

Abstract
Hypoxia-inducible factor (HIF)-1α is a master regulator of inflammatory activities of myeloid cells, including neutrophils and macrophages. These studies examine the role of myeloid cell HIF-1α in regulating asthma induction and pathogenesis, and for the first time, evaluate the roles of HIF-1α and HIF-2α in the chemotactic properties of eosinophils, the myeloid cells most associated with asthma. Wild-type (WT) and myeloid cell-specific HIF-1α knockout (KO) C57BL/6 mice were studied in an ovalbumin (OVA) model of asthma. Administration of the pharmacological HIF-1α antagonist YC-1 was used to corroborate findings from the genetic model. WT, HIF-1α, and HIF-2α KO eosinophils underwent in vitro chemotaxis assays. We found that deletion of HIF-1α in myeloid cells and systemic treatment with YC-1 during asthma induction decreased airway hyperresponsiveness (AHR). Deletion of HIF-1α in myeloid cells in OVA-induced asthma also reduced eosinophil infiltration, goblet cell hyperplasia, and levels 34 of cytokines IL-4, IL-5, and IL-13 in the lung. HIF-1α inhibition with YC-1 during asthma induction decreased eosinophilia in bronchoalveolar lavage, lung parenchyma, and blood, as well as decreased total lung inflammation, IL-5, and serum OVA-specific IgE levels. Deletion of HIF-1α in eosinophils decreased their chemotaxis, while deletion of the isoform HIF-2α led to increased chemotaxis. This work demonstrates that HIF-1α in myeloid cells plays a role in asthma pathogenesis, particularly in AHR development. Additionally, treatment with HIF-1α inhibitors during asthma induction decreases AHR and eosinophilia. Finally, we show that HIF- 1α and HIF-2α regulate eosinophil migration in opposing ways.
Keywords: Hypoxia inducible factor (HIF)-1α, Asthma, Allergic inflammation, Eosinophils, Chemotaxis, Airway hyperresponsiveness
Introduction
Asthma is an inflammatory disease of the airways, notorious for causing bronchospasm or airway hyperresponsiveness (AHR), in response to a variety of triggers. AHR is the most clinically relevant finding in asthma [1], as it causes the most asthma fatalities (3,447 per year in the USA) and emergency room visits (1.75 million per year) that contribute to high healthcare costs [2]. Eosinophils are one of the myeloid cells at the heart of this inflammatory disease, and the degree of sputum eosinophilia in an asthmatic patient correlates with disease activity [3, 4]. Many additional myeloid cells are found in the tissues and bronchoalveolar lavage (BAL) of asthmatics (macrophages, mast cells, lymphocytes, and neutrophils) and are also considered important in asthma pathogenesis [5]. Signaling between myeloid cells and non-myeloid cells, such as smooth muscle, epithelial, and goblet cells, likely drives asthma development and progression of disease.
Notably, asthma is a heterogeneous disease, with an assortment of inflammatory phenotypes [6] and a variety of responses to therapy [7, 8]. In general, inflammatory (myeloid) cells are targeted by today’s limited arsenal of therapeutic agents, such as systemic and inhaled steroids, and leukotriene inhibitors. To decrease systemic side effects of such therapies and provide more targeted treatment, suppression of inflammation via blockade of specific molecular pathways, in myeloid cells in particular, is an attractive strategy. In this study, we target hypoxia-inducible factor (HIF)-1α in myeloid cells and describe the role of HIF alpha subunits in eosinophil function.
HIF-1α is a transcription factor found in all mammalian cells and regulates over 100 functional genes. There is mounting evidence that HIF-1α is an important regulator of inflammation in asthma [9–13]. The transcription factor is upregulated in the smooth muscle, submucosa, and BAL cells of asthmatic patients [9, 12], and haploinsufficient (partial knockout) mice demonstrate decreased eosinophilia [13]. HIF-1α is known to be a master regulator of the energetics and inflammatory capacity of myeloid cells and has been studied extensively in neutrophils and macrophages [14–16]. However, it is unknown what role HIF-1α plays in eosinophils, a major effector cell in allergic inflammation and asthma.
Even less is known about the function of HIF-2α. It has been shown in some systems that HIF-2α acts in a complementary fashion to HIF-1α [17], while in other systems, researchers have identified completely separate gene regulatory roles [18, 19]. HIF-1α and HIF-2α levels are controlled by prolyl hydroxylases and oxygen levels, in a subunit-specific way [20]. Because suppression of one HIF α subunit may lead to increased effects of another (via decreased competition for their HIF-1β counterpart or via loss of regulation of one α subunit by another), we set out to tease apart the functional differences in the alpha subunits.
