

Abstract
Interleukin-22 (IL-22) is a good candidate to play a critical role in regulating gut microbiota because it is an important inducer of antimicrobial peptides and mucins in the gut. However, whether IL-22 participates into immune homeostasis by way of modulating gut microbiota remains to be elucidated. In this study, we find through 16S rRNA gene pyrosequencing analysis that healthy IL-22 deficient mice had altered colonic microbiota, notably with decreased abundance of some genera including Lactobacillus and increased levels of others. Mice harboring this altered microbiota exhibited more severe disease during experimentally-induced colitis. Interestingly, this altered gut microbiota can be transmitted to co-housed wild-type animals along with the increased susceptibility to this colitis, indicating an important role of IL-22 in shaping the homeostatic balance between immunity and colonic microbiota for host health.
Introduction
The cytokine interleukin-22 (IL-22) is an important modulator of tissue responses during inflammation in many different tissues, including the skin, lung, and gastrointestinal (GI) tract (1). We and others have previously shown that IL-22 is an important cytokine that protects the colon during inflammation (2, 3). IL-22 is highly upregulated in the sera of Crohn's disease and ulcerative colitis patients (4). In DSS-mediated colitis, IL-22 deficient mice develop more severe disease than wild-type controls and have a longer recovery period (2, 3). IL-22 has also been shown to be protective in a T cell-mediated colitis model (2) and infectious colitis, such as that caused by the pathogenic bacterium Citrobacter rodentium (5).
Barrier integrity is essential to maintain immune homeostasis within the colon. IL-22 is important to preserving the integrity of the single-layer GI epithelium during inflammation (1). Recognition of IL-22 by epithelial cells leads to activation of Stat3 signaling pathways which in turn leads to both proliferative and anti-apoptotic pathways (6). IL-22 is a potent inducer of antimicrobial molecules, including β-defensin, lipocalin-2 and RegIIIγ, as well as mucins (3, 5, 7, 8). RegIIIγ, which binds to lipotechoic acid and is therefore effective against Gram-positive bacteria, is highly regulated by IL-22 and in the absence of IL-22 there is little induction of this antimicrobial (9). Mucins are highly inducible by IL-22 and constitute the thick mucosal layer that coats the epithelium and that is impenetrable to many commensal bacteria, thereby limiting their potential to cause inflammation (3). In the absence of IL-22 signaling, levels of these molecules can be greatly diminished (5, 9) and we hypothesize this could thereby affect the composition of the microbiome.
IL-22 is upregulated in many types of lymphocytes during inflammation, but during immune homeostasis there is mainly one lymphocyte subset that expresses IL-22: the RORγt-dependent innate lymphocyte (1, 10, 11). These cells are found in the small intestine in Peyer's patches and in the colon in similar structures referred to as cryptopatches, as well as in the lamina propria (12–16). As with other aspects of the immune system, commensal bacteria play a critical role in the development and differentiation of these cells (14, 15).
Previous studies from our lab, as well as those from other labs, have shown that mice lacking specific components of the immune system, such as TLR5 or the inflammasome, have altered microbiota (17–19). Furthermore, we have shown that mice deficient in a component of the inflammasome not only have altered microbiota, but that this different microbiota can be transferred to wild-type mice and is colitogenic (19). We hypothesized that since IL-22 is important in the expression of antimicrobial peptides and mucin, its absence may alter the colonic niche altering the flora composition of the GI tract. This led us to investigate whether the absence of IL-22 could lead to altered microbiota and if this had a role in colitis.
In order to examine the effect of IL-22 on the structure of the microbiota of the colon, we performed experiments in which microbiota from IL-22 deficient mice were naturally transmitted to wild-type mice. Wild-type mice that were co-housed with IL-22 deficient mice were found by pyrosequencing analysis of 16S rRNA gene V3 region to have a microbiome more similar to IL-22 deficient mice than that of wild-type mice not exposed to IL-22 deficient mice, showing that the altered flora found in IL-22 deficient mice is transmissible. This altered microbiota was shown in part to be responsible for at least some of the increased disease observed during colitis in IL-22 deficient mice.
Materials and Methods
Mice
Il22−/− mice were described previously (2). Age- and sex-matched C57BL/6 mice were purchased from the National Cancer Institute (Frederick, MD). To re-derive our IL-22 deficient mice, IVF was performed with sperm isolated from an Il22−/− male mouse and C57BL/6 ova and the resulting embryos were implanted into a CD-1 female. The resulting offspring heterozygous were housed in a specific-pathogen free room that excluded Helicobacter and was separate from our original Il22−/− colony. The heterozygous mice were intercrossed to generate Il22−/− mice. All mice were cared for in accordance with institutional animal care and use committee-approved protocols at the Yale University animal facility. For co-housing three mice of each strain were housed together for 4–6 weeks prior to DSS treatment. Mice were 8–12 weeks old at the initiation of DSS treatment.
