

Abstract
G protein coupled receptor kinases (GRKs) phosphorylate the activated form of G protein coupled receptors (GPCRs) leading to receptor desensitization and down-regulation. We have recently shown that the chemokine receptor, CXCR2, couples to GRK6 to regulate cellular responses including chemotaxis, angiogenesis and wound healing. In this study, we investigate the role of GRK6 in tumorigenesis using murine models of human lung cancer. Mice deficient in GRK6 (GRK6−/−) exhibited a significant increase in Lewis lung cancer (LLC) growth and metastasis relative to control littermates (GRK6+/+). GRK6 deletion had no effect on the expression of proangiogenic chemokine or vascular endothelial growth factor (VEGF), but up-regulated matrix metalloproteinase (MMP)-2 and MMP-9 release, tumor-infiltrating PMNs and microvessel density. Since βarr2−/− mice exhibited increase LLC growth and metastasis similar to that of GRK6−/−we developed a double GRK6−/−/βarr2−/− mouse model. Surprisingly, GRK6−/−/βarr2−/− mice exhibited faster tumor growth relative to GRK6−/− or βarr2−/− mice. Treatment of the mice with anti-CXCR2 antibody inhibited tumor growth in both GRK6−/− and GRK6−/−/βarr2−/− animals. Altogether, the results indicate that CXCR2 couples to GRK6 to regulate angiogenesis, tumor progression and metastasis. Deletion of GRK6 increases the activity of the host CXCR2, resulting in greater PMN infiltration and MMP release in the tumor microenvironment thereby promoting angiogenesis and metastasis. Since GRK6−/−/βarr2−/− showed greater tumor growth relative to GRK6−/− or βarr2−/− mice, the data further suggest that CXCR2 couples to different mechanisms to mediate tumor progression and metastasis.
Keywords: G protein-coupled receptor kinase, Chemokine, angiogenesis, Lewis Lung carcinoma cell line, Rat Basophilic Leukemia, Neutrophils, β-arrestin, Matrix Metalloproteinases, Transgenic/Knockout Mice
Introduction
G protein coupled receptor kinases (GRKs) are a versatile family of kinases that play a critical role in G protein-coupled receptor (GPCR) homologous desensitization. GRKs phosphorylate specific serine and threonine residues in the cytoplasmic domains of the activated receptor thereby promoting receptor/arrestin interaction and uncoupling of the receptor from its G protein (1). The GRK family consists of seven members (GRK 1 to 7) with differing patterns of expression. GRK1 and GRK7 are restricted to the visual system; GRK4 is found predominantly in the testis whereas GRK 2, 3, 5, 6 are expressed in all mammalian cells (1, 2). Although, all GRKs have similar structural organization, they differ in their mechanisms of activation (2, 3). GRK2 and GRK3 are pleckstrin homology (PH) domain containing proteins, which, upon receptor activation, are recruited to the membrane by Gβγ (4–6). GRK4, GRK5 and GRK6, however, are membrane-associated proteins and are directly activated by the receptor/ligand complex (7–9).
CXCR2 is a member of the CXC subfamily of chemokine receptors that interacts with CXCL1, 2, 3, 5, 7 & 8 with high affinity (10, 11). A hallmark of this subset of chemokines is the expression of a glutamic acid-leucine-arginine (ELR) motif in their amino terminus which appears to be critical for receptor-mediated angiogenesis (12, 13). Upon activation by ligand, CXCR2 becomes desensitized and down-regulated (14–16). Desensitization of CXCR2 occurs via two mechanisms: protein kinase C (PKC)-dependent or heterologous; and GRK-dependent or homologous (14, 17, 18). We have recently shown that CXCR2 homologous desensitization occurs predominantly through phosphorylation of the receptor by GRK6 (19). Murine neutrophils and RBL-2H3 cells deficient in GRK6 expression exhibited diminished CXCR2 phosphorylation, desensitization and down-regulation(19). GRK6−/− mice also displayed greater neutrophils accumulation at the site of inflammation, enhanced angiogenesis and faster wound closure relative to wild type (GRK6+/+) animals, indicating augmentation of CXCR2 dependent functions (19). These results mirrored those previously obtained with the βarrestin-2 deficient (βarr2−/−) mouse model and suggest that CXCR2 down-modulation via the GRK6/βarr-2 axis is critical in modulating inflammatory responses (20, 21).
We have previously demonstrated that βarr2−/− mice displayed faster tumor growth and metastasis relative to wild type animals (21). This effect was shown to be mediated via increase CXCR2-mediated nuclear factor-κB (NF-κB) activation (21). Since GRK-mediated phosphorylation is critical for βarr-2 association with the receptor, we hypothesized that GRK6 inhibition could also promote CXCR2-mediated tumor progression and metastasis. To test this hypothesis, we have used murine models of heterotopic Lewis lung cancer (LLC) and tail vein lung metastasis in GRK6−/− mice, GRK6−/−/βarr2−/− double knockout and control littermates. The data herein indicated that GRK6 depletion promoted angiogenesis, tumor development and lung metastasis. These effects appear to be mediated through increased activation of the receptor leading to greater PMN accumulation and protease release in the tumor microenvironment thereby promoting angiogenesis and tumor metastasis. Interestingly, GRK6−/−/βarr2−/− animals showed greater tumor growth than that of GRK6−/− or βarr2−/−. Taken together, the data indicate that CXCR2 couples to different mechanisms to promote angiogenesis and tumor development.
