

Abstract
Androgen receptor (AR) signaling plays a pivotal role in growth and survival of prostate cancer cells. c-Jun is an important member of the activator protein 1 (AP-1) family and was shown to interact with AR. However, the role of c-Jun in AR signaling remains controversial, with being a coactivator or a corepressor reported. Here, utilizing multiple approaches, we show that c-Jun efficiently inhibits AR activity and the growth of prostate cancer cells. Overexpression of c-Jun inhibits not only the activities of various androgen-responsive promoters but also the transcripts of multiple AR target genes. Interestingly, long-term c-Jun overexpression also down-regulates AR expression at both the protein and mRNA levels. Molecular analysis suggests that c-Jun inhibits AR transactivation potential via an unknown target gene. The inhibition of AR by c-Jun occurs in both hormone naïve and castration-resistant prostate cancer cells. Our results unravel a novel mechanism by which c-Jun antagonizes the AR signaling.
Keywords: Androgen receptor, Prostate cancer, AP-1, c-Jun, LNCaP, C4-2
1. Introduction
Prostate cancer represents the most common non-cutaneous human cancer and is the second leading cause of cancer deaths among men in the US (Jemal et al., 2010). Like normal prostate gland, the proliferation and survival of prostate cancer cells rely on androgens, which signal through the androgen receptor (AR). Thus, androgen ablation therapy, also known as hormone therapy, is the most effective way to control advanced prostate cancer (Salesi et al., 2005). Despite the success of hormone therapy, most tumors eventually relapse and develop into castration-resistant prostate cancer (CRPC) due to the aberrant restoration of AR activity (Feldman and Feldman, 2001). Interestingly, numerous studies have showed that AR signaling axis remains essential for the development and maintenance of CRPC (Chen et al., 2004; Gao et al., 2006; Snoek et al., 2009; Yuan et al., 2006; Zegarra-Moro et al., 2002).
Similar to other steroid hormone receptors, AR is composed of an N-terminal domain (NTD) which contains a major activation domain, AF-1, a DNA-binding domain (DBD), a hinge region and a C-terminal ligand binding domain (LBD) containing a weak activation domain, AF-2 (Dehm and Tindall, 2007). Unliganded AR is sequestered in the cytoplasm by heat shock proteins (Marivoet et al., 1992). Upon binding to testosterone or dihydrotestosterone (DHT), the two major physiology androgens, AR dissociates from heat shock proteins and translocates to the nucleus where it functions as a transcription factor by binding as a homodimer to the androgen-response element (ARE) in the promoter and/or enhancer regions of target genes.
c-Jun is a basic region leucine zipper (bZIP) transcription factor and is an important member of the activator protein 1 (AP-1) family (Vogt, 2001). The basic region of c-Jun is required for DNA binding while the leucine zipper enables c-Jun to form a homodimer or a heterodimer with other AP-1 members, such as Fos and activating transcription factor 2 (ATF2). Depending on the dimerization partner, c-Jun/AP-1 complex binds to TPA-responsive elements (TRE) or cyclic AMP-responsive elements (CRE) in the promoter region of target genes that are involved in several cellular responses including proliferation, apoptosis and differentiation (Eferl and Wagner, 2003). Phosphorylation at residues Ser-63 and Ser-73 by c-Jun N-terminal kinases (JNKs) was shown to enhance the transactivation activity of c-Jun (Karin, 1995; Smeal et al., 1991)
Many molecular and genetic studies have provided evidence that AP-1 activity may also be implicated in the development and progression of prostate cancer. The expression of JunB and Fos were found to be up-regulated in primary prostate tumors but down-regulated in metastatic samples (Chandran et al., 2007). Conversely, Ouyang et al. reported that while both c-Jun and Fos are up-regulated in metastatic prostate cancer, only high c-Jun expression is associated with poor prognosis (Ouyang et al., 2008). However, in the same report, it was found that only few cases (3–4%) of prostate cancer showed high expression of the AP-1 proteins. On the other hand, it has also been observed that some AP-1 proteins are also down-regulated in a subset of prostate cancer patients. In fact, Edwards et al. found that while 16% of CRPC patients showed c-Jun up-regulation, 20% of CRPC patients exhibited c-Jun down-regulation (Edwards et al., 2004). Moreover, Tamura et al. showed that transcripts of both c-Jun and Fos were down-regulated in CRPC (Tamura et al., 2007). Although these studies examined the expression level of AP-1 proteins in prostate cancer tissues, it remains unclear whether and how their transcriptional activity correlates to the development and progression of prostate cancer.
In addition to functioning as an AP-1 transcription factor, AP-1 proteins also interact with other family of transcription factors such as NF-kappaB (Fujioka et al., 2004; Shyu et al., 2008), NFAT (Macian et al., 2000) and nuclear hormone receptors (Herrlich, 2001; Lamph, 1991). In fact, several studies have suggested that c-Jun may physically interact with AR and modulate the AR activity. Sato et al. reported that c-Jun can interact with the DNA-binding domain of AR via its leucine zipper region to inhibit the DNA-binding as well as the transcriptional activity of AR (Sato et al., 1997). Recently, Mulholland and the coworkers proposed that the up-regulation of c-Jun in PTEN null murine prostate cancer cells contributes to CRPC progression by suppressing AR function and thus reducing the androgen-dependence (Mulholland et al., 2011). Conversely, it was also shown that c-Jun functions as an AR coactivator by enhancing the intramolecular interaction between amino and carboxyl termini of AR (Bubulya et al., 2001; Bubulya et al., 2000; Bubulya et al., 1996; Chen et al., 2006; Shemshedini et al., 1991; Wise et al., 1998). Despite the controversy of being an AR coactivator or corepressor, it remains unclear if transcriptional activity of c-Jun is involved in these regulations. Because of the critical role of AR in prostate cancer development and progression and because of the potential regulatory role of AP-1 in the AR signaling, we took a different approach to evaluate the impact of the transcriptional activity of c-Jun on the AR signaling. We found that the DNA binding and transcriptional activities of c-Jun, rather than its physical interaction with AR, are required for the maximal inhibition of the AR signaling. Taken together, our results suggest that an unknown target gene of c-Jun is required for the inhibition of the AR activity and future identification of such a target gene will provide new insight into the regulatory role of AP-1 in the AR signaling and prostate cancer development and progression.
2. Material and Methods
2.1. Antibodies
Polyclonal anti-AR antibody (sc-816) and monoclonal anti-phospho-c-Jun antibody (sc-822) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal anti-PSA antibody (1984-1) was purchased from Epitomics (Burlingame, CA). Monoclonal Anti-β-Tubulin (T0198) and anti-Flag M2 (F3165) antibodies were from sigma. Monoclonal Anti-Human PARP antibody (4C10-5) was purchased from BD Biosciences (San Diego, CA).
2.2. Cell Culture
LNCaP and COS-1 cells were purchased from American Type Culture Collection (Manassas, VA). C4-2 cells were obtained from the University of Texas MD Anderson Cancer Center (Houston, TX). LNCaP cells were cultured in RPMI1640 medium with 10% fetal bovine serum (FBS) and C4-2 cells were maintained in T-medium with 10% FBS (Gleave et al., 1991; Wu et al., 1994). COS-1 cells were cultured in DMEM medium supplemented with 5% FBS. For androgen treatment, LNCaP or C4-2 cells were cultured in phenol red-free RPMI1640 with 10% charcoal/dextran-stripped FBS (designated androgen-depleted medium) for 24 hr before transient transfection or doxycycline (Dox) induction of c-Jun expression for another 24 hr. Cells were then treated with 10 nM R1881 for 24 hr. To determine the effect of c-Jun on the expression of endogenous AR-regulated genes, cells were cultured in regular medium (RPMI 1640 with 10% FBS for LNCaP cells; T-medium with 10% FBS for C4-2 cells) before induction of c-Jun expression for indicated periods of time.
2.3. Plasmids
Human c-Jun was amplified from a cDNA library originated from HEK 293T cells and was cloned in frame into the EcoRI/KpnI site of pFlag-CMV2 (sigma). Plasmids encoding c-Jun63A/73A, c-JunΔLZ (c-JunΔ280–317), c-Jun A→D265 In265 and TAM67 (c-JunΔ3-122) were generated by PCR or ligation PCR (Ali and Steinkasserer, 1995). To clone pLVX-Tight-Puro-Flag-c-Jun, the cDNA encoding Flag-c-Jun was amplified from pFlag-c-Jun by primer sets: BamHI-Kozak-Flag F (5′-CGG GAT CCG CCG CCA CCA TGG ACT ACA AAG ACG ATG ACG-3′) and c-Jun-stop-EcoRV R (5′-GGG ATA TCT TAA AAT GTT TGC AAC TGC TGC G-3′). The amplified cDNA was then cloned into BamHI and Klenow-blunted EcoRI sites of pLVX- Tight-Puro (Clontech). Similar strategy was used to clone all other Flag-c-Jun mutants into pLVX-Tight-Puro. All constructs generated by the PCR-based method were confirmed by DNA sequencing. For generation of c-Jun short hairpin RNA (shRNA) plasmid, annealed oligonucleotides (The RNAi Consortium TRCN0000010366) targeting TAGTACTCCTTAAGAACACAA in the 3′ untranslated region of c-Jun were cloned into pLKO-Tet-On (Wiederschain et al., 2009) to produce pLKO-Tet-On-c-JunKD.
