inadvertently fulfilled even if disadvantageous to the host, most notably with the development of autoimmunity. Clearly more work must be done to determine whether a PRR cooperation model can indeed account for when an adaptive immune response is appropriately (or inappropriately) generated, but the formation of a molecular understanding of the immunological pathways governing this process will certainly advance our ability to positively or negatively intervene in human immune responses.
Concluding remarks
Our understanding of the NLRs has grown exponentially over the past 10 years, as NLRs have been shown to have crucial functions in a wide variety of disease processes [22]. However, for many of the 22 known human NLR family members, their triggers and physiologic function remain unknown. Debate exists about which NLRs form true inflammasomes, the direct ligand regulating activity of many NLRs including NLRP3 (Figure 1a) and the function of inflammasomes in an array of adaptive immune responses ranging from vaccination with the adjuvant aluminum hydroxide to generation of influenza immunity. However, emerging studies are starting to define on a molecular level the regulation and activity of the NLRs and this will greatly aid in the answering of many of these questions. Further, other studies have suggested a new and perhaps unexpected role for NLRs in fine-tuning dendritic cell-dependent induction of adaptive immunity (Table 1). These findings in particular have led us to speculate on how the growing list of pattern recognition receptors of the innate immune system tailor an appropriate adaptive immune response to the wide array of immunological insults. As different PRR families typically induce different sets of inflammatory programs, the integration of combined PRR triggering could be one mechanism by which an appropriate immune response is tailored [104, 105].
We propose that a combination of two categorically different “hits” of molecules that are foreign with molecules released by damaged host cells (loosely categorized as PAMP and DAMP, respectively) would be sufficient to fulfil the tenets of a PRR cooperation model for adaptive immunity (Figure 1b). It is this latter, more dangerous and metabolically costly immune response that seems to require the interplay of multiple innate immune receptor pathways. Although there are some studies to support this model [90, 103], much more work will be needed to dissect the relative roles of the different PRRs in regulating innate versus adaptive immune responses.
Future Questions.
How do NLRs regulate dendritic cell movement during immune responses?
What is the direct activating ligand of NLRP3?
Synergistic activation of which PRR sets elicit an effective adaptive immune response to a particular pathogen?
Do differential expression patterns of NLRs regulate subset-specific functions of dendritic cells?
How is an immune response appropriately tailored to an invading pathogen (e.g., a PAMP) versus a self-molecule released during cell death (e.g., a DAMP) if many of the downstream signalling cascades overlap?
Acknowledgments
This work was supported by K08AI085038, UL1RR024139 and a grant from the National Blood Foundation. We would like to thank S. Calabro, D. Liu, A.M. Rhebergen, SCS. Cassel and A. Williams for helpful discussion and review of this manuscript.
Glossary
- Adaptive immunity
otherwise known as acquired immunity, is a T and B lymphocyte-mediated response against encountered antigens that generates immunological memory. The generation of memory T cells and B cells helps the body initiate robust and rapid responses on subsequent encounters with the pathogen
- Autophagy
cell intrinsic process leading to the endosomal degradation of macromolecules (microautophagy) and cell organelles (macroautophagy). Autophagic processes contribute to antigen presentation as well as activation of DCs via involvement of TLR signalling cascades
- CARD domain
caspase activation and recruitment domains are present on many PRRs, notably NLRs, ASC and caspases. One function of CARD domains is to facilitate inflammasome formation by homotypic domain interactions to create large protein complexes
- Cross presentation
presentation of extracellular antigens by MHC class I molecules to CD8+ T cells to elicit immune responses. Cross presentation plays a major role in initiating an immune response against viruses and tumors
- Damage-associated molecular patterns (DAMP)
a broad group of molecules endogenously expressed that possess the ability to initiate an inflammatory response. These molecular patterns arise from stress to cells such as damage or heat-shock
- Danger model
proposed by Polly Matzinger, it states that antigen presenting cells respond to endogenous danger signals from cells undergoing stress in order to initiate adaptive immunity
- Guard theory
first proposed to deal with plant immunology, it is the theory that the immune system responds not only to the direct detection of pathogens but also to consequences of pathogenesis such as cytoskeletal disruption or endocytic trafficking
- Human leukocyte antigen (HLA)
cell surface molecules responsible for presenting molecular epitopes to leukocytes. This antigen presentation is critical to the priming of lymphocytes by dendritic cells. In non-humans these molecules are referred to as MHC (see below)
- Inflammasome
multimolecular complex required for processing precursors of pro-inflammatory cytokines and pyroptosis. The composition of oligomers is dependent on which PRRs are activated in response to PAMPs or DAMPs. A typical inflammasome contains a caspase (caspase 1 or 11), ASC and an NLR molecule
- Innate immunity
is a body’s ability to quickly respond to a broad spectrum of pathogens. This is a general response to categories of insults, for example classes of pathogens expressing particular PAMPs (see below). Unlike the adaptive immune system, the response to pathogens does not confer long-term protection through the generation of long-lived clonal cells. Also in contrast to adaptive immunity, the innate receptors responding to particular insults are germline encoded rather than generated through DNA recombination, as is the case of lymphocyte receptors
