

Abstract
Hypertension is an epidemic health concern and a major risk factor for the development of cardiovascular disease. Although there are available treatment strategies for hypertension, numerous hypertensive patients do not have their clinical symptoms under control and it is imperative that new avenues to treat or prevent high blood pressure in these patients are developed. It is well established that increases in sympathetic nervous system (SNS) outflow and enhanced renin-angiotensin system (RAS) activity are common features of hypertension and various pathological conditions that predispose individuals to hypertension. More recently, hypertension has also become recognized as an immune condition and accumulating evidence suggests that interactions between the RAS, SNS and immune systems play a role in blood pressure regulation. This review summarizes what is known about the interconnections between the RAS, SNS and immune systems in the neural regulation of blood pressure. Based on the reviewed studies, a model for RAS/neuroimmune interactions during hypertension is proposed and the therapeutic potential of targeting RAS/neuroimmune interactions in hypertensive patients is discussed. Special emphasis is placed on the applicability of the proposed model to obesity-related hypertension.
Keywords: angiotensin, renin, inflammation, sympathetic nervous system, microglia, blood pressure
1. Introduction
Hypertension is an epidemic health concern and a major risk factor for the development of cardiovascular disease, the leading cause of death in the USA. It has long-been established that high blood pressure is often accompanied by enhanced activities of the renin-angiotensin system (RAS) and the sympathetic nervous system (SNS) and, over several decades, research efforts have led to the development and refinement of countless pharmacological antihypertensive therapeutics targeting these systems. Despite these efforts, which successfully reduce blood pressure in many hypertensive subjects, numerous patients remain unresponsive to available pharmacological interventions and are left with severely high blood pressure (Egan et al., 2010). A large proportion of these patients have increased sympathetic outflow, parasympathetic withdrawal, and norepinephrine spillover, and surgical denervation of the kidney has shown promise as an effective intervention for these patients (Krum et al., 2009; Schlaich et al., 2009a; Schlaich et al., 2009b; Esler et al., 2010; Schlaich et al., 2012). The implication is that their hypertension arises from neurogenic origins (Fisher and Fadel, 2010; Grassi, 2010; Grassi et al., 2010).
The frequent ineffectiveness of available antihypertensive pharmacotherapies coupled with the neural origins of uncontrolled hypertension underscore the urgent need to develop new avenues for the pharmacological treatment or prevention of neurogenic hypertension. One promising prospect in this regard, stems from the relatively recent realization that hypertension is also an immune condition (Harrison et al., 2008; Shi et al., 2010; Zubcevic et al., 2011; Marvar et al., 2012). The nervous and immune systems are intricately connected and their reciprocal communication may contribute to the etiology of hypertension. Here we review the intimate relationship between these systems in the regulation of blood pressure and the potential roles of the SNS and RAS in these interactions.
Before discussing this RAS/ neuroimmune connection, we first briefly review the current understanding of the roles of the SNS and RAS in blood pressure control. Then, subsequent sections will consider the reciprocity between the nervous and immune systems, and how the RAS and SNS mediate these interactions. The role of the RAS, SNS and immune system in facilitating the causal relationship between obesity and hypertension is also considered. Finally, based on the reviewed studies a model for RAS/immune interactions in the neural control of blood pressure will be proposed and the clinical relevance of these regulatory systems for the development of new antihypertensive therapeutics will be discussed.
2. The brain renin-angiotensin system, sympathetic outflow and hypertension
Before discussing how the RAS, SNS and immune systems may interact to cause hypertension, it is appropriate to first review the roles of the SNS and RAS in blood pressure regulation. The SNS plays a key role in the pathophysiology of cardiovascular disease (Goldstein, 1983; Guyenet, 2006; Fisher and Fadel, 2010; Grassi, 2010; Grassi et al., 2010). Hypertensive animals and patients exhibit elevated SNS outflow (Goldstein, 1983; Grassi, 1998; Guyenet, 2006; Joyner et al., 2008) as well as, heightened vascular reactivity, characterized by greater vasoconstrictor responses to norepinephrine (Ziegler et al., 1991).