2.4. Generation of Dox-inducible stable cell lines
LNCaP or C4-2 stable cell lines with inducible wild-type or mutant c-Jun were generated by Lenti-X Tet-On Advanced Inducible Expression System (Clontech) according to the manufacturer’s protocol with the following modifications. Cells were first infected with viral particles constitutively expressing rtTA-advanced protein (a mutant Tetracycline-repressor). To generate viral particles, HEK 293T cells cultured in 10-cm culture dish were cotransfected with 2 μg of pLVX-Tet-On, 1.5 μg of pHR′-CMV-ΔR8.20vpr, and 0.5 μg of pHR′-CMV-VSV-G using Fugene HD reagent (Roche Applied Science). The supernatant containing viruses was harvested 2 days post-transfection and was then filtered through a 0.45 μm filter to remove cell debris. Infection was carried out by applying 4 ml of viral supernatant to LNCaP or C4-2 cells cultured in 11 ml complete medium. Polybrene was added at a final concentration of 8 μg/ml to facilitate infection. Two days after infection, cells were selected with 500 μg/ml G418 for more than one week. The cells stably expressing rtTA-advanced protein (LNCaP- or C4-2-rtTA cells) were then transduced with lentiviral particles packaged with pLVX-Tight-Puro-Flag-c-Jun (wild type or mutants) using similar procedures as described above. Following transduction for 2 days, cells were then selected with 2 μg/ml puromycin for 2 additional days. To generate LNCaP cells with inducible c-Jun knockdown, cells were infected with lentiviral particles packaged with pLKO-Tet-On-c-JunKD followed by puromycin selection using similar methods described above.
2.5. Luciferase Reporter Gene Assay
LNCaP or C4-2 cells were trypsinized and washed with phosphate-buffered saline (PBS) once, followed by seeding in a 12 well plate at a density of 1 × 105 cells/well in androgen-depleted medium. Twenty-four hours later, cells were transfected with 0.5 μg of an androgen-responsive luciferase reporter construct, 100 ng of pRL-TK (Promega) along with indicated amount of plasmids encoding wild type or mutant c-Jun using Fugene HD transfection reagent (Promega). Empty vector (pFlag) was added to keep the same amount of transfected DNA per well. At 24 h post-transfection, cells were treated with ethanol (vehicle control) or 10 nM R1881 and then incubated for another 24 h. Firefly and Renilla luciferase activities were measured by the Dual Luciferase Reporter Assay kit (Promega) as previously described (Hsu and Hu, 2012). Relative luciferase activity (Firefly/Renilla) was shown as mean±S.E. from at least three independent experiments performed in duplicate. To analyze the expression level of the exogenous c-Jun proteins (Fig. 1E and 3E), 60 μg of cell lysate were precipitated by incubating with 4 volume of acetone at −20°C overnight. The protein was then pelleted by centrifugation at 14,000 rpm for 20 min at 4°C. The pelleted protein was air dried, followed by resuspension in 2X SDS sample buffer and Western blot analysis using anti-Flag-antibody.
Figure 1. c-Jun suppresses activities of multiple AR-responsive promoters.
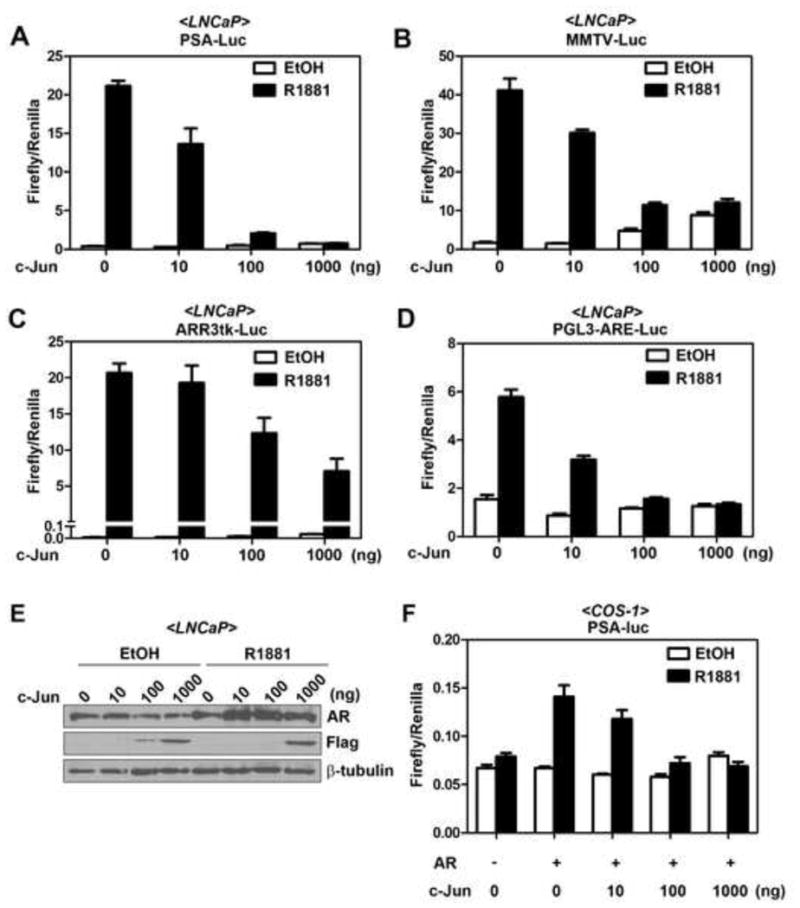
(A) to (D) Effects of c-Jun overexpression on the activities of various androgen-responsive promoters in LNCaP cells. LNCaP cells were transfected with 0.5 μg of luciferase reporter driven by PSA promoter (A), MMTV promoter (B), probasin promoter (C), or synthetic ARE (D), 100 ng of pRL-TK and increasing concentrations (0, 10, 100, and 1000 ng) of pFlag-c-Jun expression vector. After 24 hr, cells were treated with ethanol (EtOH) or 10 nM R1881 for another 24 hr. (E) Western blot analyzing the AR and Flag-c-Jun expression in luciferase lysates from (A). (F) Effect of c-Jun overexpression on AR-transactivated PSA promoter in COS-1 cells. COS-1 cells were co-transfected with 1 μg of empty vector or pHA-AR, 0.5 μg PSA promoter, 100 ng of pRL-TK and increasing concentrations (0, 10, 100, and 1000 ng) of pFlag-c-Jun expression vector. After 24 hr, cells were treated with ethanol (vehicle control) or 10 nM R1881 for another 24 hr. Error bars, S.E. (n=3 for each experiment).
Figure 3. The transcriptionally inactive c-Jun mutants fail to efficiently suppress AR activity.