DSS-induced colitis
Mice were giving ad libitum 2% (w/v) dextran sodium sulfate (DSS) (M.W. = 36,000–50,000 Da; MP Biomedicals) in their drinking water for seven days, which was then replaced with normal water.
Histology
Colons were excised, rinsed with PBS, fixed overnight in 10% formalin, and then embedded in paraffin, sectioned, and stained with H&E. Slides were prepared at the Yale University Program for Critical Technologies in Molecular Medicine, Department of Pathology. Sections were blindly analyzed by a trained gastroentero-pathologist. Each segment was given a score of 0–4. For chronicity (the degree of chronic inflammation); grade 0, no significant changes; grade 1, mildly increased inflammatory cells in the lamina propia; grade 2, moderately increased inflammation in the lamina propria (multiple foci); grade 3,high level of inflammation with evidence of wall thickening by inflammation; grade 4, maximal severity of inflammation with transmural lymphocytic infiltration and/or architectural distortion. For activity (the degree of epithelial injury); grade 0, no significant changes; grade 1, occasional epithelial injury with focal and superficial or rare cryptitis; grade 2, foci of cryptitis, including rare crypt abscess; grade 4, three plus extensive ulceration.
Colonoscopies
Colonoscopy was performed using a high resolution mouse video endoscopic system (Coloview, Karl Storz; Tuttlingen, Germany). The severity of colitis was blindly scored using MEICS (Murine Endoscopic Index of Colitis Severity) which is based on five parameters: granularity of the mucosal surface; vascular pattern; translucency of the colon mucosa; visible fibrin; and stool consistency (20).
16S rRNA Analyses
Aliquots of frozen fecal samples were processed for DNA isolation using a previously validated protocol (21). Forward 5'-NNNNNNNNATTACCGCGGCTGCT-3' and reverse 5'-NNNNNNNNCCTACGGGAGGCAGCAG-3' primers were used to amplify the 16S rRNA gene V3 region. The NNNNNNNN was the sample-unique 8-base barcode used for sorting of PCR amplicons. PCR reactions, pyrosequencing, and sequence quality controls were performed as described previously (22). High quality sequences were uploaded into QIIME and processed as described earlier (23) using Pynast (24) to align the sequences and RDP classifier (25) for the classification (cutoff 50%). OTU data were generated with 97% identity. PCA and cluster analysis were performed in the MATLAB 7.11.0 (R2010b) environment (The MathWorks, Inc.). The sequences have been deposited in GenBank Sequence Read Archive database (www.ncbi.nlm.nih.gov/Trace/sra) with accession number SRA054081.
Real time RT-PCR
RNA from colons was isolated with Trizol reagent (Invitrogen; Carlsbad, CA). RNA was subjected to reverse transcriptase with Superscript II (Invitrogen) and oligo dT primer. cDNA was semi-quantitated with commercially available primer and probe sets (Applied Biosystems; Foster City, CA) and the ΔΔCT method. Hypoxanthineguanine phosphoribosyltransferase (HPRT) was included as an internal control.
Statistical Analyses
For data of normal distribution, one-way ANOVA was performed, otherwise Mann-Whitney analysis was used. p values <0.05 were considered significant.
Results
Specific-pathogen free mice deficient in IL-22 exhibit no abnormal pathology
Previously, in the absence of inflammation we have not detected any differences in IL-22 deficient mice compared to wild-type mice (2). Since low levels of IL-22 are expressed in the colon during immune homeostasis (26), we hypothesized that a lack of the cytokine may have much longer term consequences on colonic health. Therefore we examined the colonic architecture of one year old wild-type or IL-22 deficient mice. In both wild-type and IL-22 deficient mice we found normal colonic architecture, with minimally increased inflammation in some mice, but no significant colitis in any animals (Figure 1). Thus, a long-term deficiency in IL-22 does not affect the colon morphology.
Figure 1. IL-22 deficient mice have normal colonic architecture.