Various environmental factors, such as exposure to stressful stimuli and high-fat diet-induced obesity, and humoral factors, including those synthesized by the RAS activate the SNS and lead to an increased susceptibility to hypertension (Oparil et al., 2003; Dorresteijn et al., 2012; Marvar and Harrison, 2012) and pharmacotherapies that target the SNS and RAS reduce blood pressure in many patients. Furthermore, a therapy that has recently proven effective for some individuals resistant to available antihypertensive pharmaceutics is surgical denervation of the kidney (Krum et al., 2009; Schlaich et al., 2009a; Schlaich et al., 2009b; Esler et al., 2010). This surgical intervention is minimally invasive, as it uses a percutaneous catheter-based approach to ablate both afferent and efferent renal nerves, and it is highly effective, as it reduces sympathetic outflow to the kidney, reduces plasma renin activity and increases urine output, thereby reducing blood pressure without causing long-term adverse events (Krum et al., 2009; Schlaich et al., 2009a; Esler et al., 2010; Krum et al., 2011).
The effectiveness of this surgical intervention solidifies the importance of the nervous system in uncontrolled hypertension and understanding the mechanism(s) by which the nervous system regulates blood pressure under normal conditions may therefore guide the refinement of existing and/or the development of new pharmacological therapeutics for this pathology. In this section, we review the neural circuitry controlling sympathetic outflow and blood pressure regulation and the impact of the RAS on these pathways.
2.1. The renin-angiotensin system
First, it is essential to outline the current understanding of the RAS. Classically, the RAS has been viewed as an endocrine system regulating cardiovascular function and hydromineral balance. Angiotensin-II (Ang-II) is considered the principle effector peptide of the system, which is formed from liver-derived angiotensinogen (AGT) through proteolytic cleavage first by kidney-derived renin (the rate-limiting enzyme for Ang-II synthesis) and then by lung-derived angiotensin-converting enzyme (ACE). Ang-II then acts predominantly at its type-1 receptor (AT1R), in variety of tissues to impact blood pressure regulation. Although this is the best characterized and perhaps the dominating pathway of the RAS, several additional nuances of the RAS have been revealed, that allow it to impact blood pressure in diverse ways.
This includes several endogenous mechanisms that counteract the hypertensive actions of AT1R. For example, AT1R activation can be opposed by stimulation of the angiotensin type-2 receptor (AT2R; Huang et al., 1996; Bosnyak et al., 2010; Steckelings et al., 2012) and by the ACE2-Ang-(1-7)-Mas axis (Donoghue et al., 2000; Katovich et al., 2005; Der Sarkissian et al., 2006; Diez-Freire et al., 2006; Santos et al., 2008). We have also determined that macrophage migration inhibitory factor (MIF) can oppose many of Ang-II’s pathophysiological actions mediated by AT1R, working intracellularly via its thiol-protein oxidoreductase (TPOR) activity (Busche et al., 2001; Sun et al., 2007; Li et al., 2008; Freiria-Oliveira et al., 2012).
Moreover, it is now acknowledged that the RAS is also an autocrine/ paracrine system within various tissues including adipose tissue (Yiannikouris et al., 2012a; Yiannikouris et al., 2012b) and the brain (Lenkei et al., 1997; McKinley et al., 2003; Cuadra et al., 2010). Components of the RAS are present within brain regions that control cardiovascular function, but are protected from the circulating RAS by the blood brain barrier (BBB) suggesting that brain-derived Ang-II may contribute to SNS activity and may act as a neurotransmitter in these regions (Bains et al., 1992; Li and Ferguson, 1993). Pharmacological or genetic manipulations of the RAS in these protected brain regions have profound effects on physiology and behavior, further supporting the prevalence of a functional brain-specific RAS (Morimoto et al., 2002; McKinley et al., 2003; Grobe et al., 2008; Grobe et al., 2010; de Kloet et al., 2011; Yamazato et al., 2011). However, the level of renin within the CNS is low at best (Bader and Ganten, 2002; Lavoie et al., 2004), calling into question the mechanism/source of Ang-II’s presence in the brain. In this regard, the (pro)renin receptor (PRR) which is highly expressed in the brain may mediate extracellular generation of Ang-II in the brain by binding and sequestering (pro)renin, thereby increasing its catalytic activity and propagating localized cleavage of AGT (Cuadra et al., 2010).