Utilizing a conditional KO in which HIF-1α is markedly reduced in cells of the myeloid lineage, an OVA mouse 104 model of asthma, and pharmacological inhibition of HIF-1α, we conducted a series of in vivo experiments to address the role of HIF-1α in asthma pathogenesis. Then utilizing conditional KO mice in which HIF-1α or HIF-2α is not expressed in leukocytes, we conducted a series of in vitro experiments to evaluate the role of HIF-1α and HIF-2α in regulating eosinophil chemotaxis.
Methods
Reagents
Ovalbumin (OVA), aluminum hydroxide (alum), methacholine (MCH), HIF-1α inhibitor YC-1, and HIF-1α agonist L-mimosine were purchased from Sigma. CHO-stem cell factor (SCF) cells were generously donated by Mark Kamps, University of California at San Diego (UCSD). Eotaxin-1 and MIP-1α (R&D), and IL-5 and FLT-3L (PeproTech) were purchased.
Mouse models of asthma
C57BL/6 HIF-1α lysMcre, C57BL/6 HIF-1α tie2cre, and C57BL/6 HIF-2α tie2cre mice were generated as previously described [14, 21, 22]. Tie2cre KO mice were solely used for the generation of bone marrow (BM) eosinophils, while lysM-cre KO mice were used for in vivo studies. Six- to eight-week-old female mice underwent asthma induction via intraperito-neal (i.p.) injection of 50 μg OVA and 500 μg alum in 200 μL phosphate-buffered saline (PBS) on days 0 and 12. Mice were challenged with 20 μg intranasal (i.n.) OVA in 50 μL PBS on days 26, 28, and 30, and were sacrificed on day 31. Pharmacological HIF-1α antagonist (500 μg YC-1 in 200 μL PBS with 5 % DMSO) was given i.p. on days 26–31, 1 h prior to i.n. OVA challenges, or was given one time only on day 31, 1 h prior to MCH challenge. Mice were sedated with ketamine (100 mg/kg) and xylazine (10 mg/kg) underwent tracheostomy, and flexiVent measurements of airway resistance were obtained utilizing inhaled MCH challenge. Intracardiac bleed and BAL (800 μL PBS) were performed. BM and lungs were harvested. The right lung was snap-frozen for RNA, and the left lung was fixed for histology. The Institutional Animal Care and Use Committee at UCSD approved all animal protocols.
Quantification of cell types in BAL, blood, and BM
Blood samples underwent red blood cell lysis followed by pelleting, along with BAL and BM samples, at 3,000 rpm for 4 min. All pellets were resuspended in 1 mL PBS and cytospun at 1,000 rpm for 3 min. Cells were stained with Giemsa–Wright (Protocol). Slides were deidentified and andomized. Greater or equal to 400 cells from each slide were counted via light microscopy.
Analysis of lung inflammation, eosinophilia, and goblet cell hyperplasia via histology
The left lung was inflated with 4 % paraformaldehyde (PFA) in PBS at 25 cm of water pressure for 15 min and placed in 4 % PFA at 4 °C for 24–48 h. Lungs underwent sectioning, H&E, periodic acid-Schiff (PAS), Masson’s trichrome, and toluidine blue staining. Slides were deidentified and randomized for blinded grading of lung inflammation, eosinophilia, goblet cell hyperplasia (GCH), fibrosis, and mast cell numbers. Lung inflammation score was derived from Wachtel et al. 2009 [23] and included evaluation of airway (peribronchial, mucosal, and alveolar septa), vascular (perivascular and occlusion), and subpleural inflammation utilizing a 0–3 scale (0 = none, 1 =mild, 2 = moderate, 3 = severe). Lung eosinophilia on H&E sections, GCH on PAS sections, fibrosis on Masson’s trichrome sections, and mast cell presence on toluidine blue sections were also graded on a 0–3 scale. No significant fibrosis was seen, and almost no mast cells were visualized in any lung sections.
Analysis of cytokines and NO in BAL, and IgE in serum
Undiluted BAL was assayed for IL-4, IL-5, and IL-13 using ELISA DuoSets (R&D). Concentration of NO in undiluted BAL was determined via the Sievers NO Analyzer (GE). Serum was diluted 1:100 for total IgE determination and 1:10 for OVA-specific IgE quantification (Chondrex).