One year old specific-pathogen free IL-22 deficient (Il22−/−) or wild-type (Il22+/+) control mice were euthanized. Colons were excised, fixed, sectioned and stained with H&E. (A) Shown are representative micrographs of five mice per group. (B) Activity (the degree of epithelial injury) and (C) chronicity scores (the degree of chronic inflammation) for the colon sections. Each segment was given a score of 0–4. For chronicity (the degree of chronic inflammation); grade 0, no significant changes; grade 1, mildly increased inflammatory cells in the lamina propia; grade 2, moderately increased inflammation in the lamina propria (multiple foci); grade 3,high level of inflammation with evidence of wall thickening by inflammation; grade 4, maximal severity of inflammation with transmural lymphocytic infiltration and/or architectural distortion. For activity (the degree of epithelial injury); grade 0, no significant changes; grade 1, occasional epithelial injury with focal and superficial or rare cryptitis; grade 2, foci of cryptitis, including rare crypt abscess; grade 4, three plus extensive ulceration. Each dot represent one mouse, line indicates mean.
Co-housing wild-type mice with IL-22 deficient mice increases the severity of induced colitis
Previous studies from our lab have shown that mice deficient in immune system components have altered microbiota that is transmissible to wild-type mice and can increase inflammation (19). Since we have previously shown that IL-22 deficient mice are more susceptible to DSS-mediated colitis (2) we questioned whether the microbiota of these mice could contribute to this phenotype. Thus, we designed an experiment in which wild-type C57BL/6 mice were co-housed with IL-22 deficient mice, or remained housed alone. After four weeks of co-housing (a time period previously established by our lab to be sufficient for the transmission of microbiota between mice (19)), we added 2% DSS to the drinking water for seven days and examined the resulting wasting and inflammatory diseases. We found that C57BL/6 mice co-housed with IL-22 deficient mice lost significantly more mass than C57BL/6 mice that were housed alone (Figure 2A, C, D). To examine inflammation, we performed colonoscopies at day seven post-DSS treatment and found increased severity in the mice that were co-housed with the IL-22 deficient mice (Figure 2E). We also found that the IL-22 deficient mice lost more mass than the wild-type mice they were co-housed with (Figure 2A, D) and IL-22 deficient mice were the only mice to have mortality associated with disease (Figure 2B). These data suggest that the increase in severity of DSS-mediated colitis in IL-22 deficient mice is dependent on both their microbiota as well as a biological role for IL-22 independent of the microbiota.
Figure 2. Co-housing wild-type mice with IL-22 deficient mice increases susceptibility to DSS-induced colitis.
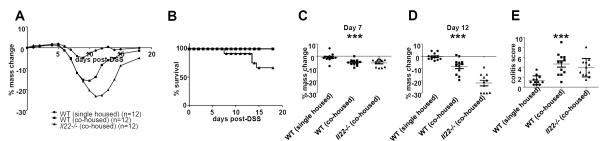
C57BL/6 mice were either housed by themselves, or co-housed with Il22−/− mice for four weeks. Mice were then given 2% DSS in their drinking water for seven days. (A) Mice were massed daily. Shown is mean change in mass for each group over time. (B) Survival of mice at different days post-DSS. Mass loss at (C) day 7 post-DSS and (D) day 12 post-DSS. Each dot presents one mouse, line indicates mean. *** p<0.001 by one-way ANOVA. (E) At day 7 post-DSS, colonoscopies were performed and blindly scored as described in the Materials and Methods. Each dot represents one mouse, line indicates mean. *** p<0.001 by one-way ANOVA. Experiment is representative of three independent experiments.
To independently confirm our findings we performed the same experiment with a distinct colony of IL-22 deficient mice obtained through IVF. Sperm collected from an IL-22 deficient male mouse was used to fertilize wild-type C57BL/6 oocytes and the resulting heterozygous embryos were implanted in CD1 mothers. After weaning, the newly derived heterozygous mice were housed under Helicobacter-excluded conditions and intercrossed to generate homozygous IL-22 deficient mice. We then performed a similar co-housing experiment in which wild-type mice were either co-housed with IL-22 deficient mice or not, and then subjected to DSS-mediated colitis. In the re-derived colony we found a similar phenotype as our original colony; wild-type mice co-housed with IL-22 deficient mice had significantly greater wasting disease than wild-type mice not exposed to IL-22 deficient mice (Supplemental Figure 1). However, in this particular experiment we did not observe a significant difference in mass loss between wild-type mice co-housed with IL-22 deficient mice and the IL-22 deficient mice, perhaps due to transfer of some protective flora from the wild-type mice to the IL-22 deficient mice. Thus, passage of our IL-22 deficient mouse line through a CD1 parent and subsequent heterozygous (Il22+/−) mice shows that IL-22 deficient mice harbor transmissible colitic microbiota that is specific to the genotype and not the specific colony.