Material and Methods
Materials
Lewis Lung carcinoma cell line (CRL-1642) was purchased from American Type Culture Collection. Anti-CD3, CD4, CD8a, CD45, NK1.1 and Ly6 were purchased from BD Pharmingen. Anti-CXCR2, CXCL1/KC, CXCL2/MIP-2, VEGF, MMP2 and MMP9 antibodies; recombinant murine CXCL1/KC, CXCL2/MIP-2, CXCL12/SDF1, VEGF, MMP2 and MMP9 proteins; MMP2 and MMP9 ELISA kits; CXCR2 inhibitor SB250002, and mouse chemokine array kits were purchased from R&D systems. Goat anti-mCXCR2 antiserum was kindly provided by Dr. Robert M. Strieter, (School of Medicine, University of Virginia, Charlottesville, VA). Monoclonal anti-β-actin and anti-factor VIII related antigen antibodies, 3, 3', 5, 5’-tetramethylbenzidine (TMB), ExtrAvidin-peroxidase, DNAse and collagenase were purchased from Sigma Life Sciences. Microtiter plates and Bouin’s fixative were purchased from VWR international. Gelatin-pre-cast Ready Gel Zymogram Gels were obtained from Bio-Rad Laboratories Inc. Novex Zymogram Developing Buffer was obtained from Invitrogen (Life Technologies). Monoclonal antiphospho-NF-κB-p65 (Ser536) and polyclonal anti-p65-NF-κB Abs were purchased from Cell Signaling Technology. All the other reagents were obtained from commercial sources.
Animals
All experiments were approved by and conformed to the guidelines of the Animal Care and Use Committee of North Carolina Central University, Durham, NC. GRK6−/− and βarr2−/− mice (C57BL/6 background) were kindly provided by Dr. Robert J. Lefkowitz (Duke University Medical Center, Durham, NC). Male and female mice were evaluated, and controls were age- and sex-matched littermates. All mice were genotyped at 21 days old; DNA samples were prepared from the tail tips with Extract-N-Amp tissue PCR kit from Sigma and subjected to triplex PCR as described (21, 22). The GRK6−/−/βarr2−/− double knockout mouse model was generated by backcrossing GRK6−/− and βarr2−/− animals. Double knockout animals were confirmed by RT-PCR.
Heterotopic Lewis lung carcinoma model
LLC was a spontaneously occurring lung cancer in C57BL/6 mice. The Lewis lung carcinoma cell line (CRL-1642) was routinely maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat inactivated fetal bovine serum, 1.5 g/L sodium bicarbonate, 4 mM/L glutamine, 100 µg/mL streptomycin, and 100 IU/mL penicillin. GRK6+/+ and GRK6−/− mice (6–8 weeks, n=14) were injected s.c. with LLC cells (5×105 cells in 100 µL). After each week, mice were euthanized by CO2 asphyxiation, tumors were dissected from mice and measured with a Thorpe caliper (VWR). Tumor volume was calculated using the formula: Volume = (d1 × d2 × d3) × 0.5236; where dn represents the three orthogonal diameter measurements (21, 23). Tumor and tissue specimens were either fixed in buffered formalin or processed for ELISA or FACS analysis.
Tail vein metastasis model
The LLC cells (3×105 viable cells per 100µL) were injected via the tail vein of GRK6+/+ and GRK6−/− mice (6–8 weeks, n=8). The mice were observed daily for any sign of respiratory distress and were euthanized by CO2 after 4 weeks. The lungs were removed and inflated with Bouin’s fixative. The number of metastatic nodules on the lungs was counted with the aid of a dissecting microscope (21, 24). For survival experiments, GRK6+/+ and GRK6−/− mice (6–8 weeks, n=14) were injected as described above and observed daily for mortality. The experiment was terminated when all the mice died from either group.
FACS analysis of single cell isolates from heterotopic LLC tumors
Tumors were isolated from mice (n=6), minced with scissors to fine slurry and incubated in digestion buffer (RPMI 1640, 5% FBS, 1mg/ml collagenase and 30µg/ml DNAse) at 37°C for 45 min. Cells were washed and cell counts and viability were determined using trypan blue exclusion on a hemocytometer. Cells (2×106) were resuspended in FACS analysis buffer and stained with PE conjugated anti-mouse CD3, CD4, CD45, CD8a, NK1.1, Ly6, or Factor VIII related Ag antibodies. Cells were also stained with rat anti-mouse CXCR2 and goat anti-rat PE conjugated secondary antibodies (21, 23). Stained cells were analyzed on a FACScan Flow Cytometer using Cell Quest software (BD Biosciences).
CXCL1, CXCL2, CXCL12, VEGF and MMPs levels in tumors
Heterotopic LLC tumors (n=10) were harvested four weeks post-inoculation. One gram of dissected tumors was homogenized in 10 ml PBS. The quantity of murine CXCL1, CXCL2, CXCL12, VEGF, MMP2 and MMP9 present in tissue homogenates was determined by specific ELISA, using a modification of the double-ligand method, as previously described (21, 23). Briefly, flat-bottom 96-well microtiter plates were coated with 100 µl/well of specific polyclonal anti-mouse CXCL1, CXCL2, CXCL12,VEGF, MMP2 and MMP9 (1µg/ml in coating buffer, pH 9.5) for 24 h at 4°C, and then washed three times with PBS, pH 7.5, plus 0.05% Tween 20 (wash buffer). Plates were blocked with 1% BSA in PBS for 90 min at 37°C and then washed three times with wash buffer. A total of 100 µl of supernatant from each homogenate was added in plates and then incubated at 37°C for 90 min. Plates were washed three times; 100 µl of biotinylated polyclonal anti-murine CXCL1, CXCL2, CXCL12, VEGF, MMP2 and MMP9 (diluted in PBS, pH 7.5, 0.05% Tween 20) was added and then incubated at 37°C for 45 min. Plates were washed three times, 100 µl of ExtrAvidin-peroxidase conjugate was added and were incubated for another 45 min at 37°C. Plates were washed again and 100 µl of 3, 3', 5, 5'-tetramethylbenzidine chromogenic substrate was added. Plates were incubated at room temperature for 20–30 min, and the reactions were terminated by the addition of 100 µl/well of 1M of H2SO4. Plates were read at 450 nm in an automated microplate reader (Perkin Elmer). The amount of mouse CXCL1, CXCL2, CXCL12, VEGF, MMP2 and MMP9 present was determined by interpolation of a standard curve generated by known amounts of recombinant mouse CXCL1, CXCL2, CXCL12, VEGF, MMP2 and MMP9 respectively.