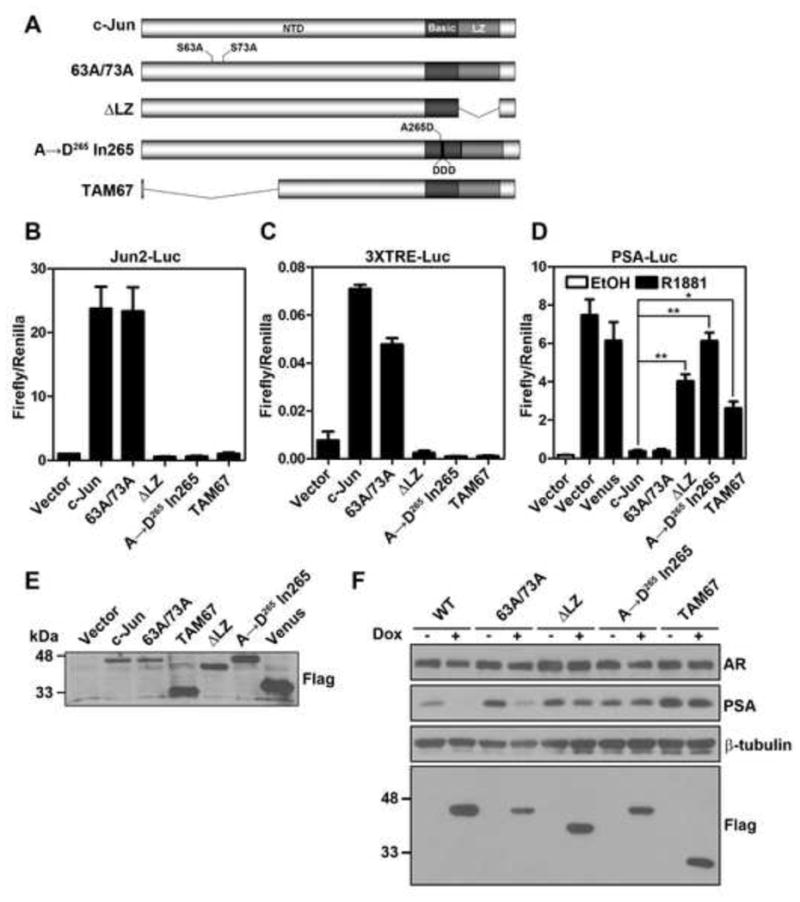
(A) Schematic view of various c-Jun mutants. NTD, N-terminal domain; LZ, leucine zipper, (B) and (C) Transcriptional activity of c-Jun mutants determined by luciferase reporters driven by the jun2 response element or three tandem repeats of TRE. LNCaP cells were transfected with 0.5 μg of jun2-luc (B) or 3XTRE-luc (C), 100 ng of pRL-TK along with 1 μg of pCMV-Flag (vector) or indicated pFlag-c-Jun (wild type or mutants) for 24 hr before assaying the luciferase activities. Error bars, S.E. (n=3). (D) Effects of c-Jun mutants on AR activity in LNCaP cells were evaluated by PSA reporter utilizing the method as described in legend of Figure 1. Error bars, S.E. (n=3~5). The asterisk and the double asterisk indicate P<0.05 and P<0.005, respectively (Student’s t-test). (E) Examination of the exogenous c-Jun expression in (D) by immunoblotting with a Flag antibody. (F) Effect of Flag-c-Jun mutants on endogenous PSA expression. LNCaP-rtTA-c-Jun cells (wild-type or mutants) were treated with 100 ng/ml Dox for 24 hr, followed by immunoblotting for AR, PSA and Flag-c-Jun proteins expression. All Flag-c-Jun mutants (Flag) were detected with the anti-Flag antibody.
2.6. Quantitative real-time PCR (qRT-PCR)
RNA was isolated from LNCaP cell lines using TRIzol (Life Technologies). One microgram of the total RNA was reverse transcribed using random primers (100 ng) and MMLV reverse transcriptase (Promega). qRT-PCR was conducted using Brilliant SYBER Green QPCR Master Mix (Stratagene) in a Mx3000P qPCR system (Stratagene). Expression levels of AR-regulated genes were normalized to GAPDH and were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001). Results are presented as mean±S.E. from at least three independent experiments performed in duplicate. The sequences of primers used are listed below. PSA F: 5′-TTG TCT TCC TCA CCC TGT CC-3′; PSA R: 5′-GAG AGG CCA CAA GCA CCT G-3′; KLK2 F:5′-TGG CTG TGT ACA GTC ATG GA-3′; KLK2 R: 5′-CCT GTG TCT TCA GGC TCA AA-3′; TMPRSS2 F: 5′-AGG TGC ATC CGG CTC AGT A-3′; TMPRSS2 R: 5′-GGG TCA AGG TGA TGC ACA GT-3′; PCDH11 F: 5′-GCG TTT CTG ACT GTG GCT ATC-3′; PCDH11 R: 5′-GGA AGG GGA ATG GAA TTT TG-3′; UGT2B15 F: 5′-TCA AAT GAT TTT CAT GGA GAG G-3′; UGT2B15 R: 5′-GCT TTC CCC ATT GTC TCA AA-3′; GAPDH F: 5′-CTG AC TCA CAC GCG ACA CC-3′; GAPDH R: 5′-CCC TGT TGC TGC AGC CAA AT-3′; AR F: 5′-GTG GAA GCT GCA AGG TCT TC-3′; AR R 5′-CGA AGA CGA CAA GAT GGA CA-3′. AR nascent RNA was measured by the following forward and reverse primers that recognize exon 1 and intron 1 of AR gene, respectively. AR nascent F: 5′-GGT GAG CAG AGT GCC CTA TC-3′; AR nascent R:5′-GCG ACA TTT CTG GAA GGA AA-3′.
2.7. Cell counting analysis
LNCaP or C4-2 stable cell lines were seeded in regular medium at a density of 5 × 105 cells/10 cm dish. On the next day, designated day 0, cells from one dish were trypsinized for determining starting cell number by trypan blue exclusion assay (Strober, 2001). At the same time, cells in other dishes were treated with or without 100 ng/ml Dox. Cell number was counted on day 1, 3 and 5 followed by Dox treatment. Medium with indicated amounts of Dox was refreshed on Day 1 and 3. Results are presented as mean±S.E. from at least three independent experiments.
2.8. Cell cycle analysis
LNCaP- or C4-2-rtTA-c-Jun cells were treated with or without Dox for 3 days using same protocol as described in cell counting analysis. Cells were then trypsinized and fixed with ice-cold 70% ethanol. Followed by 30 min incubation at 4°C, fixed cells were resuspended in PBS solution containing 100 μg/ml RNase A and 20 μg/ml propidium iodide. After 1 hr incubation at room temperature, DNA contents of cells were measured by a CytomicsTM FC 500 (Beckman Coulter). Cell cycle was then analyzed using the Dean-Jett-Fox algorithm of FlowJo software.
2.9. BrdU incorporation assay
The bromodeoxyuridine (BrdU) incorporation assay was performed using BrdU and its antibody from BrdU Cell Proliferation Assay Kit (Calbiochem Cat#QIA58). LNCaP- or C4-2-rtTA- c-Jun cells (8.3 × 104 cells/well) were seeded onto coverslips in six-well plates overnight. Followed by treatment with or without Dox for 3 days, cells were labeled with BrdU for 4 hr and then fixed by 70% ice-cold ethanol for 5 min. Cells were then incubated with 1.5N HCl for 30 min at RT. After washing with PBS, cells were blocked by 5% non-fat milk and stained by anti-BrdU antibody. Cells were then incubated with anti-mouse IgG Texas Red-conjugated secondary antibody and 2.5 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) for 1 hr. The BrdU incorporated cells were examined by a Nikon TE2000-U inverted fluorescence microscope. All images were taken at 200x magnification and percentage of BrdU positive cells were shown as mean±S.E. quantified from at least 9 randomly selected fields.
2.10. Statistical Analysis
The data were expressed as mean±S.E. Statistical analysis was performed by the unpaired two-tailed student’s t test analyzed by GraphPad Prism 5 (La Jolla, CA).
3. Results
3.1. c-Jun inhibits activities of multiple AR-responsive promoters
We examined the role of c-Jun overexpression in the activation of several AR-regulated promoters in LNCaP prostate cancer cells that express endogenous AR. As shown in Figure 1A to 1C, overexpressed c-Jun not only inhibited R1881-induced PSA promoter activity but also suppressed the activities of the mouse mammary tumor virus promoter (MMTV-Luc) and the probasin promoters (ARR3tk-luc) in a dose-dependent manner. To rule out the possibility that c-Jun inhibited the androgen-induced reporter gene activities by binding to AP-1 sites in the promoter regions, we examined the effect of c-Jun on luciferase reporter driven by tandem repeats of ARE. As shown in Figure 1D, c-Jun also suppressed the R1881-induced ARE-Luc reporter (Fig. 1D), suggesting that c-Jun inhibits the AR responsive reporter without binding to the promoter regions. Overexpression of c-Jun did not suppress R1881-induced AR protein expression (Fig. 1E), suggesting that c-Jun could inhibit AR activity without affecting its protein level. Taken together, these results suggest that c-Jun exerts a global inhibitory effect on the AR activity in LNCaP cells.
Because the AR in LNCaP cells harbors a T877A mutation in LBD (Sobel and Sadar, 2005), we next sought to determine if c-Jun also inhibits function of wild-type AR. To this end, we performed similar PSA reporter assay in AR-negative COS-1 cells. Effects of c-Jun overexpression on PSA promoter activity were evaluated in COS-1 cells with or without co-transfection of plasmids encoding wild-type AR. As shown in Figure 1F, reconstitution of AR expression is prerequisite for the induction of PSA promoter activity by R1881 in COS-1 cells. Hence, the system allows us to specifically examine the function of AR. Consistent with the results from LNCaP cells, overexpressed c-Jun also inhibited the R1881-activated PSA reporter gene in COS-1 cells (Fig. 1F), suggesting that AR inhibitory function of c-Jun is not cell type-specific or restrict to the T877A AR mutant.