IL-22 deficient mice have altered commensal microbiota
Our data support the hypothesis that IL-22 deficient mice have altered microbiota that is transmissible to wild-type mice. Thus, we undertook bacterial 16S ribosomal RNA gene pyrosequencing to examine the microbiome in three different groups of mice; (1) wild-type mice and (2) wild-type mice co-housed with (3) IL-22 deficient mice. Prior to DSS treatment we collected fecal samples from each mouse, prepared fecal DNA and then used bar-coded pyrosequencing of the 16S rRNA gene V3 region to explore the change of the microbiome.
We obtained 68,822 high-quality sequences with an average of approximately 2,000 reads per sample (Figure 3A). 657 operational taxonomic units (OTUs) were obtained and Shannon-Wiener Diversity Index was calculated for each sample (Figure 3B and C). These curves reached stable values, showing that the depth of sequencing would include most species found in the samples. Wild-type mice housed alone had much less diversity than either IL-22 deficient mice or wild-type mice housed together (Figure 3D).
Figure 3. Diversity estimation of gut microbiota.
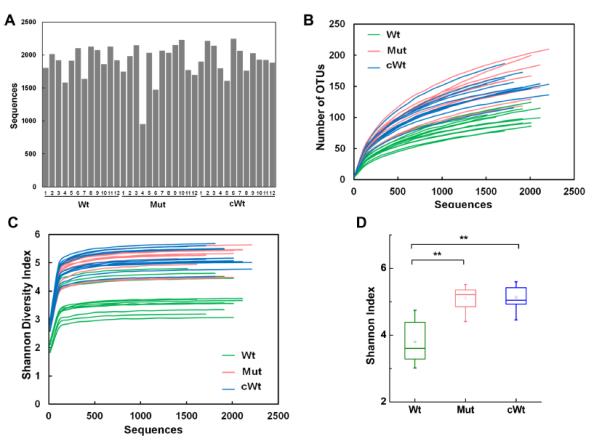
Prior to DSS-treatment, fecal samples were obtained from the mice described in Figure 1. DNA was prepared and then subjected to pyrosequencing. 68,822 total sequences were obtained from the 36 samples (n=12/group). (A) Average sequences per sample. Each bar represents one mouse. (B) OTU rarefaction curves. Each line represents one mouse. (C) Shannon-Wiener Diversity Index curves. Each line represents one mouse. (D) Difference of Shannon-Wiener Diversity among the three groups (at the depth of 910 sequences).
Multivariable analysis of the colonic microbiome structure was then performed. By principal component analysis (PCA), IL-22 deficient mice had a significantly different bacterial composition in their gut microbiota from that of wild-type mice housed alone (Figure 4A).
Figure 4. IL-22 deficient and co-housed wild-type mice have altered microbiota compared to wild-type mice by 16S rRNA analysis of fecal-derived bacterial microbiota.
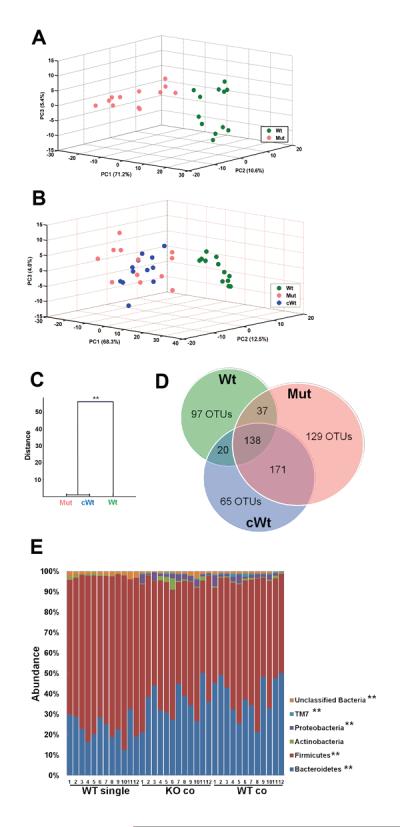
Prior to DSS-treatment, fecal samples were obtained from the mice described in Figure 2. DNA was prepared and then subjected to pyrosequencing. 68,822 total sequences were obtained from the 36 samples (n=12/group). (A) Score plot of PCA of the microbiomes of wild-type (Wt) and IL-22 deficient mice (Mut) based on the first three PCs. Each dot represents one mouse. (B) Score plot of PCA based on the first three PCs of wild-type mice (Wt), IL-22 deficient mice (Mut) and wild-type mice housed with IL-22 deficient mice (cWt). Each dot indicates one mouse. (C) Clustering of bacteria based on distances between different groups calculated with multivariate analysis of variance test of the first seven PCs of UniFrac PCoA analysis data. The Mahalanobis distances between group means are shown. ** p<0.01 (D) Venn diagram showing distribution of the shared OTUs. (E) Taxon-based analysis at phylum level among the groups. Phyla above 1% abundance are shown (Kruskal-Wallis test, **p<0.01, *p<0.05).