MMP-9 activity
MMP-9 activity was determined as previously reported (25, 26). Briefly, tumor lysates (20 µg) or recombinant murine MMP-9 (5 ng), as positive control, were electrophoresed in 10% gelatin-pre-cast Ready Gel Zymogram Gels (Bio-Rad Laboratories). The gels were washed twice in 2.5% Triton X-100 for 30 min and incubated overnight at 37°C in Novex Zymogram Developing Buffer (Life Technologies, Carlsbad, CA). The gels were stained with 0.1% Comassie Blue (R250) for 1 hour and then destained in 5% acetic acid, 10% methanol. The activity of the MMP9 relative to the control standard was determined by densitometric scanning of the gels with a UV transilluminator (BioDoc-It Imaging System, UVP, Upland, CA) and the images were analysed by Adobe Photoshop (Adobe Systems, Mountain View, CA).
Treatments in vivo
GRK6+/+ and GRK6−/− mice (6–8 weeks) were injected s.c. with LLC cells as outlined above. Mice (n=6) were injected i.p. with 500 µl of either neutralizing goat anti-mouse CXCR2, control (preimmune) serum, or no treatment every day for 4 weeks. In subsequent experiments, LLC tumor-bearing GRK6+/+ and GRK6−/− mice (n = 5) were treated i.p. with the CXCR2 inhibitor SB225002 (5mg/kg/day), control (PBS), or no treatment, every day, for four weeks. Tumor volume was measured as described above.
Quantitation of microvessel density
Paraffin-embedded tumor tissues from GRK6+/+ and GRK6−/− mice were processed for immunohistochemical localization of factor VIII-related antigen, as previously described (27). Briefly, tissue sections were dewaxed with xylene and rehydrated through graded series of alcohol. Tissue sections were treated with 0.10M citric acid buffer in a heated pressure cooker for 10mins. Slides were blocked with normal goat serum, and overlaid with 1:500 dilution of either control (rabbit) or anti-factor VIII-related antigen antibody for overnight at 4°C. Slides were then rinsed and overlaid with secondary biotinylated goat anti-rabbit IgG (1:200) and incubated for 60 min. After washing with PBS, slides were overlaid with a 1:200 dilution of ExtrAvidin-peroxidase conjugate and incubated for 60 min. 3’3 diaminobenzedine tetrahydrocloride was used for chromogenic localization of factor VIII-related antigen. After optimal color development, sections were washed with sterile water, counterstained with Mayer’s hematoxylin, and dehydrated into the graded series of alcohol and mounted. Quantitation of microvessel density was performed using the previously described method with slight modifications (27). Tumor specimens (n=10) were scanned at low magnification (20X) to identify vascular hotspots. Areas of greatest vessel density (six from each specimen) were then examined under higher magnification (200X) and counted with the help of grid eyepiece. Any distinct areas of positive staining for factor VIII-related antigen were counted as single vessels. Results were expressed as the number of microvessels per mm2.
Immunoblotting
For GRK6 expression analysis, LLC cells, zymosan-elicited PMN (1 × 106) isolated from GRK6+/+ and GRK6−/− mice, wild type RBL-2H and GRK6 depleted RBL (RBL-GRK6−/−) were lysed in radioimmune precipitation assay buffer (RIPA) and assayed for protein concentration. Twenty micrograms of proteins was resolved on 10% SDS-PAGE, transferred to nitrocellulose paper, probed with mouse monoclonal anti-GRK6 antibody, and detected with HRP-conjugated goat anti-mouse antibody and ECL. For NF-kB-p65 phosphorylation, zymosan-elicited PMN (1 × 107) were treated with or without CXCL1 (1 µM) in PBS for 10 min at 37°C. Then, cells were washed, lysed, and immunoblotted with either antiphospho-NF-κB-p65 or anti-NF-κB-p65 (21).
Chemokine measurement in LLC cells
Chemokine levels were measured using the commercially available Mouse Chemokine Array (R&D Systems) according to the protocol supplied by the manufacturer. LLC cell lysates were prepared as described in the protocol and assayed for protein concentrations. Five hundred micrograms proteins were run on each array.
Statistical analyses
Results are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Differences between groups were determined using one-way ANOVA or Student t test (two-tailed), as appropriate. A P value <0.05 was considered statistically significant.
Results
GRK6 depletion enhanced tumorigenesis and metastasis
To assess the role of GRK6 in tumor development, 6–8 week old male GRK6−/− mice and control littermates (GRK6+/+) were injected heterotopically with LLC cells (5 × 105) under the dorsal skin. Tumor size was measured 4 weeks after injection. As shown in figure 1A tumor growth occurred faster in GRK6−/− mice as compared to GRK6+/+. Time-course of tumor growth in both male (Fig 1B) and female (Fig. 1C) mice showed a significant difference in tumor size relative to control animals. This result indicates that the actions of GRK6 oppose tumor growth in our subcutaneous tumor model.
Fig 1. Knockdown of GRK6 promotes heterotopic LLC tumor growth.

GRK6+/+ and GRK6−/− mice (n=11) were injected subcutaneously with LLC cells (1×105). Heterotopic tumor growth was measured weekly with a Thorpe caliper. Tumor volume was calculated using the formula: volume = (d1 × d2 × d3) × 0.5236, where the dn represents the three orthogonal diameter measurements. Representative photographs of tumor dissected from GRK6+/+ and GRK6−/− mice (A). Heterotopic LLC tumor growth over four weeks in male (B) and female (C) mice. The experiments were repeated three times with similar results. * P<0.05, ** P<0.005.
We next determined the role of GRK6 deficiency in lung metastasis by measuring the number of metastatic nodules on the lungs. Colonization of the lung was significantly greater in GRK6−/− mice as compared to GRK6+/+ (Fig. 2A). The number of metastatic tumor nodules was ~6 fold higher in lungs from GRK6−/− mice relative to wild type animals (Fig. 2B). GRK6 deletion also caused a significant reduction in the median survival of GRK6−/− mice (~22 days) relative to the GRK6+/+ mice (~40 days) (Fig. 2C).