- Major histocompatibility complex (MHC)
cell surface molecules responsible for presenting molecular epitopes to leukocytes. This antigen presentation is critical to priming T cells by dendritic cells. Three major classes exist and are designated MHC class I, II or III. In humans these molecules are referred to as HLA (see above)
- Missing self theory
proposed theory of how cells of the immune system “recognize” the absence of the expression of a normal host molecule. The paradigm was established to describe the inhibition of natural killer (NK) cell activation to host tissue; when tumors or viral infections result in the loss of surface inhibitory molecules such as MHC, NK cells destroy the altered host cell. This paradigm in fact applies to a number of other immunologic detection methods of the innate immune system including the constitutive inhibition of complement activation on the surface of host cells and the macrophage inhibitory receptor SIRPα
- Pathogen-associated molecular patterns (PAMP)
exogenous molecules of pathogen-origin (viral, fungal or bacterial) that are composed of foreign (non-self) motifs and typify groups of pathogens (e.g., the expression of lipopolysaccharide on all Gram-negative bacteria). An alternative name for PAMPs has arisen due to the existence of molecules that stimulate innate immune receptors on microbes that are commensal rather than pathogenic – microbe associate molecular patterns (MAMPs)
- Pattern recognition theory
proposed by Charles Janeway, it lays the framework for how the innate immune system communicates with and regulates adaptive immunity. It is based on the principle of pattern recognition of PAMPs through non-clonal receptors expressed by leukocytes and other cells
- Pattern recognition receptor (PRR)
an umbrella term referring to receptors capable of detecting molecular patterns facilitating immune responses. Families of these receptors include (but are not limited to) TLRs, NLRs, RLRs, ALR, and CLRs. Again, alternative terms exist to define molecules that respond to innate immune triggers but don’t necessarily act as receptors; pattern recognition molecules (PRM) refer to the larger group of secreted and membrane-bound molecules that induce innate immune responses
- Reactive oxygen species (ROS)
chemically reactive molecules containing oxygen typically released as an innate response during infection, damage or temperature shock. ROS induce oxidative stress in the molecules it reacts with and in bacteria can cause cell death through membrane disruption
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Strowig T, et al. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 3.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 4.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 5.van den Berg TK, van der Schoot CE. Innate immune ‘self’ recognition: a role for CD47-SIRPalpha interactions in hematopoietic stem cell transplantation. Trends Immunol. 2008;29:203–206. doi: 10.1016/j.it.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Deban L, et al. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res. 2011;343:237–249. doi: 10.1007/s00441-010-1018-0. [DOI] [PubMed] [Google Scholar]
- 7.Rajalingam R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol Biol. 2012;882:391–414. doi: 10.1007/978-1-61779-842-9_23. [DOI] [PubMed] [Google Scholar]
- 8.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahrens S, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. 2012;36:635–645. doi: 10.1016/j.immuni.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Acton SE, et al. Podoplanin-Rich Stromal Networks Induce Dendritic Cell Motility via Activation of the C-type Lectin Receptor CLEC-2. Immunity. 2012;37:276–289. doi: 10.1016/j.immuni.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos HJ, Gale M. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol. 2011;1:167–176. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. 2011;243:109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- 13.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khare S, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36:464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lightfield KL, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichinohe T, et al. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nour AM, et al. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect Immun. 2009;77:1262–1271. doi: 10.1128/IAI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wickliffe KE, et al. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10:332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutterwala FS, et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao EA, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornung V, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams A, et al. The role of NOD-like Receptors in shaping adaptive immunity. Current Opinion in Immunology. 2010;22:34–40. doi: 10.1016/j.coi.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen IC, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joly S, et al. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3578–3581. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamkanfi M, et al. Fungal zymosan and mannan activate the cryopyrin inflammasome. J Biol Chem. 2009;284:20574–20581. doi: 10.1074/jbc.M109.023689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lightfield KL, et al. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79:1606–1614. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girardin SE, et al. Identification of the critical residues involved in peptidoglycan detection by Nod1. J Biol Chem. 2005;280:38648–38656. doi: 10.1074/jbc.M509537200. [DOI] [PubMed] [Google Scholar]