Quantitative RT–PCR analysis
Total RNA was extracted using TRIzol (Invitrogen), RNeasy mini kit (Qiagen), and DNase (BioRad) according to manufacturer’s instructions. RNA was evaluated for purity by 260/280 (NanoDrop, Thermo Scientific) and agarose gel electrophoresis. cDNA was prepared (Superscript III, Invitrogen) and amplified by SYBR Green (Applied Biosystems) with Qiagen primers. 18S was the housekeeping gene. Efficiencies of all primers were within 85–100 %. Relative gene expression was calculated using ΔΔCt analysis. In mouse lung tissue, this was based on no-OVA controls to demonstrate changes due to OVA challenge; OVA mouse groups were then compared to one another. For eosinophil samples, WT was compared to HIF-1α KO, and the percent knock-down was determined with (1 − ΔΔCq) × 100.
Eosinophils
BM derived eosinophils were generated as previously described [24]. In brief, BM was isolated from C57BL/6 HIF-1α tie2cre KO or HIF-2α tie2cre KO (HIF-1α or HIF-2α not expressed in leukocytes or endothelial cells, respectively) and WT mice. BM was treated with ACK lysis buffer (Invitrogen), followed by culturing at 10e6 cells per milliliter in RPMI 1640, 20 % FBS, 100 IU/mL penicillin, 10 μg/mL streptomycin, 2 mM glutamine, 25 mM HEPES, 1 × NEAA, 1 mM sodium pyruvate (Invitrogen), 50 μm 2-ME (Sigma), 1 % SCF and 100 ng/mL FLT3-ligand from days 0–4, and 10 ng/mL IL-5 from days5–13 at 37 °C with 5 % CO2. On days 11–13, 10e5 cells were cytospun, stained with Giemsa–Wright, and eosinophil percentage determined by light microscopy. Ninety to ninety eight percent pure eosinophils were used for all assays. Knockdown efficiency in HIF-1α KO eosinophils was quantified at 99.9 % via qPCR. WT and HIF-1α KO eosinophils were incubated for 18 h in a hypoxia chamber (STEMCELL; 1 % O2) followed by total protein extraction. Protein was run on NuPAGE 10 % Bis–Tris gels (Invitrogen) and underwent semi-dry transfer to nitrocellulose paper (BioRad). The blots were incubated with anti-HIF-1α (Novus) and anti-PARP (nuclear housekeeping protein; Pierce) antibodies, followed by fluorescent secondary antibodies, with the fluorescence detected with the LI-COR system.
Chemotaxis
BM eosinophils were treated with HIF-1α agonist (100 μM mimosine) or antagonist (15 μM YC- 1) for 3 h. Eotaxin-1 (CCL11; binds primarily CCR3) or MIP-1α (CCL3; binds CCR1 and CCR5) at 0, 10, and 50 ng/mL in 600 μL was placed in lower chambers with 6.5-mm transwell inserts with 5-μm pore polycarbonate membranes (Costar). The 10e5 cells in 100 μL were placed in upper wells and plates incubated at 37 °C with 5 % CO2 for 3 h. Media from lower chambers were taken and cells counted by trypan blue exclusion under light microscopy. All conditions were performed in triplicate and all experiments repeated three times.
Statistical tests
ANOVA one-way analysis and Bonferroni’s multiple comparison were used for eosinophil migration, NO levels, and ELISA. Airway resistance and BAL cellularity were evaluated with two-way repeated measures ANOVA, and qPCR by two-way ANOVA, both followed by Bonferroni post-tests. For histology scores, one-way ANOVA with Kruskal–Wallis followed by Dunn’s multiple comparison was used.
Results
HIF-1α myeloid KO OVA mice have decreased airway hyperresponsiveness
Mice rendered allergic to OVA have been widely studied, and they serve as a prototypic asthma pathogenesis model [25, 26]. OVA HIF-1α KO mice had less AHR than OVA WT mice after methacholine challenge (Fig. 1a), rather their AHR was similar to no-OVA controls. No-OVA KO and WT mouse controls were indistinguishable; thus, the results were combined. These data provide a first demonstration that HIF-1α in myeloid cells is necessary for the development of AHR.
Fig. 1.