Figure 2. GRK6 inhibition increases lung metastasis and mortality.

For lung metastasis (A) LLC cells (3×105) were injected via the tail vein of GRK6+/+ and GRK6−/− mice (n=10). The mice were euthanized by CO2 after 4 weeks; lungs were removed and inflated with Bouin’s fixative. Photographs representative of LLC cell colonization and growth in lungs of GRK6+/+ and GRK6−/− mice. B) The number of metastatic nodules on the lungs was counted with the aid of a dissecting microscope. C) For survival rate, mice (n=14) were injected LLC cells via tail vein for lung metastasis as described above and observed daily up to sixty days for the mortality. GRK6−/− mice displayed decreased survival rate compared to GRK6+/+. The experiments were repeated twice with similar results. *** P<0.0001.
We also measured the expression of GRK6 in LLC tumor cells, peritoneal neutrophils isolated from GRK6−/− and GRK6+/+ mice, control RBL-2H3 cells (RBL- GRK6+/+) and RBL-2H3 cells in which GRK6 was stably knockdown by shRNA (RBL-GRK6−/−) (19). As shown in figure 1-S, GRK6 expression in LLC cells (lane 3) was similar to that of RBL-GRK6+/+ cells (lane 4) and PMNs from GRK6+/+ mice (lane 1). GRK6 expression was inhibited in PMNs from GRK6−/− (lane 2) mice as well as RBL-GRK6−/− cells (lane 5). This result suggests that the effect of GRK6 inhibition in tumor growth is due to the host.
Effect of GRK6 depletion on chemokine, VEGF and matrix metalloproteinases (MMPs) expression
To determine the effect of GRK6 depletion on chemokine and proangiogenic factor expression, we measured the levels of CXCL1/KC, CXCL2/MIP-2, CXCL12/SDF-1, VEGF, MMP2 and MMP9 in the homogenate of the LLC tumors from GRK6−/− and control littermates. No significant changes in the levels of expression of CXCL1 (Fig. 3A), CXCL2 (Fig. 3B), CXCL12 (Fig. 3C) or VEGF (Fig. 3D) were observed. MMP-2 (Fig. 3E) and MMP-9 (Fig. 3F) expression levels, however, were significantly enhanced in tumors from GRK6−/− animals relative to GRK6+/+. The increased MMP-9 expression in GRK6+/+ was further assessed by immunoblotting. As shown in figure 3G, tumor homogenate from GRK6−/− mice (lane 2) showed a marked increase in MMP-9 expression as compared to GRK6+/+ (lane 1). MMP-9 activity in tumor homogenates was also examined using gelatin zymography. GRK6−/− mice (Fig. 3H, lane 2; and Fig. 3I, closed bar) exhibited significantly greater MMP-9 activity relative to GRK6+/+ (Fig. 3H, lane 3; and Fig. 3I, open bar). Recombinant murine MMP-9 (Fig 3H, lane 1) was used as positive control.
Figure 3. Effect of GRK6 deficiency on production of proangiogenic factors.

Levels of CXCL1/KC (A), CXCL2/MIP-2 (B), CXCL12/SDF-1 (C), VEGF (D), MMP2 (E) and MMP9 (F) in the homogenate of heterotopic LLC tumors from GRK6+/+ and GRK6−/− mice (n=6) were determined by specific double antibody sandwich ELISA as described in materials and methods.*(P<0.05), ** (P<0.005). G) Tumor lysates were assayed for MMP9 expression by immunoblotting. Twenty µg of proteins was resolved on 10% SDS-PAGE, transferred to nitrocellulose paper, and probed with rabbit polyclonal anti-MMP9 antibody. The experiment was repeated twice with similar results. H) Representative zymogram of MMP2 and MMP9 activity. Murine recombinant MMP2 and MMP9 (5 ng each, lane 1) or tumor lysates (20 µg) from GRK6−/− animals (lane 2) or wild-type GRK6+/+ were subjected to electrophoresis without heat denaturation or thiol-reduction in 10% gelatin-pre-cast Ready Gel Zymogram Gels and assayed for MMP activity as described in Materials and Methods. I) MMP activity was calculated from MMP9 and MMP2 clear band density. The data shown are average from three independent experiments. * P<0.05.
To further assess the role of GRK6 depletion in angiogenesis, tumors were stained with an antibody specific for Factor VIII-related antigen for determination of microvessels density. Tumors from GRK6−/− mice (Fig. 4B and 4C, closed bar) showed a statistically significant increase in the number of microvessels density as compared to tumors from GRK6+/+ mice (Fig. 4A and 4C, open bar).
Figure 4. GRK6 deficiency promotes angiogenesis in LLC tumors.
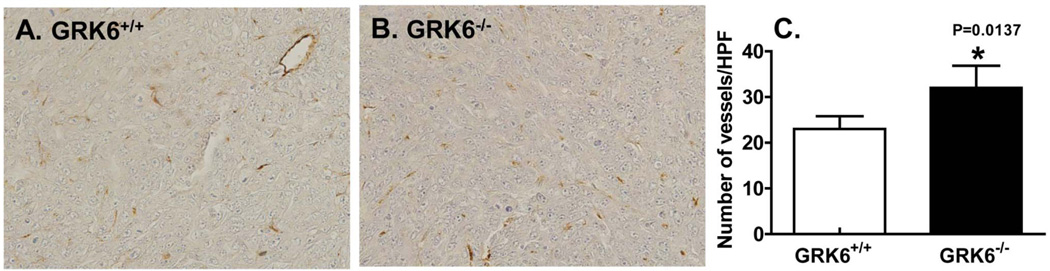
Heterotopic LLC tumors were dissected from GRK6+/+ and GRK6−/− mice (n=6) after four weeks, fixed in 10% neutral buffered formaldehyde and embedded in paraffin. Tissue sections were immunostained with factor VIII related antigen and number of microvessels in 1.0 mm2 area were counted by light microscope with a grid eyepiece. * P<0.05.