- 28.Mo J, et al. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem. 2012;287:23057–23067. doi: 10.1074/jbc.M112.344283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanabe T, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 2004;23:1587–1597. doi: 10.1038/sj.emboj.7600175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 31.Ciraci C, et al. Control of innate and adaptive immunity by the inflammasome. Microbes Infect. 2012 doi: 10.1016/j.micinf.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juliana C, et al. Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu B, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornung V, Latz E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol. 2010;40:620–623. doi: 10.1002/eji.200940185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krysko DV, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–164. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 40.Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 41.Steinman R, Inaba K. Immunogenicity: role of dendritic cells. Bioessays. 1989;10:145–152. doi: 10.1002/bies.950100503. [DOI] [PubMed] [Google Scholar]
- 42.Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 44.Randolph GJ, et al. Migration of dendritic cell subsets and their precursors. Annu Rev Immunol. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 45.Schwarz H, et al. TLR8 and NOD signaling synergistically induce the production of IL-1β and IL-23 in monocyte-derived DCs and enhance the expression of the feedback inhibitor SOCS2. Immunobiology. 2012 doi: 10.1016/j.imbio.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 46.Fritz JH, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity. 2007;26:445–459. doi: 10.1016/j.immuni.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Inohara N, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 48.Lautz K, et al. NLRP10 enhances Shigella-induced pro-inflammatory responses. Cellular microbiology. 2012 doi: 10.1111/j.1462-5822.2012.01822.x. [DOI] [PubMed] [Google Scholar]
- 49.Fritz JH, et al. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol. 2005;35:2459–2470. doi: 10.1002/eji.200526286. [DOI] [PubMed] [Google Scholar]
- 50.Magalhaes JG, et al. Essential role of Rip2 in the modulation of innate and adaptive immunity triggered by Nod1 and Nod2 ligands. Eur J Immunol. 2011;41:1445–1455. doi: 10.1002/eji.201040827. [DOI] [PubMed] [Google Scholar]
- 51.Magalhaes JG, et al. Nucleotide oligomerization domain-containing proteins instruct T cell helper type 2 immunity through stromal activation. Proc Natl Acad Sci U S A. 2011;108:14896–14901. doi: 10.1073/pnas.1015063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asano J, et al. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J Immunol. 2010;184:736–745. doi: 10.4049/jimmunol.0900726. [DOI] [PubMed] [Google Scholar]
- 53.Wagner CS, Cresswell P. TLR and nucleotide-binding oligomerization domain-like receptor signals differentially regulate exogenous antigen presentation. J Immunol. 2012;188:686–693. doi: 10.4049/jimmunol.1102214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ting JPY, et al. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–290. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneider M, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. 2012 doi: 10.1038/ni.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaki MH, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. 2011;20:649–660. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams KL, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lich JD, et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol. 2007;178:1256–1260. doi: 10.4049/jimmunol.178.3.1256. [DOI] [PubMed] [Google Scholar]
- 59.Arthur JC, et al. Heat shock protein 90 associates with monarch-1 and regulates its ability to promote degradation of NF-kappaB-inducing kinase. J Immunol. 2007;179:6291–6296. doi: 10.4049/jimmunol.179.9.6291. [DOI] [PubMed] [Google Scholar]
- 60.Fiorentino L, et al. A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1beta. J Biol Chem. 2002;277:35333–35340. doi: 10.1074/jbc.M200446200. [DOI] [PubMed] [Google Scholar]
- 61.Fontalba A, et al. NLRP2, an inhibitor of the NF-kappaB pathway, is transcriptionally activated by NF-kappaB and exhibits a nonfunctional allelic variant. J Immunol. 2007;179:8519–8524. doi: 10.4049/jimmunol.179.12.8519. [DOI] [PubMed] [Google Scholar]
- 62.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anand PK, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488:389–393. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tattoli I, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore CB, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 66.Xia X, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bauernfeind FG, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.LeibundGut-Landmann S, et al. Mini-review: Specificity and expression of CIITA, the master regulator of MHC class II genes. Eur J Immunol. 2004;34:1513–1525. doi: 10.1002/eji.200424964. [DOI] [PubMed] [Google Scholar]
- 69.Nickerson K, et al. Dendritic cell-specific MHC class II transactivator contains a caspase recruitment domain that confers potent transactivation activity. J Biol Chem. 2001;276:19089–19093. doi: 10.1074/jbc.M101295200. [DOI] [PubMed] [Google Scholar]
- 70.LeibundGut-Landmann S, et al. MHC class II expression is differentially regulated in plasmacytoid and conventional dendritic cells. Nat Immunol. 2004;5:899–908. doi: 10.1038/ni1109. [DOI] [PubMed] [Google Scholar]
- 71.Biswas A, et al. Cutting edge: impaired MHC class I expression in mice deficient for nlrc5/class I transactivator. J Immunol. 2012;189:516–520. doi: 10.4049/jimmunol.1200064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neerincx A, et al. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J Immunol. 2012;188:4940–4950. doi: 10.4049/jimmunol.1103136. [DOI] [PubMed] [Google Scholar]
- 73.Staehli F, et al. NLRC5 deficiency selectively impairs MHC class I-dependent lymphocyte killing by cytotoxic T cells. J Immunol. 2012;188:3820–3828. doi: 10.4049/jimmunol.1102671. [DOI] [PubMed] [Google Scholar]