To assess whether the altered microbiota of IL-22 deficient mice were transmissible to wild-type mice, we then further compared microbiota from co-housed wild-type mice to that of IL-22 deficient mice and wild-type mice housed alone. This revealed that the microbiome of co-housed wild-type mice was indistinguishable from that of IL-22 deficient mice (Figure 4B), but unrelated to the wild-type mice housed alone, indicating that the altered microbiota of the mice deficient in IL-22 was transmissible to wild-type mice. A multivariate analysis of variance test (MANOVA) also indicated the gut microbiota of co-housed wild-type mice was much similar to IL-22 deficient mice (Figure 4C).
We next examined the OTU distribution among the three groups to estimate shared species among the three groups of animals. Results showed that wild-type mice co-housed with IL-22 deficient mice had 65 unique OTUs and shared 309 OTUs with the IL-22 deficient mice; however they only shared 158 OTUs with the wild-type mice housed alone (Figure 4D). Taxon-based analysis revealed that the microbiome of each group was mainly composed of Bacteroidetes, Firmicutes, Proteobacteria, TM7 and Actinobacteria (Figure 4E). Ten families (Table I) and 14 genera (Table II) showed significant differences among the three groups. In all, compared to wild-type mice housed alone, seven genera were reduced in IL-22 deficient and the co-housed wild-type mice (Lactobacillus, Bacteroides, Ruminoccous, Turicibacter, Anaerobacter, Parabacteroide and Hespellia) and seven genera were increased (Coprococcus, Allobaculum, Barnesiella, Alistipes, Xylanibacter, Butyricimonas and Helicobacter).
Table I.
Taxon-based analysis at the family level among the groups
| Family | P Value (Kruskal-Wallis test) | Wt | Mut | cWt | |||
|---|---|---|---|---|---|---|---|
| Median | Range | Median | Range | Median | Range | ||
| Lactobacillaceae | 0.0055224 | 43.45 | 24.4–52.4 | 25.44 | 5.3–49.7 | 22.38 | 8.9–43.9 |
| Bacteroidaceae | 0.0000083 | 13.48 | 7.8–30.6 | 1.09 | 0.6–2.2 | 1.18 | 0.5–3.6 |
| Clostridiaceae | 0.0000821 | 0.98 | 0.4–2.5 | 0.06 | 0–1.5 | 0.05 | 0–0.8 |
| Peptococcaceae | 0.0000001 | 0.91 | 0.4–2.3 | 0.00 | 0-0 | 0.00 | 0-0 |
| Porphyromonadaceae | 0.0001488 | 5.22 | 0.7–17.9 | 20.94 | 11.4–34.8 | 21.60 | 12.0–29.6 |
| Prevotellaceae | 0.0000029 | 0.00 | 0–1.3 | 8.06 | 4.0–14.8 | 10.66 | 4.5–20.9 |
| Rikenellaceae | 0.0000031 | 0.00 | 0–0.1 | 1.12 | 0.3–2.9 | 2.16 | 0.4–3.0 |
| Coriobacteriaceae | 0.0193184 | 0.26 | 0–0.6 | 0.32 | 0.2–1.4 | 0.47 | 0.2–1.1 |
| Helicobacteraceae | 0.0000111 | 0.00 | 0-0 | 0.33 | 0.1–1.5 | 0.41 | 0–0.6 |
| Desulfovibrionaceae | 0.0000230 | 0.00 | 0-0 | 0.11 | 0–0.7 | 0.25 | 0.1–1.2 |
The number in the tables shows the abundance (%) of each family.
Table II.