Effect of GRK6 depletion on intratumor leukocyte infiltrations
We next analyze the intratumor leukocyte infiltration of cell isolates from LLC tumors of GRK6−/− and GRK6+/+ mice. As shown in Table 1, no significant difference was found in tumor infiltrating NK+CD3+CD4+CD45+ and CD8a+ in GRK6−/− as compared to GRK6+/+ mice. However, significant increases were observed in PMN, CXCR2 and Factor VIII-related antigen-expressing cells in GRK6−/− relative to GRK6+/+ (Table 1).
Table 1. FACS analysis of intratumor-infiltrating leukocytes subpopulations.
Single cell isolates from heterotopic LLC tumors from GRK6+/+ and GRK6−/− mice (n=6) were stained for different leukocyte subpopulations (NK, CD3, CD4, CD45, CD8a, PMN, CXCR2 and factor-VIII) and analyzed by FACScan Flow Cytometer using Cell quest software.
| NK+ | CD3+ | CD4+ | CD45+ | CD8+ | PMN+ | CXCR2+ | Factor VIII | |
|---|---|---|---|---|---|---|---|---|
| GRK6+/+ | 2.71±0.30 | 3.22±0.569 | 3.53±0.56 | 18.81±1.30 | 1.68±0.341 | 1.91±0.42 | 17.92±1.39 | 21.87±2.29 |
| GRK6−/− | 2.92±0.29 | 4.01±0.52 | 3.73±0.69 | 26.78±3.36 | 1.787±0.50 | 4.88±0.24** | 30.09±6.15* | 37.44±3.35** |
(P<0.05),
(P<0.005
Inhibition of CXCR2 diminished the effect of GRK6 depletion in LLC tumor development
To assess the role of CXCR2 in GRK6 mediated tumor progression, a CXCR2 inhibitor SB225002 and a CXCR2-neutralizing antibody were used to block the receptor in the heterotopic model. GRK6−/− mice treated with SB225002 showed a significant decrease (~45%) in tumor size relative to non-treated animals (Fig. 5A, closed bars). Treatment of GRK6+/+ animals with SB225002, however, had no significant effect in tumor size (Fig. 5A, open bars). Treatment of both GRK6+/+ (Fig 5B, open bars) and GRK6−/− (Fig 5B, closed bars) mice with the anti-mouse CXCR2 antibody caused a significant decrease (~85% for GRK6+/+ and ~73% for GRK6−/−) in tumor size relative to untreated animals. The inhibitory effect of the murine CXCR2 antibody in tumor growth was greater than that of the pharmacological inhibitor SB225002 (~85% versus 45%), relative to wild type animals. This is likely due to de dose of SB225002 used in these experiments (5 mg/kg/day). Greater inhibitory effects in tumor volume were obtained with higher dose 7.5 and 10 mg/kg/day (data not shown). However, because of the reduced survival rates (80–100 % of the mice died between week 2–3) and the animals aggressive behavior, we were limited to 5 mg/kg/day.
Figure 5. Blockade of CXCR2 inhibits heterotopic LLC tumor growth in mice.

GRK6+/+ and GRK6−/− mice were injected s.c. with LLC cells (1×105). For CXCR2 inhibition, mice were treated with (A) SB225002 or vehicle (n=10) and (B) neutralizing anti-mCXCR2 serum (n=6) or goat preimmune serum (n=6) daily. After four weeks mice were euthanized by CO2 and tumor volume was measured with a Thorpe caliper. ** P<0.05, *** P<0.001.
Are the effects of GRK6 and βarrestin-2 inhibition in LLC growth additive?
We have previously shown that deletion of βarr2 promoted CXCR2-mediated angiogenesis, tumor progression and metastasis (19). To determine whether the effects of βarr2 and GRK6 deficiency are additive, we generated a double knockout (GRK6−/−/βarr2−/−) animal model (Fig 6A). Six-8 weeks old male GRK6−/−/βarr2−/−βarr2−/− and wild type (WT) mice were injected heterotopically with LLC cells (5 × 105) under the dorsal skin and tumor size was measured 3 weeks after injection. As shown in figure 6B and 6C, GRK6−/−/βarr2−/− mice exhibited faster tumor growth relative to βarr2−/− or WT animals.
Figure 6. Effects of GRK6 and βarr2 knockdown in heterotopic LLC tumor growth.

A) Double knockout GRK6−/−/βarr2−/− mice were generated by backcrossing GRK6−/− with βarr2−/− animals and genotyped by PCR analysis. B) GRK6−/−/βarr2−/−GRK6+/+/βarr2−/−and wild type (GRK6+/+/βarr2+/+) mice (n=6–9) were injected subcutaneously with LLC cells (1×105). Heterotopic LLC tumors were grown over four weeks and dissected. Shown are representative photographs of 3 experiments. C) After four weeks mice were euthanized by CO2 and tumor volume was measured with a Thorpe caliper. * P<0.05, ** P<0.005, *** P<0.0001.
We also examined the effect of CXCR2-neutralizing antibody in GRK6−/−/βarr2−/ tumor growth. GRK6−/−/βarr2−/ mice were injected heterotopically with LLC cells (2.5 × 105; the number of cells was reduced in half due to tumor size and concern by the animal facility) and treated daily with murine CXCR2 antiserum or goat serum (control). As shown in figure 7, mCXCR2 treated animals exhibited a significant decreased (~8%) in tumor growth as compared to control animals. These suggest that the effects of βarr2 and GRK6 deletion in CXCR2-mediated LLC tumor growth are additive.
Figure 7. Anti-mCXCR2 inhibits tumor growth in GRK6−/−/βarr2−/− animals.

Male double knockout GRK6−/−/βarr2−/− mice were injected s.c. with LLC cells (0.5×105). For CXCR2 inhibition, mice were treated with neutralizing anti-mCXCR2 serum (n=5) or goat preimmune serum (n=5) daily. A) Representative photographs of tumor dissected after four weeks. B) Heterotopic LLC tumor growth was measured weekly with a Thorpe caliper. The experiments were repeated twice with similar results. * P<0.05.