2.2. Neural circuitry controlling the sympathetic nervous system and blood pressure
Sympathetic stimulation of the heart, vasculature and kidneys increases blood pressure by elevating cardiac output, vascular resistance and fluid retention, respectively, while inhibiting the SNS produces the opposite effects (Goldstein, 1983; Grassi, 1998; Grassi et al., 1998; Burke et al., 2012). Several forebrain and hindbrain nuclei control sympathetic outflow to these tissues, and are regulated by the RAS. Of particular importance are the paraventricular nucleus of the hypothalamus (PVN), subfornical organ (SFO), rostral ventral lateral medulla (RVLM) and nucleus of the solitary tract (NTS), all of which contain receptors for the RAS; however, there are many other brain regions that influence blood pressure via projections to and from these key cardiovascular regulatory nuclei. For more comprehensive reviews of the neural circuits regulating sympathetic outflow please refer to (Dampney, 2004; Guyenet, 2006).
The circulating RAS can influence the SNS and blood pressure by acting peripherally at presynaptic AT1R to augment norepinephrine release onto specific tissues (Abboud, 1974; Cassis and Dwoskin, 1991; Cassis, 1993), and by acting centrally at receptors in circumventricular organs (CVOs), such as the SFO. The SFO contains receptors for numerous circulating factors and is densely populated with AT1R, enabling the brain to monitor and respond to changes in systemic RAS activity. The levels of AT1R within the SFO are regulated by hypertensive stimuli and increased AT1R within CVOs reduces baroreflex sensitivity, increases sympathetic tone and elevates blood pressure (Zucker et al., 2009; Braga, 2011; Nunes and Braga, 2011; Hilzendeger et al., 2012). Moreover, lesion of the SFO or interference with pathways activated by Ang-II/AT1R specifically within the SFO (e.g., ER stress or oxidative stress) attenuate the elevated SNS activity and blood pressure that accompany experimental hypertension (Zimmerman et al., 2004a; Zimmerman et al., 2004b; Hendel and Collister, 2005; Cao et al., 2012; Osborn et al., 2012; Young et al., 2012). The SFO then sends direct projections to the PVN, as well as other cardiovascular regulatory sites to impact blood pressure (Lind et al., 1984; Li and Ferguson, 1993; Krause et al., 2008; Marvar et al., 2010; Krause et al., 2011).
The PVN lies at the heart of the regulation of many homeostatic systems, including those controlling blood pressure, energy balance and stress responsiveness. Neurons of the PVN are divided into distinct magnocellular and parvocellular sub-nuclei (Swanson and Kuypers, 1980). Magnocellular neurons express vasopressin or oxytocin and, although activation of these neurons primarily leads to the release of these neuropeptides from the posterior pituitary into the systemic circulation, some of these neurons are spinally-projecting and influence blood pressure by regulating SNS activity. Parvocellular PVN neurons are classified as preautonomic or neurosecretory and regulate sympathetic tone and neuroendocrine secretion, respectively (Swanson and Kuypers, 1980). PVN neurons receive and integrate signals from CVOs, brainstem nuclei, and from cortico-limbic brain regions that are sensitive to various stimuli such as changes in the systemic milieu or psychological stressors, and respond to them by regulating SNS activity and neuroendocrine secretion.
In addition to influencing the PVN via activating CVOs with excitatory afferents in the PVN, Ang-II can also act locally within the PVN to enhance SNS activity through many mechanisms. AT1R are located post-synaptically on preautonomic glutamate neurons of the PVN and Ang-II can directly activate these neurons to perpetuate increases in SNS activity (Jiang et al., 2009; Kleiber et al., 2010). Additionally, AT1R are located presynaptically within the PVN, stimulation of which reduces GABA release, thereby activating RVLM and spinally-projecting preautonomic neurons through presynaptic disinhibition (Li et al., 2003; Li and Pan, 2005). Ang-II also acts at AT1R, possibly on immune cells of the PVN (Shi et al., 2010), to induce oxidative stress and inflammation which feed-forward to further enhance sympathetic outflow and hypertension. Evidence that Ang-II acts at the immune cells of the PVN to perpetuate increases in SNS activity is discussed in detail in subsequent sections. AT1R activation in the PVN is opposed by the endogenous counter-regulatory ACE2-Ang(1-7)-Mas axis via the overexpression of ACE2 specifically within the PVN (Sriramula et al., 2011) and by the overexpression of MIF specifically within the PVN (Sun et al., 2004; Li et al., 2006), both of which reduce blood pressure in spontaneously hypertensive rats (SHR; a widely utilized model of hypertension).