3.2. Overexpression of c-Jun suppresses multiple AR-regulated gene expression at endogenous level
To confirm these observations in a more physiological relevant context, we examined the effect of c-Jun on androgen-induced PSA protein expression. Consistent with the observation in the promoter activity assay, transient expression of c-Jun suppressed R1881-induced PSA protein expression (Fig. 2A). To gain more insight into the effects of c-Jun on AR function, we utilized lentivirus to generate an inducible system in which c-Jun expression is controlled in a doxycycline (Dox)-dependent manner in LNCaP cells. Cells with inducible c-JunΔLZ were generated as a negative control based on a previous report that LZ region is responsible for its interaction with AR (Sato et al., 1997). The purpose of using Dox-inducible system was to prevent artifacts during clonal selection of c-Jun overexpressing cells. Treatment of cells with Dox (100 ng/ml) for 2 days induced the expression of exogenous Flag-c-Jun in ~70% of cells (data now shown). We first examined the time course of c-Jun induction on endogenous PSA protein levels. As shown in Figure 2B, the endogenous PSA protein expression was reduced within 6 hr upon c-Jun induction. The down-regulation of PSA sustained at least to 24 hr, at which time point no obvious change in AR expression was observed. Induction of c-Jun, but not c-JunΔLZ, down-regulated both steady state and androgen-induced PSA protein expression (Fig. 2C and D). Furthermore, induction of c-Jun, but not c-JunΔLZ, also significantly down-regulated steady state mRNA levels of and two other androgen-regulated genes, KLK2 and transmembrane protease, serine 2 (TMPRSS2) (Fig. 2. E–G), suggesting that c-Jun globally inhibits AR transactivation function. Because AR was also shown to function as a transcription repressor, we then examined the effects of c-Jun on two AR-repressed genes, PCDH11 (Yang et al., 2005) and UGT2B15 (Bao et al., 2008). Although c-Jun overexpression caused a ~2-fold increase of PCDH11 transcripts (Fig. 2H), it did not affect the expression of UGT2B15 (Fig. 2I), suggesting that c-Jun alleviates AR transrepression function in a gene context-dependent manner. Taken together, our results strongly suggest that c-Jun is a potent inhibitor for the AR transactivation function. To determine if endogenous c-Jun suppresses AR activity, we generated an LNCaP stable cell line in which c-Jun expression can be inducibly knocked down by Dox. Interestingly, no significant change in PSA protein level was observed in c-Jun knockdown cells (Fig. 2J), suggesting that basal level of c-Jun in LNCaP cells is not sufficient to inhibit AR function. In fact, the steady state c-Jun level in LNCaP cells is relatively low compared with PC3 or DU145 cells, two other human prostate cancer cell lines with little AR expression (Ouyang et al., 2008; Sun et al., 1999).
Figure 2. c-Jun suppresses multiple AR target genes at endogenous level.
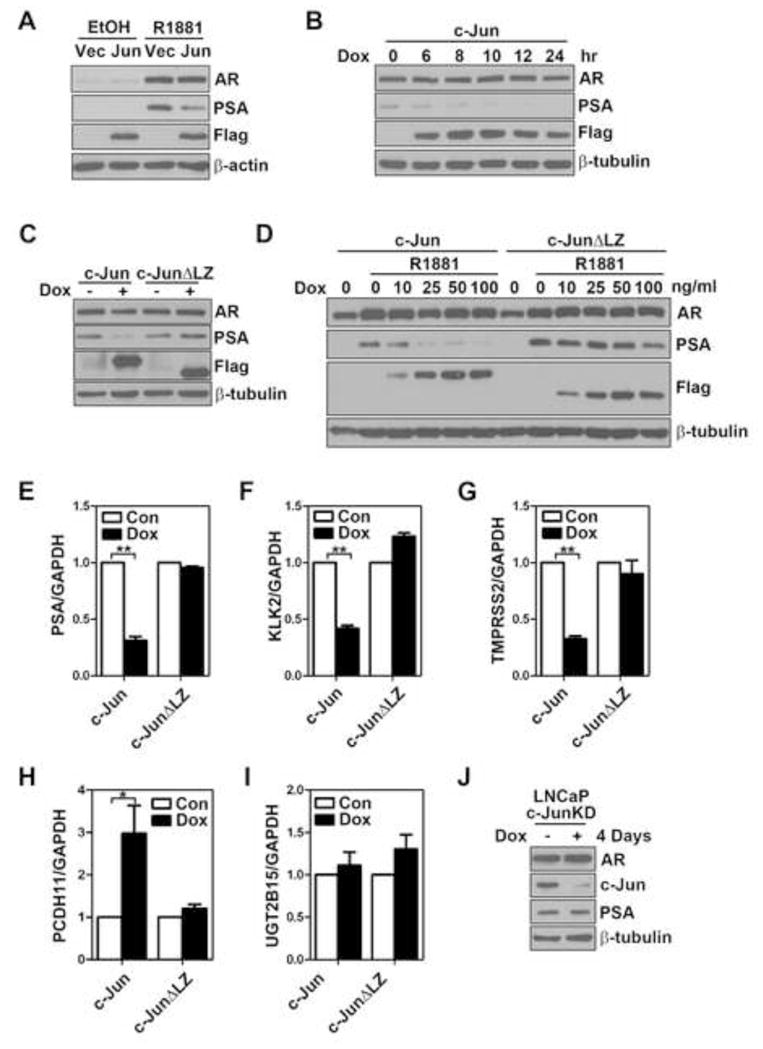
(A) Effect of c-Jun overexpression on endogenous PSA level in LNCaP cells. LNCaP cells cultured in androgen-depleted medium for 24 hr, followed by transient transfection with empty vector (Vec) or plasmids encoding Flag-c-Jun (Jun). After 24 hr, cells were treated with 10 nM R1881 or the ethanol control (EtOH) for another 24 hr before harvesting for immunoblotting analysis of AR, PSA and Flag-c-Jun (Flag). (B) Time course of c-Jun induction on endogenous PSA protein expression in LNCaP cells. LNCaP-rtTA-c-Jun cells were treated with 100 ng/ml doxycycline (Dox) for indicated periods of time. AR, PSA and Flag-c-Jun (Flag) were immunoblotted. (C) Effect of LZ deletion on c-Jun-inhibited PSA expression in LNCaP cells. LNCaP-rtTA-c-Jun or c-JunΔLZ cells were treated with 100 ng/ml Dox for 24 hr. AR, PSA and Flag-c-Jun (Flag) were immunoblotted. (D) Wild-type but not LZ-deleted c-Jun efficiently suppresses androgen-induced PSA expression in LNCaP cells. LNCaP-rtTA-c-Jun or c-JunΔLZ cells were cultured in androgen-depleted medium for 24 hr, followed by induction of wild-type or mutant c-Jun with indicated concentrations of Dox. Twenty-four hours post c-Jun induction, cells were treated with 10 nM R1881 for another 24 hr. AR, PSA and Flag-c-Jun (Flag) were then immunoblotted. (E) to (I) Effect of c-Jun or c-JunΔLZ expression on steady state mRNA levels of PSA, KLK2,TMPRSS2, PCDH11 and UGT2B15 in LNCaP cells. LNCaP-rtTA-c-Jun or c-JunΔLZ cells were treated with 100 ng/ml Dox for 24 hr. Transcripts of PSA (E, n=4), KLK2 (F, n=3), TMPRSS2 (G, n=3), PCDH11 (H, n=3), and UGT2B15 (I, n=3) were determined by qRT-PCR. The single and the double asterisk indicate the P value between control (Con) and Dox groups is less than 0.05 and 0.001, respectively (Student’s t-test). (J) Effect of c-Jun knockdown on PSA expression in LNCaP cells. LNCaP-c-JunKD cells were treated with Dox (100 ng/ml) for 4 days followed by immunoblotting analysis of AR, PSA and c-Jun expression.