Taxon-based analysis at the genus level among the groups
| Genus | P value (Kruskal-Wallis test) | Wt | Mut | cWt | |||
|---|---|---|---|---|---|---|---|
| Median | Range | Median | Range | Median | Range | ||
| Lactobacillus | 0.0055224 | 43.45 | 24.40–52.32 | 25.44 | 5.34–49.62 | 22.38 | 8.92–43.88 |
| Bacteroides | 0.0000083 | 13.48 | 7.77–30.61 | 1.09 | 0.57–2.24 | 1.18 | 0.52–3.64 |
| Ruminococcus | 0.0000004 | 1.68 | 0.84–2.83 | 0.00 | 0-0 | 0.00 | 0-0 |
| Turicibacter | 0.0001339 | 1.10 | 0.43–3.73 | 0.19 | 0–1.75 | 0.00 | 0–2.8 |
| Anaerobacter | 0.0000220 | 0.93 | 0.43–2.44 | 0.00 | 0–1.49 | 0.00 | 0–0.63 |
| Parabacteroides | 0.0053751 | 0.85 | 0.52–1.81 | 0.52 | 0.25–1.16 | 0.51 | 0.19–1.8 |
| Hespellia | 0.0108047 | 0.88 | 0.3–1.88 | 0.34 | 0.1–1.15 | 0.37 | 0.16–1.00 |
| Coprococcus | 0.0456690 | 3.09 | 1.49–8.34 | 8.93 | 1.98–25.04 | 6.80 | 2.27–28.38 |
| Allobaculum | 0.0005015 | 0.00 | 0-0 | 2.94 | 0–14.39 | 0.65 | 0–12.27 |
| Barnesiella | 0.0000046 | 0.00 | 0–0.14 | 2.77 | 1.72–5.31 | 3.29 | 1.59–7.59 |
| Alistipes | 0.0000031 | 0.00 | 0–0.1 | 0.94 | 0.3–2.69 | 1.99 | 0.1–2.74 |
| Xylanibacter | 0.0000583 | 0.00 | 0-0 | 0.39 | 0–1.48 | 0.25 | 0–1.28 |
| Butyricimonas | 0.0000386 | 0.00 | 0-0 | 0.34 | 0–0.83 | 0.25 | 0.09–1.43 |
| Helicobacter | 0.0000111 | 0.00 | 0-0 | 0.33 | 0.1–1.49 | 0.41 | 0–0.62 |
The number in the tables shows the abundance (%) of each family.
Wild-type mice co-housed with IL-22 deficient mice have altered colonic antimicrobial peptide expression
Since IL-22 is an important regulator of certain antimicrobial peptides, we hypothesized that deficiency in IL-22 would decrease their expression. We examined expression of the genes encoding RegIIIβ and RegIIIγ in the colons of IL-22 deficient mice, wild-type mice co-housed with IL-22 deficient mice or control wild-type mice. As previously observed by us and others (5, 9) expression of both RegIIIβ and RegIIIγ were decreased in IL-22 deficient mice compared to wild-type mice housed alone (Figure 5). In contrast to the levels we observed in these wild-type mice, we noticed significantly reduced expression of RegIIIβ and RegIIIγ in wild-type mice co-housed with IL-22 deficient mice. Thus, co-housing of wild-type mice with IL-22 deficient mice reduced their expression of IL-22-regulated antimicrobial proteins.
Figure 5. Wild-type mice co-housed with IL-22 deficient mice have reduced levels of RegIIIβ and RegIIIγ.
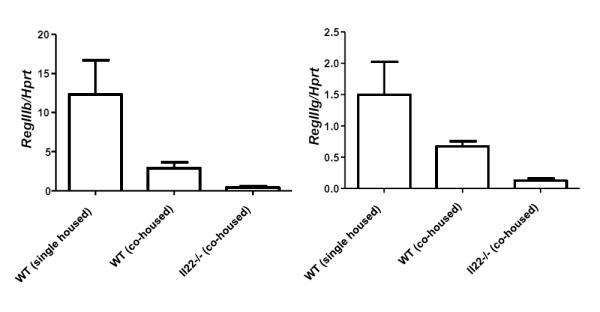
C57BL/6 mice were either housed by themselves, or co-housed with Il22−/− mice for four weeks. Level of RegIIIβ and RegIIIγ mRNA in the colon tissue was semi-quantitated by real time RT-PCR. Bars indicate mean±SD (n=5). Experiment was performed twice with similar results.