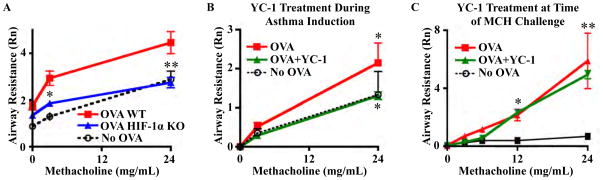
Knocking out HIF-1α expression in myeloid cells and systemic treatment during asthma induction with HIF-1α inhibitor YC-1 decreases AHR. a AHR was decreased in OVA HIF-1α KO mice (*P < 0.01, **P < 0.001). b AHR was the same in YC-1-treated mice as no-OVA controls (P > 0.05). Non-treated OVA controls had higher AHR than controls and YC-1-treated mice (*P < 0.01). c Treatment of OVA mice with YC-1 once, 1 h prior to MCH challenge, did not alter AHR. Both OVA and OVA + YC-1 × 1 groups had significantly higher AHR than no- OVA controls (*P < 0.05, **P < 0.001). Eight WT and 14 KO mice were used in our WT versus KO OVA mouse model of asthma, and four mice per group were used in our treatment models
Mice treated with HIF-1α antagonist during asthma induction have decreased airway hyperresponsiveness
The suppression of one of the most important pathologic findings in asthma—AHR—via the KO of HIF-1α in myeloid cells led us to consider whether pharmacological targeting of HIF-1α could ameliorate asthma pathophysiology. YC-1 specifically inhibits HIF-1α by blocking expression at the post-transcriptional level [27] and by stimulating factor inhibiting HIF binding to the COOH-terminal transactivation domain of HIF-1α [28]. We observed that C57BL/6 OVA mice treated with the HIF-1α antagonist YC-1 during asthma induction had AHR identical to control no-OVA mice (Fig. 1b), establishing that pharmacological HIF-1α suppression is sufficient to block OVA-induced AHR. However, one dose of YC-1 given 1 h prior to MCH challenge was not adequate to prevent AHR (Fig. 1c).
OVA-induced asthma in HIF-1α myeloid KO mice
H&E staining showed decreased inflammation in OVA HIF-1α KO lung parenchyma compared to OVA WT (Fig. 2a). OVA HIF-1α KO lung inflammation scores tended to be lower than OVA WT (5.3 versus 6.8, p > 0.05; Fig. 2b). OVA HIF-1α KO mice had lower GCH by PAS stain (Fig. 2a) and histology score (Fig. 2b). OVA HIF-1α KO mice developed mild lung eosinophilia, not significantly different to those of no-OVA controls, while OVA WT mice had significantly higher lung eosinophilia (Fig. 2c). There were no significant differences in blood eosinophilia between WT and HIF-1α KO mice in the no-OVA (1.83 and 1.05 %, respectively) or OVA (3.8 and 5 %, respectively) groups. There were no differences in BM eosinophilia between WT and HIF-1α KO mice. We did not observe differences in fibrosis on trichrome stain, mast cell numbers on toluidine blue stain, nor in BAL cellularity. BAL IL-5 and IL-13 levels were similar between OVA groups, but BAL IL-4 levels were decreased in OVA HIF-1α KO mice (Fig. 2d). Total IgE levels were similar in OVA groups (Fig. 2e). Gene expression of IL-5 was threefold lower and IL-13 twofold lower in OVA HIF-1α KO relative to OVA WT (1.8 versus 5.4 and 1.3 versus 3.8, respectively), while extrinsic NO synthase and intrinsic NO synthase expressions did not differ between groups (Fig. 2f). Moreover, C57BL/6 mice did not develop elevated NO levels in their BAL following OVA asthma induction (Fig. 2g). C57BL/6 HIF-1α KO mice tended to have higher baseline NO levels than WT, but these levels did not increase with OVA exposure (Fig. 2g). Thus, in addition to preventing AHR development, the lack of HIF-1α in myeloid cells led to decreased lung eosinophilia and GCH, as well as small decreases in associated inflammatory markers.
Fig. 2.

Knocking out HIF-1α expression in myeloid cells decreases allergic inflammation. a Lung inflammation and goblet cell hyperplasia (GCH) were decreased in OVA HIF-1α KO mice. b Lung inflamma- tion and GCH tended to be lower in OVA HIF-1α KO mice (*P<0.05). c OVA mice had higher lung eosinophilia (*P < 0.01). d OVA HIF-1α KO mice had lower BAL IL-4 levels (*P00.007). e Total IgE levels were similar in both OVA groups. f OVA HIF-1α KO gene expression of IL-5 and IL-13 was twofold lower than OVA WT mice. g BAL NO is similar in C57BL/6 WT and HIF-1α KO mice (P00.25), and NO levels did not increase with OVA asthma induction. Eight WT and 14 KO mice were used for histology grading, and eight samples per group were used for in vitro assays.