3.3. The AR inhibitory function correlates to the transactivational activity of c-Jun
It was reported that c-Jun may inhibit the AR activity by its physical interaction with AR via its leucine zipper region (Sato et al., 1997). This conclusion was largely based on the observation that deletion of the zipper region abrogated the AR inhibitory function. Since transcriptional function of AP-1 proteins is dependent on dimerization, c-Jun mutant without LZ region is also transcriptionally inactive. To further determine whether the inhibition of the AR activity by c-Jun is mediated by its AP-1 activity or by its physical interaction with AR, we tested the AR-inhibitory function of several c-Jun mutants with impaired transactivation function. The mutants used include: c-Jun63A/73A, in which both serines 63 and 73 were replaced with non-phosphorylatable alanines; c-JunΔLZ, in which LZ region was deleted; c-Jun A→D265 In265 whose DNA binding activity is abrogated by introduction of aspartic acids in the DBD (Brown et al., 1996); c-JunΔ3-122 (TAM67), in which the N-terminal transactivation domain was deleted (Fig. 3A) (Brown et al., 1996). It is important to note that TAM67 and the DNA binding-deficient mutant retain the ability to dimerize. The transcriptional activity of these mutants was first evaluated using two different c-Jun responsive promoters: luciferase reporters driven by a jun2 response element or by three tandem repeats of TRE. We confirmed that c-JunΔLZ, c-Jun A→D265 In265 and TAM67 are transcriptionally inactive (Fig. 3B and C). Surprisingly, the 63A/73A mutation only partially reduced (~30% reduction) the transactivation potential of c-Jun on 3XTRE promoter activity. Importantly, consistent with our previous observation (Liu et al., 2006a), the non-phosphorylatable mutant activated the jun2-promoter activity as well as wild-type c-Jun. These results suggest that the effect of the double alanine mutation on c-Jun transcriptional activity is promoter context-dependent and that this mutation does not always affect the transactivation potential of c-Jun. We then examined the effect of these mutants on R1881-induced PSA promoter activity. A Venus fluorescent protein was used as a negative control for the effect of protein overexpression on PSA promoter activity. Overexpression of Venus slightly reduced androgen-induced PSA promoter activity, which may result from nonspecific competition for transcription and translation machinery (Fig 3D). The c-Jun63A/73A mutant inhibited AR activity as well as wild-type one (Fig. 3D and S1), suggesting that the phosphorylation event at these two serine residues is dispensable for the AR inhibitory function. Consistent with the finding of Sato et al.(Sato et al., 1997), deletion of the LZ region significantly alleviated the c-Jun inhibitory activity. Remarkably, the AR inhibitory function of c-Jun was completely lost in DNA binding-deficient mutant (c-Jun A→D265 In265) and was significantly attenuated in the transactivation domain-deleted mutant (TAM67). Immunoblotting analysis verified that the lack of the AR inhibitory role was not due to lower expression of these mutants. In fact, the expression level of these transcriptionally inactive Jun proteins was even slightly higher than wild-type c-Jun (Fig 3E). To further confirm the effects of these mutants on AR function in a more physiologically relevant context, we examined PSA protein levels in LNCaP stable transfectants expressing various inducible c-Jun mutants. As shown in Figure 3F, endogenous PSA protein of LNCaP cells was significantly down-regulated by induction of c-Jun or c-Jun63A/73A, but not c-JunΔLZ, c-Jun A→D265 In265 or TAM67. Taken together, these results suggest that transcriptional activity of c-Jun is critical for its AR inhibitory role.
3.4. Long-term c-Jun overexpression leads to down-regulation of AR protein
We next determined the long-term effect of c-Jun expression on AR signaling. Interestingly, AR protein was significantly decreased 3 days after c-Jun induction (Fig. 4A). To determine the mechanism underlying the down-regulation of AR protein, we evaluated if c-Jun regulates AR expression at the levels of transcription or protein stability in LNCaP cells. We found that AR mRNA level was significantly down-regulated (~37% reduction) at 24 hr post-c-Jun induction (Fig. 4B). A significant down-regulation of nascent AR RNA transcript was also observed in c-Jun-overexpressing cells (Fig. 4C), suggesting that c-Jun reduces AR mRNA level by suppressing AR transcription. We then determine AR protein stability by treating cells with cycloheximide, a protein synthesis inhibitor. The cycloheximide treatment (for up to 9 hr) did not significantly alter endogenous AR protein level in cells with c-Jun induction (Fig. 4D), suggesting that AR protein remained stable under the condition of c-Jun overexpression. Taken together, our results suggest that long-term c-Jun induction diminishes AR protein level by inhibiting AR transcription.
Figure 4. Long-term c-Jun induction down-regulates AR expression at transcription level.
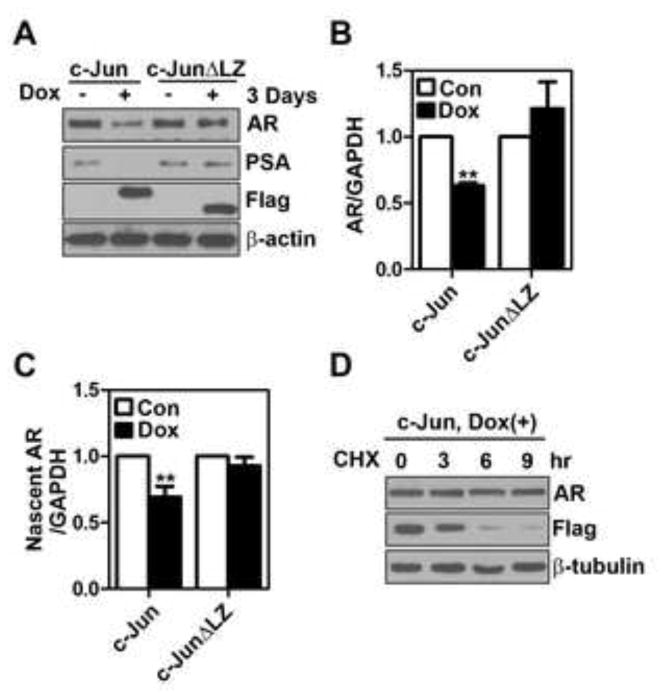
(A) LNCaP-rtTA-c-Jun or c-JunΔLZ cells were treated with 100 ng/ml doxycycline (Dox) for 72 hr, followed by immunoblotting analysis of AR, PSA and Flag-c-Jun expression. (B) and (C) LNCaP-rtTA-c-Jun or c-JunΔLZ cells were treated with Dox (100 ng/ml) for 24 hr. Transcripts of AR mRNA (B, n=4) and nascent AR RNA (C, n=4) were determined by qRT-PCR. The double asterisk indicates the P value between control (Con) and Dox groups is less than 0.01 (Student’s t-test). (D) LNCaP-rtTA-c-Jun cells were treated with Dox (100 ng/ml) for 24 hr followed by cycloheximide (10 μg/ml) treatment for indicated periods of time. AR and Flag-c-Jun were immunoblotted.
3.5. Overexpression of c-Jun inhibits proliferation of LNCaP cells
It is well known that AR signaling axis is required for growth of androgen-dependent prostate cancer cells. Our result that c-Jun inhibits AR function prompted us to test if induction of c-Jun affects proliferation of LNCaP cells. LNCaP-rtTA-c-Jun, c-JunΔLZ or c-Jun A→D265 In265 cells were treated with or without Dox (100 ng/ml) and cell numbers were counted on day 0, 1, 3 and 5. As shown in Figure 5A, treatment of LNCaP-rtTA-c-Jun with 100 ng/ml Dox for 5 days significantly reduced the cell number from 43.9±8.2 to 15.7±2 × 105. In contrast, neither c-JunΔLZ nor c-Jun A→D265 In265 induction significantly reduced cell number. Flow cytometry analysis showed that c-Jun induction for 3 days suppressed cell cycle progression, as evidenced by increased G1 population and reduced S and G2/M populations (Fig. 5B). Moreover, c-Jun induction also suppressed the Brdu incorporation rate (Fig 5C), confirming the suppressive role of c-Jun in LNCaP cell proliferation. On the other hand, we did not observe poly (ADP-ribose) polymerase (PARP) cleavage, an apoptosis marker, in LNCaP cells that overexpressed c-Jun (Fig. S2), suggesting that the reduced cell number of cells is primarily due to the inhibition of cell proliferation rather than the induction of apoptosis. Taken together, c-Jun not only inhibits AR activity but also cell proliferation of androgen-dependent prostate cancer cells.
Figure 5. Overexpression of c-Jun inhibits proliferation of LNCaP cells.
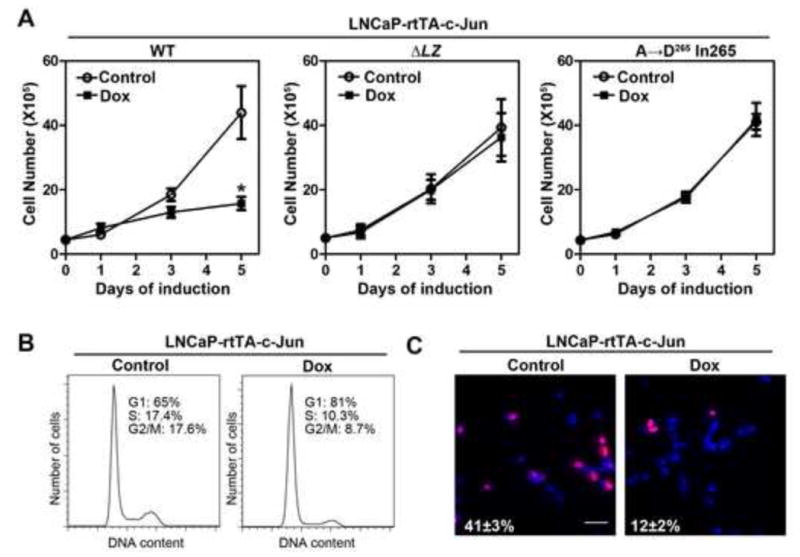
(A) Effect of c-Jun overexpression on cell number of LNCaP cells. LNCaP-rtTA-c-Jun cells (wild-type or mutants) were treated with or without 100 ng/ml of doxycycline (Dox) for 5 days. Cell numbers were counted on day 0, 1, 3, and 5. The asterisk indicates a significant difference (P<0.05) when compared with the control group (Student’s t-test). Error bars, S.E. (n=3). (B) Effect of c-Jun overexpression on cell cycle progression of LNCaP cells. LNCaP-rtTA-c-Jun cells were treated with or without Dox for 3 days, followed by flow cytometry analysis of cell cycle progression. (C) Effect of c-Jun overexpression on DNA synthesis of LNCaP cells. LNCaP-rtTA-c-Jun cells were treated with or without Dox for 3 days and followed by Brdu incorporation assay. Shown are representative merged images of BrdU labeling (red) and DAPI staining (blue). Bar, 100 μm. The number indicates the percentage of Brdu incorporated cells±S.E.