- 74.Eisenbarth SC, et al. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe H, et al. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956–1963. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 76.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 77.Li H, et al. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allen IC, et al. Analysis of NLRP3 in the development of allergic airway disease in mice. J Immunol. 2012;188:2884–2893. doi: 10.4049/jimmunol.1102488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKee AS, et al. Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J Immunol. 2009;183:4403–4414. doi: 10.4049/jimmunol.0900164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomas PG, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Franchi L, Núñez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. 2008;38:2085–2089. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kool M, et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 83.Kool M, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–540. doi: 10.1016/j.immuni.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 84.Inoue M, et al. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109:10480–10485. doi: 10.1073/pnas.1201836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gris D, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–981. doi: 10.4049/jimmunol.0904145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shaw PJ, et al. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4614. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ichinohe T, et al. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sallusto F, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 89.Ohl L, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21:279–288. doi: 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 90.Eisenbarth SC, et al. NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature. 2012;484:510–513. doi: 10.1038/nature11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li J, et al. Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat Cell Biol. 2007;9:276–286. doi: 10.1038/ncb1541. [DOI] [PubMed] [Google Scholar]
- 92.Ippagunta SK, et al. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization. Nat Immunol. 2011;12:1010–1016. doi: 10.1038/ni.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ippagunta SK, et al. Addendum: defective Dock2 expression in a subset of ASC-deficient mouse lines. Nat Immunol. 2012;13:701–702. doi: 10.1038/ni.2389. [DOI] [PubMed] [Google Scholar]
- 94.Arthur JC, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol. 2010;185:4515–4519. doi: 10.4049/jimmunol.1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Allen IC, et al. Characterization of NLRP12 during the development of allergic airway disease in mice. PLoS ONE. 2012;7:e30612. doi: 10.1371/journal.pone.0030612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eisenbarth SC, et al. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schnare M, et al. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 98.Krishnaswamy JK, et al. Toll-like receptor-2 agonist-allergen coupling efficiently redirects th2 cell responses and inhibits allergic airway eosinophilia. Am J Respir Cell Mol Biol. 2012;47:852–863. doi: 10.1165/rcmb.2011-0414OC. [DOI] [PubMed] [Google Scholar]
- 99.Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999;96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.My father’s (G.S.E.) analogy for a similar situation potentially underlying autoimmune processes is that aspects of the immune system (such as germ line encoded T cell receptor segments specific for self peptides) are like having a loaded gun in your house – it can be used to fend off invaders but can also hurt those within the household; therefore safety locks must be built in to prevent inadvertent firing.
- 102.Nish S, Medzhitov R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity. 2011;34:629–636. doi: 10.1016/j.immuni.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Garaude J, et al. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Sci Transl Med. 2012;4:120ra116. doi: 10.1126/scitranslmed.3002868. [DOI] [PubMed] [Google Scholar]
- 104.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nature Reviews Immunology. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 105.Kufer TA, Sansonetti PJ. Sensing of bacteria: NOD a lonely job. Curr Opin Microbiol. 2007;10:62–69. doi: 10.1016/j.mib.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 106.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 107.Medzhitov R. Approaching the asymptote: 20 years later. Immunity. 2009;30:766–775. doi: 10.1016/j.immuni.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 108.Karre K, et al. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 109.Lee SJ, et al. The IFN-gamma-induced transcriptional program of the CIITA gene is inhibited by statins. Eur J Immunol. 2008;38:2325–2336. doi: 10.1002/eji.200838189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pashenkov MV, et al. Muropeptides trigger distinct activation profiles in macrophages and dendritic cells. Int Immunopharmacol. 2010;10:875–882. doi: 10.1016/j.intimp.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 111.Franchi L, et al. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Levinsohn JL, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vladimer GI, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bruey JM, et al. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J Biol Chem. 2004;279:51897–51907. doi: 10.1074/jbc.M406741200. [DOI] [PubMed] [Google Scholar]
- 115.Cui J, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ting JP, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Margulis L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth. Yale University Press; 1970. [Google Scholar]
- 118.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 119.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 121.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 122.Jounai N, et al. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646–1655. doi: 10.4049/jimmunol.1001654. [DOI] [PubMed] [Google Scholar]
- 123.Turley SJ, et al. The stromal and haematopoietic antigen-presenting cells that reside in secondary lymphoid organs. Nat Rev Immunol. 2010;10:813–825. doi: 10.1038/nri2886. [DOI] [PubMed] [Google Scholar]