Treatment of OVA-induced asthma in C57BL/6 mice with HIF-1α antagonist YC-1 during asthma induction
C57BL/6 OVA mice treated with YC-1 had less inflammation than untreated mice by histology and lung inflammation score (Fig. 3a, b). Parenchymal inflammation was predominantly eosinophilic in 25 % of C57BL/6 OVA mice versus 0 % of YC-1-treated OVA animals. There were no differences in fibrosis on trichrome stain or mast cell numbers on toluidine blue stain. BAL cell counts in YC-1-treated mice showed a fourfold decrease in eosinophilia and fivefold increase in macrophages compared to control mice (Fig. 3c). BAL IL-4 and IL-13 levels were similar between the two groups; however, BAL IL-5 levels were higher in non-treated OVA mice (Fig. 3d). OVA-specific IgE levels in C57BL/6 OVA YC-1-treated mice did not significantly differ from levels in no-OVA controls, while untreated OVA mice had significantly higher levels (Fig. 3e). Eosinophilia in blood was significantly decreased by treatment of C57BL/6 OVA mice with YC-1, and eosinophilia in BM of mice treated with YC-1 was similar to no-OVA controls (Fig. 3f). Thus, pharmacological HIF-1α inhibition eliminates not only AHR but also decreases BAL, lung, blood, and BM eosinophilia, and overall markers of lung inflammation.
Fig. 3.

HIF-1α antagonist YC-1 decreases eosinophilia and allergic inflammatory markers in a C57BL/6 OVA asthma model. a Lung histology demonstrates decreased inflammation in YC-1- treated OVA mice. b Total lung inflammation was decreased in YC-1-treated OVA mice (*P < 0.001, **P < 0.01). c YC-1-treated OVA mice had fewer eosinophils and more macrophages in BAL (*P < 0.001). d BAL IL-5 was lower in YC-1-treated OVA mice (*P < 0.01, **P < 0.05). e OVA-specific IgE levels were no different in YC-1-treated OVA mice compared to controls and OVA mice, while OVA mice had higher levels than controls (*P < 0.05). f Eosinophilia in blood was de- creased in YC-1-treated OVA mice (*P < 0.001) and was no dif- ferent than no-OVA controls. Eight mice were used per experimental group. ELISAs and NO measurements were done in triplicate and repeated once.
Eosinophil chemotaxis
The significant decrease in asthma lung eosinophilia in the myeloid HIF-1α KO model and the decrease in lung and BAL eosinophilia in the YC-1 treatment model led us to study the role of HIF-1α in regulating eosinophil chemotaxis. We first compared chemotactic properties of WT and HIF-1α KO eosinophils, which were phenotypically indistinguishable under microscopy by Giemsa–Wright stain (Fig. 4a). HIF-1α KO eosinophils had much decreased HIF-1α protein levels compared to WT eosinophils by Western blot (Fig. 4b). WT exhibited a twofold higher migration toward eotaxin-1 and a sevenfold higher migration toward MIP-1α, compared to HIF- 1α KO (Fig. 4c). Treatment of WT eosinophils with HIF-1α antagonist YC-1 led to a 60 % decrease in migration toward eotaxin-1. Conversely, treatment with HIF-1α agonist mimosine increased migration 30–38 % (Fig. 4d). Confirming specificity of action, HIF-1α KO eosinophils treated with YC-1 or mimosine showed no change in chemotaxis (Fig. 4d). HIF-2α is another α subunit, which is regulated in a similar fashion to HIF-1α. HIF-2α KO eosinophils migrated 336 more than WT eosinophils toward both eotaxin-1 and MIP- 1α (Fig. 4e). These findings suggest that HIF-2α counterregulates HIF-1α pro-chemotaxis activities.
Fig. 4.

HIF-1α and HIF-2α regulate eosinophil chemotaxis. a Giemsa–Wright stain of C57BL/6 WT and HIF-1α KO eosinophils. b HIF-1α KO eosinophils have much decreased HIF-1α levels com- pared to WT eosinophils by Western blot, with PARP as a nuclear housekeeping protein control. c Transwell chemotaxis assay demon- strating that HIF-1α KO eosinophils migrate less than WT toward eotaxin-1 and MIP-1α (*P < 0.001). d HIF-1α antagonist YC-1 and agonist mimosine alter migration of WT but not HIF-1α KO eosino- phils (*P < 0.001). e HIF-2α KO eosinophils migrate more than WT toward eotaxin-1 and MIP-1α (*P < 0.01). Each condition was done in duplicate during the experiment, and each experiment was repeated three times
Discussion
By utilizing HIF-1α lysMcre KO mice as a genetic tool, we were able to study the specific role of HIF-1α in myeloid cells in asthma pathogenesis. Myeloid HIF-1α KO mice did not develop AHR, demonstrating that HIF-1α in myeloid cells is necessary for AHR induction. Thus, decreasing HIF-1α in myeloid cells, which would include eosinophils, macrophages, neutrophils, and mast cells, leads to decreased smooth muscle responses to allergic stimuli. Knocking out HIF-1α in myeloid cells did not prevent the development of all allergic inflammatory findings, suggesting that HIF-1α activation in additional cells (epithelial, smooth muscle, endothelial, etc.) may be contributing to asthma pathogenesis and/or that certain aspects of asthma pathophysiology are propagated largely independently of HIF-1α. In addition, it is estimated that 5–10 % of myeloid cells in the lysMcre KO model retain HIF-1α; thus, the presence of HIF-1α in even a small set of myeloid cells may have sufficient impact to influence the development of allergic inflammation in the lung and systemically.