Discussion
CXCR2 binds the proangiogenic ELR-positive (ELR+) chemokines to mediate cellular responses including chemotaxis and angiogenesis. Elevated expression levels of CXCR2 were observed in several cancer cell types including melanoma, lung, gastric and colon carcinoma cells, and were shown to be associated with enhanced angiogenesis (28–32). Knockdown of CXCR2 in animal models of cancer was also shown to inhibit tumor growth and metastasis in several mouse models of cancers including melanoma, renal cell carcinoma and LLC (23, 33, 34). We have previously shown that deletion of βarr-2, a key regulator of CXCR2 functions, decreased receptor desensitization but promoted LLC progression and metastasis in mice (21). Our recent studies using GRK6 deficient RBL-2H3 cells and mouse models of inflammation have shown that GRK6-mediated phosphorylation of CXCR2 plays an important role in receptor desensitization and down-regulation as well as receptor-induced angiogenesis and wound closure (19). In this study we investigated the role of GRK6 depletion in CXCR2-mediated tumorigenesis. The results herein clearly demonstrated that, as was the case for βarr2−/− mice, deletion of GRK6 promoted CXCR2 mediated tumor progression and metastasis. First, heterotopically implanted LLC tumors in GRK6−/− mice showed a significant increase in tumor growth compared with wild-type mice (Fig. 1A–C). Second, in the tail vein metastasis assay, the number of metastatic nodules significantly increased in the lung of GRK6−/− mice (Fig. 2A–B). Third, survival rate of GRK6−/− mice decreased very significantly relative to GRK6+/+ (Fig. 2-C). Fourth, tumors from GRK6−/− mice showed significantly greater microvessels density as compared to wild type animals (Fig. 4). And, fifth, treatment of the mice with anti-CXCR2 antibody or the CXCR2 specific inhibitor, SB225002, abolished the effect of GRK6 depletion (Fig. 5).
Up-regulation of ELR+ chemokines in the tumor microenvironment is known to promote tumor angiogenesis (35, 36). In barr2−/− animals, the increased tumor development and metastasis correlated with enhanced production of CXCL1, CXCL2 and vascular endothelial growth factor (VEGF) (21). Surprisingly, tumors from GRK6−/− animals displayed levels of CXCL1, CXCL2 and VEGF similar to those of wild type GRK6+/+ mice (Fig. 3-A, B and D). These results contrast with the βarr2−/− LLC model and suggest that the increased tumor growth and metastasis caused by GRK6 inhibition likely occurs via a mechanism different than that of induction of pro-angiogenic chemokine production. Indeed, while βarr2−/− mice displayed significant increase in NF-κB activity relative to βarr2+/+ (21), CXCL1-induced NF-κB p65 phosphorylation in peritoneal neutrophils from GRK6−/− animals was similar to that of control GRK6+/+ (data not shown). Interestingly, both matrix metalloproteinase-2 (MMP-2) and MMP-9 were up-regulated in GRK6−/− tumors relative to wild type GRK6+/+ (Fig. 3E, F & G). Several studies have shown that degradation of the extracellular matrix by MMPs is critical for endothelial cells angiogenesis (37–39). The CXCL8/CXCR2 axis was also shown to mediate tumor angiogenesis and metastasis via up-regulation of the MMP2 and MMP9 (40, 41). Peritoneal neutrophils and RBL-2H3 cells deficient in GRK6 exhibited increased CXCR2-mediated G protein activation and exocytosis (19). Therefore, it is possible that the increase in tumor angiogenesis and metastasis observed in GRK6 deficient animals is due to increased production of MMPs mediated by the host CXCR2 (Fig. 3E, F & G). Supporting that contention is that western blot analysis and gelatin zymography showed significant increase in MMP-9 expression and activity in tumor homogenate from GRK6−/− mice relative to GRK6+/+ animals (Fig. 3G, H & I)
The infiltration of immune cells into the tumor microenvironment is a key component of the tumor immunity (42). GRK6−/− depletion had no effect on T cells infiltration into the tumors but caused a significant increase in the level of PMNs (Table 1). These observations also contrast with the βarr2−/− model of LLC which displayed a significant decrease in T cell infiltration but no effect in PMN levels in the tumor microenvironment, relative to tumors from βarr2+/+ mice (21). The role of PMNs in tumor infiltrates remains unclear. They have been associated with both better prognosis due to their phagocytic activities and poorer outcomes due to their abilities to release pro-angiogenic factors such as chemokines, VEGF and MMPs (43, 44). Interestingly, the elevated levels of PMNs observed in GRK6−/− tumor infiltrates in this study, correlated with enhanced expression of MPP-2 and MPP-9 (Fig 3-E and F). Thus, the increased tumor growth and decreased survival rate of GRK6−/− animals compared to control GRK6+/+ could be a consequence of PMNs increased activity resulting in greater secretion of proteases and tissue digestion thereby promoting angiogenesis.
LLC cells express both GRK6 and CXCR2 (Fig. 1S and Ref 23). Mice deficient in CXCR2 showed decreased tumor progression and metastasis (23). Thus, as was the case for the βarr2 knockout model, the enhanced tumor growth and metastasis exhibited by GRK6−/− animals is likely due to increased CXCR2 activity of the host endothelial cells. Supporting that contention is that immunostaining of tumor sections with anti-factor VIII-related antigen (specific for endothelial cells) showed a significant increase in microvessel density in GRK6−/− mice relative to GRK6+/+ mice (Fig 4). Keane et al (23) have also shown that in LLC tumors, the cells expressing CXCR2 are predominantly the vascular endothelial cells.
Chemokine profiling of LLC cells using a mouse chemokine array revealed that these cells predominantly express KC/CXCL1 (Fig. 2S). Whether LLC produced chemokines' play a role in the enhanced PMN infiltrations and/or increased tumor growth in GRK6−/− animals remains unclear. However, both GRK6−/− and GRK6+/+ tumors showed similar levels of CXCL1 and CXCL2 expression (Fig 3A & B). Furthermore, mice deficient in CXCR2 or treated with a CXCR2 blocking antibody, displayed decrease chemokine expression and tumor growth, but similar levels of PMN in the tumor infiltrates (23). Raghuwanshi et al (21) have also shown that tumors from βarr2−/− animals displayed increase CXCL1 and CXCL2, but similar level of PMNs in the tumor infiltrates. Taken together, these results further suggest that the effect of GRK6 deletion in tumor growth is due to increase CXCR2 activity in the host but not the LLC cells.