Discussion
In this study we have shown that the cytokine IL-22 contributes to the composition of the colonic microbiota. In the absence of IL-22, the commensal flora homeostasis could be tipped in favor of different species or one particular keystone species that can stabilize the altered flora (27). IL-22 is a potent inducer of several antimicrobial peptides and mucins, which help protect the epithelial barrier. RegIIIγ is active against Gram-positive bacteria due to its binding to the peptidoglycan surface (28) and is important for microbiota localization in the small intestine, but not the colon (29). Changes we observed in the colonic microbiome in IL-22 deficient and co-housed wild-type mice could be explained by the observed reduced levels of RegIIIβ and RegIIIγ. In the absence of IL-22, commensals that are normally suppressed by RegIIIβ and/or RegIIIγ may now expand. Upon co-housing, these altered flora are then transmitted to wild-type mice, the persistence of which is unknown. In turn these mice have suppression of the genes whose encoded proteins may have generated the altered flora in the first place. The mechanism by which this occurs is unknown, but we speculate that the altered flora may be inhibitory to IL-22 or upstream IL-23 signaling, or alternatively, MyD88-mediated signaling pathways (30).
We found many differences in the altered microbiota generated by the absence of IL-22 compared to microbiota in immune-intact mice. The bacteria that were decreased in the absence of IL-22 and therefore appear to require IL-22 to maintain their niche in the microbiota, included the family Lactobacillacae, a Gram-positive family of lactic acid producing bacteria that is thought to generally be part of the healthy flora (31). A reduced population of beneficial microbiota may have allowed the increase in colitis severity of the IL-22 deficient and co-housed wild-type mice. However, the more notable trend was that in IL-22 deficient mice and co-housed wild-type mice we found the appearance of low levels of bacterial phyla that were generally absent or at extremely low levels in wild-type mice. This included TM7 and Proteobacteria, and the genus Barnseiella. TM7 is an unculturable phyla that was identified by 16S ribosomal RNA sequencing in environmental soil samples and has been since found in the feces of mice (19) and human oral cavity (32). Barnesiella, which was recently found to be one the most abundant of newly identified taxa by metagenomic sequence analysis of human stool samples and still lacks a known role in the GI tract (33). The phylum Proteobacteria are Gram-negative bacteria and include such notable genera as Escherichia, Salmonella, Vibrio and Helicobacter. These findings suggest that the absence of IL-22 allows for the colonization or outgrowth of these organisms.
Our study examines the microbiota composition, but we did not address whether IL-22 alters the localization of commensal bacteria within the lumen. In the absence of IL-22 and the accompanying decrease in expression of antimicrobial proteins and mucin, commensal bacteria may encroach closer to the epithelium, where they are normally excluded. This may have further affected the course of DSS-mediated colitis in the IL-22 deficient and co-housed wild-type mice.
Colonic flora is easily transmitted between mice living in the same cage due to their copraphagia. Why does the altered bacteria transmit from the IL-22 deficient mice to the wild-type mice and not the other way around? Our data suggest that once the altered flora has been established, it could potentially be more persistent and/or stable than the normal microbiota. In addition, since we found that the co-housed mice had lower expression levels of IL-22-regulated genes than non-co-housed mice, it appears that the altered flora may create a niche in which it is favored.
From our previous studies, we know that the role of IL-22 in colitis can be independent of commensal microbiota. Our experiments using a CD4 T cell-mediated model of disease used cohorts of the same Rag1−/− mice and only differed in the genotype of T cells adoptively transferred into these hosts (2). We also observed a similar protective role for IL-22 in DSS-mediated colitis that was independent of adaptive immunity (2). DSS is thought to disrupt the colonic epithelium, allowing commensal bacteria to come into contact with, and possibly translocate, the epithelial barrier (34). We now show that in this innate colitis model, that the microbiota play at least a partial role in the severity of disease in the absence of IL-22. This agrees with findings that mice treated with neutralizing IL-22 Ab during colitis, and therefore have not developed altered homeostatic flora, have a less pronounced phenotype than Il22 gene deficient mice, although these studies have not yet been performed contemporaneously (2, 3).
Our findings in experimental colitis models are relevant to human disease. IBD patients are known to have altered gut microbiota although it is unclear as to whether this is a cause or an outcome of disease (35). As we have shown for the absence of IL-22 in mice, an increase in IL-22 may play a similar role in altering the composition of the microbiome. The upregulation of IL-22 that accompanies disease may be responsible for altering patient microbiota due to changes in levels of antimicrobial molecules or through other mechanisms. This shows that altered microbiota of IBD patients may result from the changes in cytokine expression that accompany disease. If this proves true, then therapies that try to return cytokine levels in IBD patients to those of healthy people may be beneficial not only from an immune system viewpoint, but also from the perspective of the microbiota.