We complimented our genetic studies with a pharmacologic model in which we treated OVA mice systemically with HIF-1α inhibitor YC-1. This systemic treatment during asthma induction also prevented AHR development. Indeed, it had a more profound effect than the myeloid specific genetic KO on eosinophilia in the lung, BAL, and blood. We were interested in determining whether HIF-1α in eosinophils was the driving force behind these findings versus HIF-1α in other cells that then signaled the eosinophils to join in the allergic inflammation in the lung.
Eosinophils are a hallmark of allergic inflammation and increased eosinophil levels in the BAL or sputum of asthma patients which signify ongoing inflammation in the lung. Eosinophils subjected to hypoxia have increased HIF-1α and decreased apoptosis, leading to more eosinophil accumulation in hypoxic (inflamed) tissues [29], which is one mechanism by which HIF-1α may induce eosinophilia in asthmatic lungs. Eosinophils are targeted to sites of allergic inflammation by the release of chemokines. CCR1 and CCR5 are activated by MIP-1α, and CCR3 is activated by eotaxin-1, leading to eosinophil migration.
Our in vitro eosinophil studies demonstrate that signaling to induce eosinophil chemotaxis in response to eotaxin-1 and MIP-1α, therefore likely involving CCR1, CCR3, and/or CCR5, requires HIF-1α. Also, if HIF-2α is removed from eosinophils, chemotaxis increases in response to eotaxin-1 and MIP-1α, suggesting that whereas HIF-1α promotes eosinophil chemotaxis, HIF- 2α acts as a negative (counter) regulator. These findings demonstrate another mechanism by which HIF-1α and HIF-2α influence eosinophilia in the lung. Further studies of the roles of HIF- 1α and HIF-2α in eosinophil function will be enlightening, to determine if these proteins regulate additional eosinophil functions, and thus impact antimicrobial, allergic, or other inflammatory pathways.
Another myeloid cell type that is important in asthma development is the macrophage. Transfer of alveolar macrophages can transmit susceptibility or resistance to the devel- opment of AHR in rats [30, 31]. Our finding that alveolar macrophage numbers increase in the setting of HIF-1α antagonist treatment suggests that these alveolar macrophages may be acting to suppress airway inflammation and AHR. Experiments in which varying numbers of WT and HIF-1α KO alveolar macrophages are transferred into asthmatic mice may elucidate the role of the quantity of macrophages and HIF-1α expression in macrophages in controlling airway inflammation and AHR.
Exhaled NO is an important marker of disease activity in asthma patients [32]. We show here that NO is not elevated in the C57BL/6 OVA model of asthma. Thus, this mouse model may not be the ideal system for evaluation of NO targeted therapies. Also, treatment of OVA mice with one dose of the HIF-1α inhibitor YC-1 immediately prior to MCH challenge was not adequate to abort the AHR response. These data suggest that suppression of HIF-1α does not have an immediate effect on AHR and thus would not be helpful in the first hours of an acute asthma attack. However, because HIF-1α regulates gene transcription, YC-1 may have therapeutic effects within 4–6 h of an asthma attack.
In these studies, we describe the novel observations that eosinophil chemotaxis is regulated by HIF-1α and HIF-2α, and that AHR does not develop in the absence of HIF-1α in myeloid cells, a phenotype that is elicited further by systemic pharmacological suppression of HIF-1α.
Acknowledgments
This research was supported by a Stephen Silberstein Senior Fellowship Award from the American Asthma Foundation (V Nizet) and a Veterans Affairs Career Development Award-2 (LE Crotty Alexander). SCF producing CHO cells were donated by Dr. Mark Kamps, UCSD. Thank you to Katherine Sladewski for counting many blood and BM slides. Dr. James Hagood generously donated his flexiVent machine and lab space. Dr. Nissi Varki contributed her cytology and histopathology expertise, as well as microscopes and cytospin machine at the UCSD Histology Core. Dr. Timothy Bigby contributed his asthma and research expertise.
Footnotes
Conflict of interest
The authors declare that they have no competing financial interests that might influence the results and discussion in this paper.