Since the effects of GRK6 and βarr2 in CXCR2-induced tumor growth appeared to be mediated via distinct mechanisms we hypothesized that deletion of the two molecules could have in additive effect. Indeed, the double knockout GRK6−/−βarr2−/− displayed tumor growth at a faster rate than those of the single knockout animals (Fig. 6). The size of the GRK6−/−βarr2−/− tumors at week three was similar to those of βarr2−/− or GRK6−/− at week four (data not shown). These results further indicate that CXCR2 modulate LLC tumor development through different mechanisms. One could be through βarr-2-mediated regulation of NF-κB and expression of proangiogenic factors (21), and another through activation of GRK6 and production of MMPs.
In summary, the data herein demonstrate that GRK6 plays a feedback regulatory role in CXCR2-mediated tumor development, invasion, and metastasis. In contrast to βarr2, these processes appear to be mediated through increased activity of CXCR2 resulting in greater production of matrix MMPs and enhanced angiogenesis. Both CXCR1 and CXCR2 were shown to mediate angiogenesis via increase MMPs production (40, 41). Thus, an unanswered question is that whether GRK6 deletion would also enhance CXCR1 mediated tumor growth? To date, conflicting reports exist concerning the expression and function of a murine homologue of CXCR1 (45–47). Fan and al (47) have recently identified a possible mouse CXCR1 that binds CXCL6 and CXCL8 with high affinity. Studies in RBL-2H3 cell stably expressing CXCR1, however, have shown that the receptor couples predominantly to GRK2 to mediate and regulate cellular responses (19). Therefore, it is unlikely that deletion of GRK6 would have a significant effect in CXCR1-mediated tumor growth and metastasis.
Supplementary Material
Acknowledgments
We thank Dr. Robert J. Lefkowitz (Howard Hughes Medical Institute, Duke University Medical Center, Durham, NC) for providing the GRK6 and βarr2 deficient mouse models; Dr. Robert M. Strieter (School of Medicine, University of Virginia, Charlottesville, VA) for anti-mouse CXCR2 antisera; and Dr. Ann Richmond (Vanderbilt University) for critical reading of the manuscript. We are very thankful to Ms. Kimberly M. Malloy (JLC Biomedical/Biotechnology Research Institute, North Carolina Central University, Durham, NC) for technical support.
This work was supported by NIH Grants AI38910, CA156735, NIMHD P20 MD00175 and USAMRMC 07-1-0418 to R.M.R.; NIH Grant CA34590; Historically Black College and Universities Grant, Department of Veterans Affairs, to R.M.R and North Carolina Biotechnology Center (2009-BRG-1213 to TCL).
Abbreviations used in this paper
- GRK
G protein coupled receptor kinase
- GPCR
G protein-coupled receptor
- LLC
Lewis lung cancer
- βarr2
β-arrestin-2
- CXCL8/IL-8
interleukin-8
- CXCR2
IL-8 receptor B
- ELR
Glu-Leu-Arg
- KC or CXCL1
keratinocyte-derived chemokine
- VEGF
vascular endothelial growth factor
- MMPs
matrix metalloproteinases
References
- 1.Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- 2.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 4.Koch WJ, Inglese J, Stone WC, Lefkowitz RJ. The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J Biol Chem. 1993;268:8256–8260. [PubMed] [Google Scholar]
- 5.Luttrell LM, Hawes BE, Touhara K, van Biesen T, Koch WJ, Lefkowitz RJ. Effect of cellular expression of pleckstrin homology domains on Gi-coupled receptor signaling. J Biol Chem. 1995;270:12984–12989. doi: 10.1074/jbc.270.22.12984. [DOI] [PubMed] [Google Scholar]
- 6.Eichmann T, Lorenz K, Hoffmann M, Brockmann J, Krasel C, Lohse MJ, Quitterer U. The amino-terminal domain of G-protein-coupled receptor kinase 2 is a regulatory Gbeta gamma binding site. J Biol Chem. 2003;278:8052–8057. doi: 10.1074/jbc.M204795200. [DOI] [PubMed] [Google Scholar]
- 7.Premont RT, Macrae AD, Stoffel RH, Chung N, Pitcher JA, Ambrose C, Inglese J, MacDonald ME, Lefkowitz RJ. Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. J Biol Chem. 1996;271:6403–6410. doi: 10.1074/jbc.271.11.6403. [DOI] [PubMed] [Google Scholar]
- 8.Premont RT, Koch WJ, Inglese J, Lefkowitz RJ. Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem. 1994;269:6832–6841. [PubMed] [Google Scholar]
- 9.Stoffel RH, Randall RR, Premont RT, Lefkowitz RJ, Inglese J. Palmitoylation of G protein-coupled receptor kinase, GRK6. Lipid modification diversity in the GRK family. J Biol Chem. 1994;269:27791–27794. [PubMed] [Google Scholar]
- 10.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 11.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 12.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 13.Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42:768–778. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J Biol Chem. 1998;273:23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 15.Richardson RM, Marjoram RJ, Barak LS, Snyderman R. Role of the cytoplasmic tails of CXCR1 and CXCR2 in mediating leukocyte migration, activation, and regulation. J Immunol. 2003;170:2904–2911. doi: 10.4049/jimmunol.170.6.2904. [DOI] [PubMed] [Google Scholar]
- 16.Fan GH, Yang W, Wang XJ, Qian Q, Richmond A. Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry. 2001;40:791–800. doi: 10.1021/bi001661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasser MW, Marjoram RJ, Brown SL, Richardson RM. Cross-desensitization among CXCR1, CXCR2, and CCR5: role of protein kinase C-epsilon. J Immunol. 2005;174:6927–6933. doi: 10.4049/jimmunol.174.11.6927. [DOI] [PubMed] [Google Scholar]
- 18.Richardson RM, DuBose RA, Ali H, Tomhave ED, Haribabu B, Snyderman R. Regulation of human interleukin-8 receptor A: identification of a phosphorylation site involved in modulating receptor functions. Biochemistry. 1995;34:14193–14201. doi: 10.1021/bi00043a025. [DOI] [PubMed] [Google Scholar]