3.6. c-Jun inhibits AR function in castration-resistant prostate cancer cells
Several previous studies showed that a subset of castration-resistant prostate cancer patients also showed c-Jun down-regulation (Edwards et al., 2004; Tamura et al., 2007), though its clinical significance remains unclear. To determine whether c-Jun overexpression may have a differential effect on hormone naïve prostate cancer cells and CRPC cells, we next examined the role of c-Jun in CRPC cells. For the purpose of comparison, we chose the C4-2 cell line, which is widely used as a CRPC line and is derived from LNCaP cells (Wu et al., 1994). We found transfection of c-Jun dose-dependently inhibited R1881-indeuced PSA promoter activity in C4-2 cells as well (Fig 6A). We similarly generated C4-2 stable cell lines with inducible expression of c-Jun proteins and found that like in LNCaP cells, c-Jun, but not c-JunΔLZ or c-Jun A→D265 In265, suppressed endogenous PSA expression (Fig. 6B). Interestingly, overexpression of c-Jun, but not c-JunΔLZ or c-Jun A→D265 In265, for 5 days also reduced cell number of C4-2 cells (Fig. 6C). Overexpression of c-Jun inhibited cell cycle progression (Fig. 6D) and Brdu incorporation (Fig. 6E) in C4-2 cells, albeit to a lesser extent compared to LNCaP cells. The decreased anti-proliferative effect in C4-2 cells as opposed to LNCaP cells was likely due to lower induction of Flag-c-Jun expression in C4-2 cells (data not shown). Our observations are consistent with previous reports that AR signaling remains important for proliferation of CRPC cells (Chen et al., 2004; Snoek et al., 2009; Yuan et al., 2006; Zegarra-Moro et al., 2002). Taken together, these results suggest that c-Jun is capable of suppressing AR function, through a mechanism that depends on the transcriptional activity of c-Jun, in prostate cancer cells that acquired castration resistance.
Figure 6. c-Jun suppresses AR function in castration-resistant prostate cancer cells.
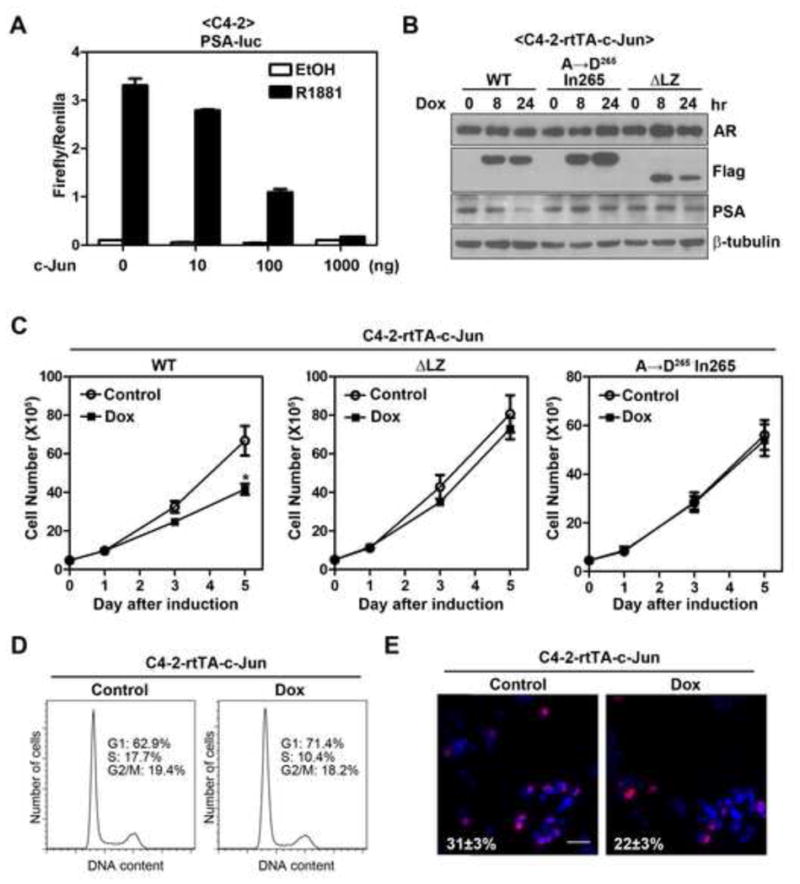
(A) Effect of c-Jun overexpression on R1881-induced PSA promoter activity in C4-2 cells. PSA reporter assay was performed in C4-2 cells using method as described in Figure 1A. Error bars, S.E. (n=3). (B) Induction of c-Jun inhibits endogenous PSA expression in C4-2 cells. C4-2-rtTA-c-Jun cells (wild-type or mutants) were treated with 100 ng/ml of doxycycline (Dox) for indicated periods of time. AR, PSA and Flag-c-Jun (Flag) proteins were immunoblotted. (C) Overexpression of c-Jun reduces cell number of C4-2 cells. Effects of c-Jun (wild-type or mutants) induction on cell number of C4-2 cells were determined using the method as describe in Figure 5A. The asterisk indicates a significant difference (P<0.05) when compared with the control group (student’s t-test). Error bars, S.E. (n=4 for c-Jun or c-JunΔLZ; n=3 for c-Jun A→D265 In265). (D) Effect of c-Jun overexpression on cell cycle progression of C4-2 cells. C4-2-rtTA-c-Jun cells were treated with or without Dox for 3 days, followed by flow cytometry analysis of cell cycle progression. (E) Effect of c-Jun overexpression on DNA synthesis of C4-2 cells. C4- 2-rtTA-c-Jun cells were treated with or without Dox for 3 days and followed by Brdu incorporation assay. Shown are representative merged images of BrdU labeling (red) DAPI staining (blue). Bar, 100 μm. The number indicates the percentage of Brdu incorporated cells±S.E.
4. Discussion
The interplay between AP-1 and steroid hormone receptors has been extensively studied (Herrlich, 2001; Karin and Chang, 2001; Kushner et al., 2000; Pfahl, 1993). With regard to the impact of c-Jun on AR signaling, both stimulatory (Bubulya et al., 2001; 2000; 1996; Chen et al., 2006; Shemshedini et al., 1991; Wise et al., 1998) and inhibitory (Chung et al., 2001; Lobaccaro et al., 1999; Murtha et al., 1997; Sato et al., 1997; Yuan et al., 2010) effects were reported and these observations suggested that c-Jun may act as a coactivator or corepressor of AR by physically interacting with AR. In the present study, we provide evidence here supporting that c-Jun is a potent inhibitor for AR function and the transcriptional activity of c-Jun, rather than the AR/c-Jun interaction, plays a major role for this inhibition. First, c-Jun dose-dependently inhibited activity of four AR responsive luciferase reporters. Second, c-Jun overexpression suppressed steady state and androgen-induced PSA protein expression in LNCaP cells. Third, c-Jun down-regulated mRNA levels of multiple AR target genes, including PSA, KLK2 and TMPRSS2. Forth, the inhibitory effect of c-Jun on PSA promoter activity and protein expression was significantly alleviated in transactivation-deficient mutants, including the transactivation domain-deleted (TAM67), dimerization- (c-JunΔLZ) and DNA binding-deficient (c-Jun A→D265 In265) mutants.
The impact of c-Jun on AR activity has been a controversial issue. Although c-Jun was reported to potentiate AR activity (Bubulya et al., 2001; 2000; 1996; Chen et al., 2006; Shemshedini et al., 1991; Wise et al., 1998), the opposite effects were observed by others (Chung et al., 2001; Lobaccaro et al., 1999; Murtha et al., 1997; Sato et al., 1997; Yuan et al., 2010). Interestingly, the use of AR ligands is the major discrepancy, with DHT and R1881 been utilized by groups reported that c-Jun positively and negatively regulates AR activity, respectively. Because DHT, but not R1881, is known to be rapidly converted to other inactive metabolites in many cell lines (Bjelfman et al., 1997; Martini, 1982; Negri-Cesi and Motta, 1994), DHT does not stimulate AR activity as effectively as R1881. Indeed, DHT only weakly stimulated MMTV promoter activity in reports showing that c-Jun enhanced AR activity (Bubulya et al., 1996; Shemshedini et al., 1991; Wise et al., 1998). Interestingly, we found that c-Jun stimulated MMTV promoter activity in the absence of androgen (Fig. 1B), presumably due to the binding to the putative AP-1 sites reported (Vacca et al., 1989). Thus, a more careful examination is needed when using MMTV promoter to determine the impact of c-Jun on AR function. On the other hand, it was reported that LNCaP cells stably overexpressing c-Jun exhibited higher AR activity (Chen et al., 2006). Our results that c-Jun induction suppressed growth of LNCaP cells raised the concern that generation of stable cell line with constitutive c-Jun overexprsssion could likely confer a selection pressure which favors the growth of clones that are less dependent on AR signaling for proliferation. Hence, we believe our strategy of utilizing the inducible expression system allowed us to investigate the impact of c-Jun on AR function more accurately.