Contributor Information
Laura E. Crotty Alexander, Email: lecrotty@gmail.com, Pulmonary Critical Care Section, Department of Veterans Affairs San Diego Healthcare System, La Jolla, CA, USA. Department of Medicine, University of California at San Diego, La Jolla, CA, USA
Kathryn Akong-Moore, Department of Pediatrics, University of California at San Diego, La Jolla, CA, USA.
Stephanie Feldstein, Department of Pediatrics, University of California at San Diego, La Jolla, CA, USA.
Per Johansson, Department of Medical and Health Sciences, Linköping University, Linköping, Sweden.
Anh Nyugen, Department of Pediatrics, University of California at San Diego, La Jolla, CA, USA.
Elisa K. McEachern, Pulmonary Critical Care Section, Department of Veterans Affairs San Diego Healthcare System, La Jolla, CA, USA
Shari Nicatia, Pulmonary Critical Care Section, Department of Veterans Affairs San Diego Healthcare System, La Jolla, CA, USA. School of Pharmacy, Utrecht University, Utrecht, the Netherlands.
Andrew S. Cowburn, Department of Medicine, University of Cambridge School of Clinical Medicine, Cambridge, UK
Joshua Olson, Department of Pediatrics, University of California at San Diego, La Jolla, CA, USA.
Jae Youn Cho, Department of Medicine, University of California at San Diego, La Jolla, CA, USA.
Hart Isaacs, Jr., Department of Pathology, University of California at San Diego, La Jolla, CA, USA
Randall S. Johnson, Department of Physiology, Development, and Neuroscience, University of Cambridge, Cambridge, UK
David H. Broide, Department of Medicine, University of California at San Diego, La Jolla, CA, USA
Victor Nizet, Department of Pediatrics, University of California at San Diego, La Jolla, CA, USA. Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, La Jolla, CA, USA.
References
- 1.Lemanske RF, Jr, Busse WW. Asthma. JAMA. 1997;278:1855–1873. [PubMed] [Google Scholar]
- 2.Akinbami LJ, Moorman JE, Liu X. National Health Statistics Reports no 32. Hyattsville, MD: National Center for Health Statistics; 2011. Asthma prevalence, Q2 health care use, and mortality: United States, 2005–2009 CDC. [PubMed] [Google Scholar]
- 3.Lemiere C, Ernst P, Olivenstein R, Yamauchi Y, Govindaraju K, Ludwig MS, Martin JG, Hamid Q. Airway inflammation assessed by invasive and noninvasive means in severe asthma: eosinophilic and noneosinophilic phenotypes. J Allergy Clin Immunol. 2006;118:1033–1039. doi: 10.1016/j.jaci.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Bime C, Nguyen J, Wise RA. Measures of asthma control. Curr Opin Pulm Med. 2012;18:48–56. doi: 10.1097/MCP.0b013e32834db0f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balkissoon R. Asthma overview. Prim Care. 2008;35:41–60. doi: 10.1016/j.pop.2007.09.008 454. [DOI] [PubMed] [Google Scholar]
- 6.Bhakta NR, Woodruff PG. Human asthma phenotypes: from the clinic, to cytokines, and back again. Immunol Rev. 2011;242:220–232. doi: 10.1111/j.1600-065X.2011.01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin RJ, Szefler SJ, King TS, Kraft M, Boushey HA, Chinchilli VM, Craig TJ, Dimango EA, Deykin A, Fahy JV, et al. The Predicting Response to Inhaled Corticosteroid Efficacy (PRICE) trial. J Allergy Clin Immunol. 2007;119:73–80. doi: 10.1016/j.jaci.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szefler SJ, Martin RJ, King TS, Boushey HA, Cherniack RM, Chinchilli VM, Craig TJ, Dolovich M, Drazen JM, Fagan JK, et al. Significant variability in response to inhaled corticosteroids for persistent asthma. J Allergy Clin Immunol. 2002;109:410–418. doi: 10.1067/mai.2002.122635. [DOI] [PubMed] [Google Scholar]
- 9.Huerta-Yepez S, Baay-Guzman GJ, Bebenek IG, Hernandez-Pando R, Vega MI, Chi L, Riedl M, Diaz-Sanchez D, Kleerup E, Tashkin DP, et al. Hypoxia inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy. 2011;66:909–918. doi: 10.1111/j.1398-9995.2011.02594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huerta-Yepez S, Baay-Guzman GJ, Garcia-Zepeda R, Hernandez-Pando R, Vega MI, Gonzalez-Bonilla C, Bonavida B. 2- Methoxyestradiol (2-ME) reduces the airway inflammation and remodeling in an experimental mouse model. Clin Immunol. 2008;129:313–324. doi: 10.1016/j.clim.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Kim SR, Lee KS, Park HS, Park SJ, Min KH, Moon H, Puri KD, Lee YC. HIF-1alpha inhibition ameliorates an allergic air- way disease via VEGF suppression in bronchial epithelium. Eur J Immunol. 2010;40:2858–2869. doi: 10.1002/eji.200939948. [DOI] [PubMed] [Google Scholar]