- 19.Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J Immunol. 2012;189:2824–2832. doi: 10.4049/jimmunol.1201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. Altered CXCR2 signaling in beta-arrestin-2-deficient mouse models. J Immunol. 2005;175:5396–5402. doi: 10.4049/jimmunol.175.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghuwanshi SK, Nasser MW, Chen X, Strieter RM, Richardson RM. Depletion of beta-arrestin-2 promotes tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2008;180:5699–5706. doi: 10.4049/jimmunol.180.8.5699. [DOI] [PubMed] [Google Scholar]
- 22.Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proc Natl Acad Sci U S A. 2002;99:7478–7483. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172:2853–2860. doi: 10.4049/jimmunol.172.5.2853. [DOI] [PubMed] [Google Scholar]
- 24.Elkin M, Vlodavsky I. Tail Vein Assay of Cancer Metastasis. In: Bonifacino MDJS, Harford JB, Lippincott-Schwartz J, Yamada KM, editors. Current Protocols in Cell Biology. Vol. 19. John Wiley & Sons; New York: 2001. pp. 19.12.11–19.12.17. [DOI] [PubMed] [Google Scholar]
- 25.Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, Lengyel E. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001;7:2396–2404. [PubMed] [Google Scholar]
- 26.Murase S, McKay RD. Matrix metalloproteinase-9 regulates survival of neurons in newborn hippocampus. J Biol Chem. 2012;287:12184–12194. doi: 10.1074/jbc.M111.297671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD, Strieter RM. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J Clin Invest. 1996;97:2792–2802. doi: 10.1172/JCI118734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varney ML, Johansson SL, Singh RK. Distinct expression of CXCL8 and its receptors CXCR1 and CXCR2 and their association with vessel density and aggressiveness in malignant melanoma. Am J Clin Pathol. 2006;125:209–216. doi: 10.1309/VPL5-R3JR-7F1D-6V03. [DOI] [PubMed] [Google Scholar]
- 29.Varney ML, Li A, Dave BJ, Bucana CD, Johansson SL, Singh RK. Expression of CXCR1 and CXCR2 receptors in malignant melanoma with different metastatic potential and their role in interleukin-8 (CXCL-8)-mediated modulation of metastatic phenotype. Clin Exp Metastasis. 2003;20:723–731. doi: 10.1023/b:clin.0000006814.48627.bd. [DOI] [PubMed] [Google Scholar]
- 30.Eck M, Schmausser B, Scheller K, Brandlein S, Muller-Hermelink HK. Pleiotropic effects of CXC chemokines in gastric carcinoma: differences in CXCL8 and CXCL1 expression between diffuse and intestinal types of gastric carcinoma. Clin Exp Immunol. 2003;134:508–515. doi: 10.1111/j.1365-2249.2003.02305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu YM, Webster SJ, Flower D, Woll PJ. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br J Cancer. 2004;91:1970–1976. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, Tang H, Burdick MD, Sato M, Minna JD, Mao L, Wistuba I, Strieter RM, Kurie JM. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66:4198–4207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 33.Singh S, Varney M, Singh RK. Host CXCR2-dependent regulation of melanoma growth, angiogenesis, and experimental lung metastasis. Cancer Res. 2009;69:411–415. doi: 10.1158/0008-5472.CAN-08-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mestas J, Burdick MD, Reckamp K, Pantuck A, Figlin RA, Strieter RM. The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J Immunol. 2005;175:5351–5357. doi: 10.4049/jimmunol.175.8.5351. [DOI] [PubMed] [Google Scholar]
- 35.Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 36.Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- 37.Sang QXA. Complex role of matrix metalloproteinases in angiogenesis. Cell Res. 1998;8:171–177. doi: 10.1038/cr.1998.17. [DOI] [PubMed] [Google Scholar]
- 38.Rundhaug JE. Matrix metalloproteinases, angiogenesis, and cancer: commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin Cancer Res. 2003;9:551–554. [PubMed] [Google Scholar]
- 39.Rundhaug JE. Matrix metalloproteinases and angiogenesis. Journal of Cellular and Molecular Medicine. 2005;9:267–285. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 41.Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71. doi: 10.1007/s10456-005-5208-4. [DOI] [PubMed] [Google Scholar]
- 42.Chiou SH, Sheu BC, Chang WC, Huang SC, Hong-Nerng H. Current concepts of tumor-infiltrating lymphocytes in human malignancies. J Reprod Immunol. 2005;67:35–50. doi: 10.1016/j.jri.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 44.Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo E, Tanghetti E, Albini A, Lowell C, Berton G, Noonan DM, Cassatella MA. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol. 2004;172:5034–5040. doi: 10.4049/jimmunol.172.8.5034. [DOI] [PubMed] [Google Scholar]
- 45.Fu W, Zhang Y, Zhang J, Chen W-F. Cloning and characterization of mouse homolog of the CXC chemokine receptor CXCR1. Cytokine. 2005;31:9–17. doi: 10.1016/j.cyto.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Moepps B, Nuesseler E, Braun M, Gierschik P. A homolog of the human chemokine receptor CXCR1 is expressed in the mouse. Molecular Immunology. 2006;43:897–914. doi: 10.1016/j.molimm.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 47.Fan X, Patera AC, Pong-Kennedy A, Deno G, Gonsiorek W, Manfra DJ, Vassileva G, Zeng M, Jackson C, Sullivan L, Sharif-Rodriguez W, Opdenakker G, Van Damme J, Hedrick JA, Lundell D, Lira SA, Hipkin RW. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282:11658–11666. doi: 10.1074/jbc.M607705200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.