Excitation of preautonomic PVN neurons increases blood pressure, via synapses onto RVLM neurons and preganglionic neurons in the intermediolateral cell column (IML) of the spinal cord (Swanson and Kuypers, 1980; Cechetto and Saper, 1988; Shafton et al., 1998; Pyner and Coote, 2000) that receive inputs from many brain structures and regulate sympathetic outflow to cardiovascular tissues (Dean et al., 1992; Ding et al., 1993; Madden and Sved, 2003). Upon excitation, RVLM neurons release glutamate, catecholamines and other transmitters to regulate sympathetic outflow via their actions on preganglionic IML neurons (Guyenet, 2006; Bourassa et al., 2009). Moreover, barosensitive sympathetic efferents are regulated in large part by the RVLM (Dampney et al., 2002) and hyperactivity of RVLM barosensitive neurons controlling renal sympathetic nerve activity leads to hypertension, by permitting blood pressure to increase without decreasing heart rate. Hypertensive stimuli, such as Ang-II and high-salt intake, modulate the excitability of these neurons (Stocker et al., 2006; Adams et al., 2008; Bourassa et al., 2009). RVLM AT1R activation potentiates increases in blood pressure, while ACE2 acts to reduce hypertension in SHRs. Importantly, salt intake tonically activates RVLM AT1R and enhances the sympathoexcitatory responses to RVLM administered Ang-II, highlighting an important interaction between the humoral and environmental regulators of sympathetic outflow (Adams et al., 2008).
The NTS is also an important integrative site for many homeostatic systems, including the regulation of circulation. It allows for the monitoring of the peripheral milieu as it receives input from sympathetic afferents, arterial baroreceptors, chemoreceptors, volume receptors and more. This site then mediates the sympathetic chemo- and baroreflexes. In hypertension arterial baroreceptors are reset to a higher pressure. Accordingly, abnormalities in the receipt, processing or integration of this input within the NTS could then contribute to enhanced blood pressure. AT1R are densely localized to the NTS and AT1R activation within the NTS stimulates GABAB receptor expression and dampens the baroreflex, thereby contributing to hypertension (Paton and Kasparov, 1999; Polson et al., 2007; Yao et al., 2008). Conversely, utilizing virally-mediated gene transfer techniques we have recently determined that counteracting AT1R activation via the NTS-specific overexpression of ACE2 (Yamazato et al., 2011) or MIF (Freiria-Oliveira et al., 2012) improves baroreceptor heart-rate reflex in SHRs, further corroborating protective roles for these systems in blood pressure control.
Neurons of the PVN, NTS, RVLM and IML receive projections from many other brain regions to regulate SNS activity and cardiovascular function and dysregulation of these neural circuits by genetic, humoral and environmental factors then contributes to the onset of hypertension.
3. Brain RAS, SNA and mobilization of the peripheral immune system
There is considerable cross-talk between the immune and nervous systems, such that alterations in the immune system can influence signaling within the nervous system while the brain can regulate immune function. This reciprocal communication occurs via several mechanisms and both the CNS’s ability to alter immune function and the immune system’s ability to regulate neuronal function can impact cardiovascular and metabolic regulation. In this section, we focus on the impact of peripheral immune activation on cardiovascular and metabolic homeostasis, as well as the potential role of the brain RAS and the SNS in mediating the mobilization of the peripheral immune system during these pathologies.