- 12.Lee SY, Kwon S, Kim KH, Moon HS, Song JS, Park SH, Kim YK. Expression of vascular endothelial growth factor and hypoxia-inducible factor in the airway of asthmatic patients. Ann Allergy Asthma Immunol. 2006;97:794–799. doi: 10.1016/S1081-1206(10)60971-4. [DOI] [PubMed] [Google Scholar]
- 13.Guo J, Lu W, Shimoda LA, Semenza GL, Georas SN. En- hanced interferon-gamma gene expression in T cells and reduced ovalbumin-dependent lung eosinophilia in hypoxiainducible factor-1-alpha-deficient mice. Int Arch Allergy Immunol. 2009;149:98–102. doi: 10.1159/000189191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madjdpour C, Jewell UR, Kneller S, Ziegler U, Schwendener R, Booy C, Klausli L, Pasch T, Schimmer RC, Beck-Schimmer B. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284:L360–L367. doi: 10.1152/ajplung.00158.2002. [DOI] [PubMed] [Google Scholar]
- 16.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF- 1alpha expression regulates the bactericidal capacity of phago- cytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N- terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18:4528–4542. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF- 2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic devel- opment, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita N, Markova D, Anderson DG, Chiba K, Toyama Y, Shapiro IM, Risbud MV. Expression of prolyl hydroxylases (PHDs) s selectively controlled by HIF-1 and HIF-2 in nucleus pulposus cells of the intervertebral disc: distinct roles of PHD2 and PHD3 in controlling HIF-1alpha activity in hypoxia. Journal of biological chemistry. 2012;287(20):16975–16986. doi: 10.1074/jbc.M111.334466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. 520. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–2306. 523. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wachtel MS, Shome G, Sutherland M, McGlone JJ. Derivation and validation of murine histologic alterations resembling asthma, with two proposed histologic grade parameters. BMC Immunol. 2009;10:58. doi: 10.1186/1471-2172-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, Rosenberg HF. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol. 2008;181:4004–4009. doi: 10.4049/jimmunol.181.6.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bochner BS, Hamid Q. Advances in mechanisms of allergy. J Allergy Clin Immunol. 2003;111:S819–S823. doi: 10.1067/mai.2003.149. [DOI] [PubMed] [Google Scholar]
- 26.Zosky GR, Sly PD. Animal models of asthma. Clin Exp Allergy J British Soc Allergy Clin Immunol. 2007;37:973–988. doi: 10.1111/j.1365-2222.2007.02740.x. [DOI] [PubMed] [Google Scholar]
- 27.Chun YS, Yeo EJ, Choi E, Teng CM, Bae JM, Kim MS, Park JW. Inhibitory effect of YC-1 on the hypoxic induction of erythropoietin and vascular endothelial growth factor in Hep3B cells. Biochem Pharmacol. 2001;61:947–954. doi: 10.1016/s0006-2952(01)00564-0. [DOI] [PubMed] [Google Scholar]
- 28.Li SH, Shin DH, Chun YS, Lee MK, Kim MS, Park JW. A novel mode of action of YC-1 in HIF inhibition: stimulation of FIH-dependent p300 dissociation from HIF-1{alpha} Mol Cancer Ther. 2008;7:3729–3738. doi: 10.1158/1535-7163.MCT-08-0074. [DOI] [PubMed] [Google Scholar]
- 29.Nissim Ben Efraim AH, Eliashar R, Levi-Schaffer F. Hypoxia modulates human eosinophil function. Clin Mol Allergy. 2010;8:10. doi: 10.1186/1476-7961-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Careau E, Bissonnette EY. Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2004;31:22–27. doi: 10.1165/rcmb.2003-0229OC. [DOI] [PubMed] [Google Scholar]
- 31.Careau E, Proulx LI, Pouliot P, Spahr A, Turmel V, Bissonnette EY. Antigen sensitization modulates alveolar macrophage functions in an asthma model. Am J Physiol Lung Cell Mol Physiol. 2006;290:L871–L879. doi: 10.1152/ajplung.00219.2005. [DOI] [PubMed] [Google Scholar]
- 32.Spitale N, Popat N, McIvor A. Update on exhaled nitric oxide in pulmonary disease. Expert Rev Respir Med. 2012;6:105–115. doi: 10.1586/ers.12.1. [DOI] [PubMed] [Google Scholar]