It was also proposed that AP-1 inhibits AR activity by competing with limited amount of CREB binding protein (CBP), which serves as a coactivator for both AR and c-Jun (Fronsdal et al., 1998). The amino-terminal region, especially the Ser-63/73, of c-Jun is required for the interaction with CBP (Bannister et al., 1995). Because AR activity was efficiently inhibited by c- Jun63A/73A but not by the bZIP mutants whose N-terminal domains remain intact, the repression is less likely due to the competition with limited amount of CBP. Instead, our finding that AR inhibition was significantly attenuated in c-Jun mutants that lose the transcriptional activity suggests that c-Jun may indirectly inhibit AR function via activating a downstream target gene. Since cyclin D1 was shown to be a c-Jun target gene (Albanese et al., 1995; Bakiri et al., 2000; Herber et al., 1994) and an AR corepressor (Comstock et al., 2011; Petre et al., 2002; Petre-Draviam et al., 2003), we considered the possibility that cyclin D1 may mediate the inhibitory effect. However, we did not observe a significant increase in cyclin D1 expression at both mRNA and protein levels in LNCaP cells with c-Jun induction (data not shown). Thus, c-Jun may inhibit AR through transactivating downstream target genes other than cyclin D1. A microarray analysis of differentially expressed genes in LNCaP cells with or without c-Jun induction may help identify the critical target genes that are responsible for the AR inhibition.
One interesting finding is that c-Jun also down-regulated the expression of AR at the protein level after three days induction (Fig. 4A), whereas induction of c-Jun for one day only affect the expression of AR at the transcript level (Fig. 2B and 4B). Because of high stability of AR protein in LNCaP cells cultured in regular medium (Fig. 4D), it may take longer time (more than 24 hr) to see any significant change in protein level. Because PSA expression is reduced as early as 6 hr followed by c-Jun induction when AR protein remains unchanged (Fig. 2B), the down-regulation of AR protein is not essential for the inhibition of AR function. Nevertheless, sustained elevation of c-Jun expression could further suppress AR signaling by inhibiting AR expression. Thus, the elevated c-Jun level may inhibit AR signaling axis via a dual mechanisms; with a suppression of AR activity first and a down-regulation of AR protein level later. In addition to functioning as a transcriptional activator, c-Jun was also reported to suppress gene expression by binding to the promoter region (Ivanov et al., 2001; Schreiber et al., 1999). Further experiments are needed to clarify if c-Jun inhibits AR transcription directly via binding to the AP-1 sites identified in human AR promoter (Mizokami et al., 1994). Alternatively, c-Jun per se does not inhibit AR transcription. Instead, c-Jun target genes may inhibit AR transcription.
In conclusion, our results suggest that the transcriptional activity of c-Jun is required for its efficient inhibition of AR function. Given that both the AR activity and the proliferation of hormone naïve prostate cancer cells and CRPC cells are suppressed by c-Jun overexpression, identification of the critical c-Jun downstream target genes will provide a novel therapeutic strategy aimed at treating a subset of prostate cancer in which c-Jun down-regulation may underly the progression of the disease.
Supplementary Material
Highlights.
Overexpression of c-Jun efficiently inhibits AR activity.
The transcriptional activity of c-Jun is important for the AR inhibition.
c-Jun overexpression suppresses AR signaling and cell proliferation of hormone naïve and castration-resistant prostate cancer cells.
c-Jun may inhibit AR function indirectly through induction of a unknown target gene.
Acknowledgments
We thank Dr. Xiaoqi Liu (Purdue University) for kindly sharing lentiviral packaging vectors pHR′-CMV-ΔR8.20vpr and pHR′-CMV-VSV-G (Liu et al., 2006b). We thank Dr. Haojie Huang (Mayo Clinic, Rochester, MN) for kindly providing AR expression vector and Dr. Donald Tindall for providing the luciferase reporter driven by 5.8-kilobase pair PSA promoter (PSA-Luc). We also thank Dr. Bryce M. Paschal (University of Virginia Charlottesville, VA) for generously sharing luciferase reporters driven by MMTV promoter (MMTV-Luc), probasin promoter (ARR3tk-Luc) or synthetic androgen response elements (PGL3-ARE-Luc) (Ni et al., 2010). This work was supported, in part, by grants from the U.S. Army Medical Research Acquisition Activity, Prostate Cancer Research Program grant PC073098, and Purdue University Center for Cancer Research Small Grants Program. DNA sequencing was conducted in the Purdue University Center for Cancer Research Genomic Core Facility supported by NCI CCSG GA23168 to Purdue University Center for Cancer Research. CC Hsu was a recipient of Ronal W. Dollens Graduate Scholarship in the Life Sciences, Purdue Research Foundation Graduate Fellowship and Bilsland Dissertation Fellowship.
Abbreviations
- AP-1
activator protein 1
- CRPC
castration-resistant prostate cancer
- DBD
DNA-binding domain
- ARE
androgen-response element
- DHT
dihydrotestosterone
- bZIP
basic region leucine zipper
- TRE
TPA-responsive elements
- CRE
cyclic AMP-responsive elements
- Dox
doxycycline
- TMPRSS2
transmembrane protease, serine 2
- CBP
CREB binding protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Ali SA, Steinkasserer A. PCR-ligation-PCR mutagenesis: a protocol for creating gene fusions and mutations. Biotechniques. 1995;18:746–750. [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- Bao BY, Chuang BF, Wang Q, Sartor O, Balk SP, Brown M, Kantoff PW, Lee GS. Androgen receptor mediates the expression of UDP-glucuronosyltransferase 2 B15 and B17 genes. Prostate. 2008;68:839–848. doi: 10.1002/pros.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelfman C, Soderstrom TG, Brekkan E, Norlen BJ, Egevad L, Unge T, Andersson S, Rane A. Differential gene expression of steroid 5 alpha-reductase 2 in core needle biopsies from malignant and benign prostatic tissue. J Clin Endocrinol Metab. 1997;82:2210–2214. doi: 10.1210/jcem.82.7.4080. [DOI] [PubMed] [Google Scholar]
- Brown PH, Kim SH, Wise SC, Sabichi AL, Birrer MJ. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ. 1996;7:1013–1021. [PubMed] [Google Scholar]
- Bubulya A, Chen SY, Fisher CJ, Zheng Z, Shen XQ, Shemshedini L. c-Jun potentiates the functional interaction between the amino and carboxyl termini of the androgen receptor. J Biol Chem. 2001;276:44704–44711. doi: 10.1074/jbc.M107346200. [DOI] [PubMed] [Google Scholar]
- Bubulya A, Zhou XF, Shen XQ, Fisher CJ, Shemshedini L. c-Jun targets amino terminus of androgen receptor in regulating androgen-responsive transcription. Endocrine. 2000;13:55–62. doi: 10.1385/ENDO:13:1:55. [DOI] [PubMed] [Google Scholar]
- Bubulya A, Wise SC, Shen XQ, Burmeister LA, Shemshedini L. c-Jun can mediate androgen receptor-induced transactivation. J Biol Chem. 1996;271:24583–24589. doi: 10.1074/jbc.271.40.24583. [DOI] [PubMed] [Google Scholar]
- Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen SY, Cai C, Fisher CJ, Zheng Z, Omwancha J, Hsieh CL, Shemshedini L. c-Jun enhancement of androgen receptor transactivation is associated with prostate cancer cell proliferation. Oncogene. 2006;25:7212–7223. doi: 10.1038/sj.onc.1209705. [DOI] [PubMed] [Google Scholar]
- Chung BH, Mitchell SH, Zhang JS, Young CY. Effects of docosahexaenoic acid and eicosapentaenoic acid on androgen-mediated cell growth and gene expression in LNCaP prostate cancer cells. Carcinogenesis. 2001;22:1201–1206. doi: 10.1093/carcin/22.8.1201. [DOI] [PubMed] [Google Scholar]
- Comstock CE, Augello MA, Schiewer MJ, Karch J, Burd CJ, Ertel A, Knudsen ES, Jessen WJ, Aronow BJ, Knudsen KE. Cyclin D1 is a selective modifier of androgen-dependent signaling and androgen receptor function. J Biol Chem. 2011;286:8117–8127. doi: 10.1074/jbc.M110.170720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855–2863. doi: 10.1210/me.2007-0223. [DOI] [PubMed] [Google Scholar]
- Edwards J, Krishna NS, Mukherjee R, Bartlett JM. The role of c-Jun and c-Fos expression in androgen-independent prostate cancer. J Pathol. 2004;204:153–158. doi: 10.1002/path.1605. [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J Biol Chem. 1998;273:31853–31859. doi: 10.1074/jbc.273.48.31853. [DOI] [PubMed] [Google Scholar]
- Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. 2004;24:7806–7819. doi: 10.1128/MCB.24.17.7806-7819.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A. 2006;103:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res. 1991;51:3753–3761. [PubMed] [Google Scholar]
- Herber B, Truss M, Beato M, Muller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:1295–1304. [PubMed] [Google Scholar]
- Herrlich P. Cross-talk between glucocorticoid receptor and AP-1. Oncogene. 2001;20:2465–2475. doi: 10.1038/sj.onc.1204388. [DOI] [PubMed] [Google Scholar]
- Hsu CC, Hu CD. Critical Role of N-terminal End-localized Nuclear Export Signal in Regulation of Activating Transcription Factor 2 (ATF2) Subcellular Localization and Transcriptional Activity. J Biol Chem. 2012;287:8621–8632. doi: 10.1074/jbc.M111.294272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov VN, Bhoumik A, Krasilnikov M, Raz R, Owen-Schaub LB, Levy D, Horvath CM, Ronai Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol Cell. 2001;7:517–528. doi: 10.1016/s1097-2765(01)00199-x. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Karin M, Chang L. AP-1--glucocorticoid receptor crosstalk taken to a higher level. J Endocrinol. 2001;169:447–451. doi: 10.1677/joe.0.1690447. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Lamph WW. Cross-coupling of AP-1 and intracellular hormone receptors. Cancer Cells. 1991;3:183–185. [PubMed] [Google Scholar]
- Liu H, Deng X, Shyu YJ, Li JJ, Taparowsky EJ, Hu CD. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO J. 2006a;25:1058–1069. doi: 10.1038/sj.emboj.7601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006b;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lobaccaro JM, Poujol N, Terouanne B, Georget V, Fabre S, Lumbroso S, Sultan C. Transcriptional interferences between normal or mutant androgen receptors and the activator protein 1--dissection of the androgen receptor functional domains. Endocrinology. 1999;140:350–357. doi: 10.1210/endo.140.1.6418. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marivoet S, Van Dijck P, Verhoeven G, Heyns W. Interaction of the 90-kDa heat shock protein with native and in vitro translated androgen receptor and receptor fragments. Mol Cell Endocrinol. 1992;88:165–174. doi: 10.1016/0303-7207(92)90021-w. [DOI] [PubMed] [Google Scholar]
- Martini L. The 5alpha-reduction of testosterone in the neuroendocrine structures. Biochemical and physiological implications. Endocr Rev. 1982;3:1–25. doi: 10.1210/edrv-3-1-1. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Yeh SY, Chang C. Identification of 3′,5′-cyclic adenosine monophosphate response element and other cis-acting elements in the human androgen receptor gene promoter. Mol Endocrinol. 1994;8:77–88. doi: 10.1210/mend.8.1.8152432. [DOI] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtha PE, Zhu W, Zhang J, Zhang S, Young CY. Effects of Ca++ mobilization on expression of androgen-regulated genes: interference with androgen receptor-mediated transactivation by AP-I proteins. Prostate. 1997;33:264–270. doi: 10.1002/(sici)1097-0045(19971201)33:4<264::aid-pros7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Negri-Cesi P, Motta M. Androgen metabolism in the human prostatic cancer cell line LNCaP. J Steroid Biochem Mol Biol. 1994;51:89–96. doi: 10.1016/0960-0760(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol Cell Biol. 2010;30:1243–1253. doi: 10.1128/MCB.01891-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, Reuter VE, Scardino PT, Shen MM, Aronow BJ, Vickers AJ, Gerald WL, Abate-Shen C. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008;68:2132–2144. doi: 10.1158/0008-5472.CAN-07-6055. [DOI] [PubMed] [Google Scholar]
- Petre-Draviam CE, Cook SL, Burd CJ, Marshall TW, Wetherill YB, Knudsen KE. Specificity of cyclin D1 for androgen receptor regulation. Cancer Res. 2003;63:4903–4913. [PubMed] [Google Scholar]
- Petre CE, Wetherill YB, Danielsen M, Knudsen KE. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J Biol Chem. 2002;277:2207–2215. doi: 10.1074/jbc.M106399200. [DOI] [PubMed] [Google Scholar]
- Pfahl M. Nuclear receptor/AP-1 interaction. Endocr Rev. 1993;14:651–658. doi: 10.1210/edrv-14-5-651. [DOI] [PubMed] [Google Scholar]
- Salesi N, Carlini P, Ruggeri EM, Ferretti G, Bria E, Cognetti F. Prostate cancer: the role of hormonal therapy. J Exp Clin Cancer Res. 2005;24:175–180. [PubMed] [Google Scholar]
- Sato N, Sadar MD, Bruchovsky N, Saatcioglu F, Rennie PS, Sato S, Lange PH, Gleave ME. Androgenic induction of prostate-specific antigen gene is repressed by protein-protein interaction between the androgen receptor and AP-1/c-Jun in the human prostate cancer cell line LNCaP. J Biol Chem. 1997;272:17485–17494. doi: 10.1074/jbc.272.28.17485. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemshedini L, Knauthe R, Sassone-Corsi P, Pornon A, Gronemeyer H. Cell-specific inhibitory and stimulatory effects of Fos and Jun on transcription activation by nuclear receptors. EMBO J. 1991;10:3839–3849. doi: 10.1002/j.1460-2075.1991.tb04953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu YJ, Suarez CD, Hu CD. Visualization of AP-1 NF-kappaB ternary complexes in living cells by using a BiFC-based FRET. Proc Natl Acad Sci U S A. 2008;105:151–156. doi: 10.1073/pnas.0705181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Snoek R, Cheng H, Margiotti K, Wafa LA, Wong CA, Wong EC, Fazli L, Nelson CC, Gleave ME, Rennie PS. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39–47. doi: 10.1158/1078-0432.CCR-08-1726. [DOI] [PubMed] [Google Scholar]
- Sobel RE, Sadar MD. Cell lines used in prostate cancer research: a compendium of old and new lines--part 1. J Urol. 2005;173:342–359. doi: 10.1097/01.ju.0000141580.30910.57. [DOI] [PubMed] [Google Scholar]
- Strober W. Trypan blue exclusion test of cell viability. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.ima03bs21. Appendix 3, Appendix 3B. [DOI] [PubMed] [Google Scholar]
- Sun SY, Yue P, Lotan R. Induction of apoptosis by N-(4-hydroxyphenyl)retinamide and its association with reactive oxygen species, nuclear retinoic acid receptors, and apoptosis-related genes in human prostate carcinoma cells. Mol Pharmacol. 1999;55:403–410. [PubMed] [Google Scholar]
- Tamura K, Furihata M, Tsunoda T, Ashida S, Takata R, Obara W, Yoshioka H, Daigo Y, Nasu Y, Kumon H, Konaka H, Namiki M, Tozawa K, Kohri K, Tanji N, Yokoyama M, Shimazui T, Akaza H, Mizutani Y, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 2007;67:5117–5125. doi: 10.1158/0008-5472.CAN-06-4040. [DOI] [PubMed] [Google Scholar]
- Vacca A, Screpanti I, Maroder M, Petrangeli E, Frati L, Gulino A. Tumor-promoting phorbol ester and ras oncogene expression inhibit the glucocorticoid-dependent transcription from the mouse mammary tumor virus long terminal repeat. Mol Endocrinol. 1989;3:1659–1665. doi: 10.1210/mend-3-10-1659. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, Sellers WR, Benson JD. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- Wise SC, Burmeister LA, Zhou XF, Bubulya A, Oberfield JL, Birrer MJ, Shemshedini L. Identification of domains of c-Jun mediating androgen receptor transactivation. Oncogene. 1998;16:2001–2009. doi: 10.1038/sj.onc.1201697. [DOI] [PubMed] [Google Scholar]
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- Yang X, Chen MW, Terry S, Vacherot F, Chopin DK, Bemis DL, Kitajewski J, Benson MC, Guo Y, Buttyan R. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005;65:5263–5271. doi: 10.1158/0008-5472.CAN-05-0162. [DOI] [PubMed] [Google Scholar]
- Yuan H, Young CY, Tian Y, Liu Z, Zhang M, Lou H. Suppression of the androgen receptor function by quercetin through protein-protein interactions of Sp1, c-Jun, and the androgen receptor in human prostate cancer cells. Mol Cell Biochem. 2010;339:253–262. doi: 10.1007/s11010-010-0388-7. [DOI] [PubMed] [Google Scholar]
- Yuan X, Li T, Wang H, Zhang T, Barua M, Borgesi RA, Bubley GJ, Lu ML, Balk SP. Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am J Pathol. 2006;169:682–696. doi: 10.2353/ajpath.2006.051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.