3.1. Peripheral immune activation during hypertension and obesity
Hypertension and predisposing conditions, such as obesity, have independently become acknowledged as chronic inflammatory conditions, with immune factors accumulating in the brain and peripheral tissues. In the periphery, hypertensive and obesigenic stimuli initiate innate and adaptive immune responses in cardiovascular (e.g., vasculature and kidney) and metabolic (e.g., adipose tissue and liver) tissues that exacerbate the pathologies (Guzik et al., 2007a; Guzik et al., 2007b; Crowley et al., 2010; De Miguel et al., 2010; Madhur et al., 2010; Dorresteijn et al., 2012; Marvar et al., 2012; Sell et al., 2012). Hypertensive stimuli increase the levels of macrophages and activate T-cells that secrete pro-inflammatory cytokines, promote vasoconstriction and enhance sodium retention (Guzik et al., 2007a; Crowley et al., 2010; De Miguel et al., 2010; Madhur et al., 2010; De Miguel et al., 2011a; Harrison et al., 2011; Harrison et al., 2012; Marvar et al., 2012). Moreover, macrophage and lymphocyte infiltration of adipose tissue and ectopic fat storage in liver and other tissues are hallmarks of obesity that lead to elevated circulating levels of immune factors which impair blood pressure and glucose regulation (Hotamisligil, 2003; Weisberg et al., 2003; Xu et al., 2003; Hotamisligil, 2006; Caspar-Bauguil et al., 2009; Bennett et al., 2012).
A causal relationship between activation of the peripheral immune system and hypertension is reflected by evidence that interfering with immune responses leads to perturbations in metabolic and cardiovascular homeostasis. In this regard, immune factors, such as TNFα (Sriramula et al., 2008; Tran et al., 2009), IL-6 (Brands et al., 2010) and T-lymphocytes (Guzik et al., 2007a; Marvar et al., 2010) are necessary for Ang-II-induced hypertension. Disruption of the innate immune system by eliminating macrophages and neutrophils via the Cre-lox-mediated specific expression of diphtheria toxin receptor in all LysM cells reduces oxidative stress, improves vascular function and reduces blood pressure (Wenzel et al., 2011). Pharmacological interference with adaptive immunity by administration of mycophenolate mofetil, which suppresses T-cells, decreases blood pressure (De Miguel et al., 2010). Further, mice with genetic deficiencies in the adaptive immune system (i.e., SCID mice or mice that lack lymphocytes [RAG1-/- mice]) are resistant to many forms of experimental hypertension (Guzik et al., 2007a; Crowley et al., 2010; Marvar et al., 2010). Adoptive transfer of T-cells, not B-cells in the RAG1-/- mice restores hypertension (Guzik et al., 2007a; Marvar et al., 2010).
Collectively, these studies point to an integral role for both the adaptive and innate immune systems in hypertension and accumulating evidence suggests that the brain, via the SNS, mediates this immune activation during hypertension.
3.2. A role for the brain RAS and sympathetic nervous system in the control of the peripheral immune system during hypertension
The brain can control peripheral immune function via neural or neuroendocrine mechanisms. The SNS innervates both primary and secondary lymphoid organs and thereby regulates the immune system (Elenkov et al., 2000). Additionally, the immune cells express receptors for and are influenced by many neuroendocrine systems, including the hypothalamic pituitary adrenal axis and the RAS. The RAS facilitates brain-immune system cross-talk via a neuroendocrine mechanism and by acting in specific brain nuclei to enhance the SNS mobilization of the peripheral immune system and these interactions impact blood pressure regulation. As discussed above, hypertension and pre-disposing obesity are often associated with increased activities of the SNS, the RAS and the peripheral immune system and this can have deleterious effects on cardiovascular and metabolic homeostasis. It is possible that a brain RAS-mediated SNS mobilization of the peripheral immune system represents a facilitating mechanism for hypertension and obesity.