This study is a striking example of how the absence of one specific cytokine can have effects on the commensal microbiota, and in turn, the course of inflammation. Although IL-22 is well-known to be involved during inflammation, it clearly can also play a role in immune homeostasis by altering the commensal microbiota. The transmission of the altered gut microbiota from IL-22 deficient mice to co-housed wild-type mice along with increased susceptibility to DSS-induced colitis indicate that the altered gut microbiota may work as a contributing factor for, rather than a consequence of, the disease. Future studies will investigate if some particular species of bacteria could be responsible for mediating these effects.
Supplementary Material
Acknowledgments
We thank Cindy Hughes for re-derivation of our IL-22 deficient mouse colony.
L.A.Z. was supported by an American Cancer Society fellowship. E.E. was supported by Cancer Research Institute (2010–2012) and the Israel-US Educational Foundation (2009) and was the recipient of the Claire and Emmanuel G. Rosenblatt Award from the American Physicians for Medicine in Israel Foundation. R.A.F. is an Investigator of the Howard Hughes Research Institute.
Abbreviations used in this manuscript
- (IL-22)
interleukin-22
- (GI)
gastrointestinal
- (DSS)
dextran sodium sulfate
- (OTU)
operational taxonomic unit
- (PC)
principal component
- (PCA)
principal component analysis
References
- 1.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nature immunology. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 2.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. The Journal of clinical investigation. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, Shimizu N, Fujiyama Y. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 5.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 6.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. The Journal of experimental medicine. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, Baumler AJ. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell host & microbe. 2009;5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinnebrew MA, Ubeda C, Zenewicz LA, Smith N, Flavell RA, Pamer EG. Bacterial flagellin stimulates Toll-like receptor 5-dependent defense against vancomycin-resistant Enterococcus infection. The Journal of infectious diseases. 2010;201:534–543. doi: 10.1086/650203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature immunology. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 12.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luci C, Reynders A, Ivanov, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, Dalod M, Littman DR, Vivier E, Tomasello E. Influence of the transcription factor RORγt on the development of NKp46+ cell populations in gut and skin. Nature immunology. 2009;10:75–82. doi: 10.1038/ni.1681. [DOI] [PubMed] [Google Scholar]
- 14.Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A. RORγt and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nature immunology. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, Eberl G, Di Santo JP. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Satoh-Takayama N, Lesjean-Pottier S, Sawa S, Vosshenrich CA, Eberl G, Di Santo JP. Lymphotoxin-beta receptor-independent development of intestinal IL-22-producing NKp46+ innate lymphoid cells. European journal of immunology. 2011;41:780–786. doi: 10.1002/eji.201040851. [DOI] [PubMed] [Google Scholar]
- 17.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nature protocols. 2006;1:2900–2904. doi: 10.1038/nprot.2006.446. [DOI] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, Mao Y, Zhang X, Pang X, Wei C, Zhao G, Chen Y, Zhao L. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. The ISME journal. 2010;4:312–313. doi: 10.1038/ismej.2009.112. [DOI] [PubMed] [Google Scholar]
- 23.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nature reviews. Microbiology. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cash HL, Whitham CV, Hooper LV. Refolding, purification, and characterization of human and murine RegIII proteins expressed in Escherichia coli. Protein expression and purification. 2006;48:151–159. doi: 10.1016/j.pep.2006.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-mediated signals induce the bactericidal lectin RegIIIγ and protect mice against intestinal Listeria monocytogenes infection. The Journal of experimental medicine. 2007;204:1891–1900. doi: 10.1084/jem.20070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders ME. Impact of probiotics on colonizing microbiota of the gut. Journal of clinical gastroenterology. 2011;(45 Suppl):S115–119. doi: 10.1097/MCG.0b013e318227414a. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD, Gibbons TR, Treangen TJ, Chang YC, Li S, Stine OC, Hasturk H, Kasif S, Segre D, Pop M, Amar S. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PloS one. 2012;7:e37919. doi: 10.1371/journal.pone.0037919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wylie KM, Truty RM, Sharpton TJ, Mihindukulasuriya KA, Zhou Y, Gao H, Sodergren E, Weinstock GM, Pollard KS. Novel bacterial taxa in the human microbiome. PloS one. 2012;7:e35294. doi: 10.1371/journal.pone.0035294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pizarro TT, Arseneau KO, Bamias G, Cominelli F. Mouse models for the study of Crohn's disease. Trends in molecular medicine. 2003;9:218–222. doi: 10.1016/s1471-4914(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 35.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.