Models of hypertension are associated with enhanced SNS driven immune responses. In this regard, Harwani et al. recently ascertained that the pre-hypertensive SHR exhibits proinflammatory, while the normotensive WKY rat exhibits anti-inflammatory, TLR7/8 or 9 responses to cholinergic stimulation (Harwani et al., 2012). Further, Ang-II modulates immune function by activating receptors expressed on peripheral immune cells and by acting in the brain to promote SNS outflow. Elevated brain Ang-II enhances pro-inflammatory splenic cytokines, through an SNS dependent mechanism (Ganta et al., 2005). During obesity and hypertension, the SNS is also positioned to contribute to the rise in immune factors specifically within metabolic and cardiovascular tissues, which then act locally to exacerbate dysfunction. Metabolic tissues, such as fat, express adrenergic receptors and activation of these receptors initiates local inflammation (Bartness and Song, 2007; Wang et al., 2011). Of relevance, perivascular adipose tissue is particularly highly innervated by the SNS and it is possible that adrenergic induced immune activation in perivascular fat that occurs during obesity and contributes to the progression of co-morbid hypertension (Guzik et al., 2007b). Similarly, SNS activation of cardiovascular tissue initiates local inflammatory responses that exacerbate pathology (Rodriguez-Iturbe et al., 2002; Levick et al., 2010; De Miguel et al., 2011a; De Miguel et al., 2011b).
CVOs are in an ideal position to mediate reciprocal communication between the nervous and immune systems as they can monitor peripheral levels of inflammation, Ang-II and other hypertensive stimuli and then facilitate the activation of the immune system. These brain regions are potent regulators of cardiovascular function and hydromineral balance and they likely play a causal role in the SNS mobilization of the peripheral immune system (peripheral activation of T-cells and vascular inflammation) during hypertension (Lob et al., 2010; Marvar et al., 2010; Marvar et al., 2012). Commensurate with this, electrolytic lesion of the anterioventral third ventricle region prevents experimental hypertension (Marvar et al., 2010) and blocks the activation and vascular infiltration of T-cells in response to chronic Ang-II infusion (Marvar et al., 2010). Marvar and colleagues have further delineated a role of oxidative stress specifically within the SFO, as the loss of superoxide dismutase 3 within this brain region leads to T-cell activation, enhanced inflammation and reactive oxygen species in peripheral vessels, and enhanced sympathetic outflow and hypertension (Lob et al., 2010).
Furthermore, our group has recently published studies supporting a brain-SNS-bone marrow connection, originating in the PVN that plays a facilitating role in the progression of Ang-II-induced neurogenic hypertension (Jun et al., 2012). Specifically, we have found that elevations in Ang-II decrease bone marrow-derived endothelial progenitor cells and increase bone marrow-derived inflammatory cells, thereby compromising vascular repair and facilitating the progression of hypertension. It is likely that this pathway not only activates the peripheral immune system, but also mobilizes brain immune cell progenitors during hypertension.
Based on these studies, it is clear that the SNS mobilization of the peripheral immune system is involved in various forms of experimental hypertension, including stress-related high blood pressure (Marvar and Harrison, 2012; Marvar et al., 2012). Since obesity can also be considered a stressor, it is possible that a similar mechanism contributes to obesity-related hypertension.
4. Recruitment and activation of immune cells within the CNS during neurogenic hypertension
Another mechanism by which the immune and nervous systems interact is via the activation of resident immune cells and/or the recruitment of peripheral immune cells to the neural parenchyma. The healthy CNS is thought to be “immune-privileged,” in that there is a lack of parenchymal dendritic cells, which are critical for antigen uptake, migration to draining lymph nodes and presentation to naïve T-cells. Despite this immune-privilege that protects the brain from inflammatory reactions that may damage non-regenerating neurons, the immune system impacts the brain during several pathologies. For instance, the nervous system clearly responds to acute systemic infections by initiating sickness behavior, which is characterized by decreased locomotor activity and food intake (Langhans, 2007) and is considered an adaptive response to facilitate survival during the challenge (Dantzer and Kelley, 2007). Additionally, when the brain becomes injured, for example, by a stroke or a neurodegenerative disease, resident and recruited exogenous immune cells act to remove damaged tissue and protect the remaining neurons. The magnitude of the insult likely governs whether the immune cells are of central origin or are recruited from the periphery and although the immune system is clearly in place to protect the host from antigens, immune cells also release factors that exacerbate pathologies, including hypertension and metabolic disease.
4.1. Hypertension and brain inflammation
Several lines of evidence suggest that immune factors are capable of acting in the brain to intensify hypertension. In this regard, models of hypertension are associated with increased brain immune factors while models of neuroinflammation are coupled with increased blood pressure (Wu et al., 2012). Ang-II is a potent regulator of the immune system centrally and peripherally and induces brain inflammatory responses that are critical for the development and progression of hypertension. Ang-II increases PVN expression of proinflammatory cytokines (Shi et al., 2010; Shi et al.), such as IL-1β, that activate PVN neurons (Yang et al., 1997) and increase sympathetic outflow (Kannan et al., 1996). Ang-II also activates NFκB within the PVN and this is required for Ang-II’s hypertensive effects (Kang et al., 2009; Cardinale et al., 2012). On the other hand, expression of the anti-inflammatory cytokine, IL-10, is reduced during Ang-II infusion, and its overexpression attenuates Ang-II-induced hypertension (Shi et al., 2010). Similarly, the cytokine MIF, working intracellularly via its intrinsic TPOR activity, opposes Ang-II actions and reduces blood pressure in hypertensive animals (Busche et al., 2001; Sun et al., 2004; Li et al., 2006; Li et al., 2008; Freiria-Oliveira et al., 2012). Other models of hypertension, such as the SHR, as well as predisposing conditions such as obesity or stress-related disorders, are also associated with increased hypothalamic expression of inflammatory factors that may represent a causal link among these pathologies(Jankord et al., 2010; Agarwal et al., 2011). Moreover, during hypertension, different brain regions important for the regulation of blood pressure also exhibit a proinflammatory milieu. For example, within the brainstem, microarray comparison between SHR and WKY NTS (Waki et al., 2007; Waki et al., 2008b, a) revealed an upregulation of junctional adhesion molecule-1 [JAM-1], overexpression of which is sufficient to increase blood pressure in normotensive rats. In addition, the LPS-induced model of systemic inflammation is associated with neuroinflammation and oxidative stress specifically in the RVLM that leads to neurogenic hypertension (Wu et al., 2012). The implication is that in addition to influencing blood pressure via direct actions on neurons, many hypertensive stimuli also initiate a rise in proinflammatory factors that then augment sympathetic outflow.
4.2. Blood brain barrier
The blood brain barrier (BBB), which on one hand protects the CNS from deleterious peripheral immune reactions, on the other hand regulates much of the neuroimmune exchange by serving as an intermediary for the peripheral immune system’s ability to impact the brain (Banks, 2005; Banks and Erickson, 2010). Incidentally, one important mechanism by which the peripheral immune system can directly interact with the CNS is through a compromised blood brain barrier (BBB). Certain threats, including homeostatic challenges that cause hypertension or obesity, compromise the integrity of the BBB which can then lead to the recruitment of peripheral immune cells to the brain parenchyma. For example, chronic administration of hypertensive doses of Ang-II or consumption of high-fat diet increase the permeability of the BBB (Kanoski et al., 2010; Zhang et al., 2010; Davidson et al., 2012).
Also of relevance, peripheral immune system macrophages are associated with the perivascular space, the CVOs, the choroid plexus and the meninges of the brain. Certain brain regions that are key regulators of metabolism and blood pressure, such as the PVN and the arcuate nucleus of the hypothalamus (ARC), are highly vascularized and it is possible that these regions are particularly susceptible to factors that jeopardize the BBB. Further, it is possible that the abundance of immune factors within these regions in response to hypertensive and obesigenic stimuli occurs secondary to a threatened integrity of the BBB.
Importantly, several cytokines can also effectively be transported bi-directionally across the intact BBB. Moreover, peripheral cytokines can activate receptors situated on the BBB, thereby disrupting transport systems of the BBB and/or leading to the release of paracrine factors, such as prostaglandins, that can impact various cell-types of the CNS (Yu et al., 2010).
4.3. Resident Immune Cells of the Brain
Other important sources for the elevations in central sympathoexcitatory pro-inflammatory factors during hypertension are the resident immune cells of the brain, which are capable of both sensing and responding to hypertensive stimuli. Microglia and astrocytes are the brain’s resident innate immune cells (reviewed in Ransohoff and Brown (2012)) that respond to various threats to homeostasis, including hypertensive stimuli (Shi et al., 2010; Thaler et al., 2012). While it is clear that the interactions among the cell-types of the nervous system contribute to the maintenance of homeostasis, it is also possible that these exchanges contribute to metabolic and cardiovascular pathologies.
Microglia
Microglia are of mesodermal/mesenchymal origin, originating most likely from the bone marrow, and are distributed throughout the healthy and injured CNS (Kettenmann et al., 2011). Under normal conditions, ‘resting’ microglia have a small soma and long fine processes that act to survey their microenvironment for pathogens and other disturbances (Badoer, 2010; Kettenmann et al., 2011). They sense threats to homeostasis, and respond to them by rapidly becoming activated and migrating to the damaged tissue (Nguyen et al., 2002; Shi et al., 2010; Thaler et al., 2012). Upon activation microglia undergo a phenotypic change, transforming into cells with enlarged cell bodies and ramified processes (Kreutzberg, 1996; Nakajima and Kohsaka, 2001; Shi et al., 2010). Activated microglial cells then facilitate neuroprotection by removing debris, clearing dead cells and secreting neurotrophic factors. However, chronic recruitment and activation of microglia can also be maladaptive and contribute to impaired neuronal function (Nakajima and Kohsaka, 2001; Badoer, 2010; Thaler et al., 2012)
There are extensive communications between microglia and neurons, facilitating the ability of microglia to sense disturbances to the nervous system, and to regulate the development, structure and function of neural connections (Kettenmann et al., 2011). Gap junctions allow the bidirectional exchange of ions and small molecules between neurons and microglia (Eugenin et al., 2001; Dobrenis et al., 2005). Microglia make contact with synaptic structures (Wake et al., 2009) and thereby have the capability to sense synaptic activity. Further, upon injury, microglia play a role in reactive synaptogenesis (Bessis et al., 2007).
Microglia express toll-like receptors TLRs, which are receptors of the innate immune system that recognize molecules that are conserved among many pathogens. Importantly, a number of TLRs are known to recognize lipid-containing motifs and TLR-4 is well-known to recognize lipopolysaccharides (LPS). Microglia also express receptors for numerous chemokines, cytokines, neurotransmitters (e.g., glutamatergic, GABAergic and adrenergic) and neurohormones (e.g., angiotensin-II, glucocorticoids and mineralocorticoids) (reviewed by Kettenmann et al. (2011)), aiding their ability to sense disturbances to homeostasis. Additionally, various factors are released by microglia that either support the survival of remaining neurons or induce neuronal death. Of relevance, microglia are known to respond to and produce factors that influence cardiovascular and metabolic homeostasis (Shi et al., 2010; Thaler and Schwartz, 2010; Thaler et al., 2012; Yi et al., 2012a; Yi et al., 2012b).
Under normal conditions, microglia lie in close proximity to preautonomic PVN neurons (Figure 1), placing them in an ideal position to regulate sympathetic outflow. During experimental hypertension, brain regions important for regulating sympathetic outflow, such as the PVN and NTS, are infiltrated with additional microglia and the levels of proinflammatory factors and reactive oxygen species within these regions rise (Waki et al., 2008b, a; Shi et al., 2010; Zubcevic et al., 2011; Cardinale et al., 2012), and have the potential to influence PVN neuron activity. Several lines of evidence suggest that the RAS facilitates these interactions. Hypertension produced by chronic infusion of Ang-II is associated with AT1R-mediated increases in microglial activation and cytokine levels in the PVN (Shi et al., 2010). Further, the central administration of minocycline, an anti-inflammatory antibiotic that reduces microglial activation, decreases Ang-II-induced hypertension (Shi et al., 2010). This has led us to hypothesize that microglial activation in response to Ang-II stimulation results in an inflammatory response (increases in cytokine, chemokine, ROS production) that can, via a paracrine action, modulate the activity of associated PVN preautonomic neurons (Shi et al., 2010). Our preliminary data, generated from microglia isolated from rat hypothalamus, support this idea and suggest that there are two components to the Ang-II induction of microglia in the PVN during hypertension. A direct effect of Ang-II at AT1R located on microglia to elicit their activation, and an indirect effect via neuronal AT1R to cause secretion of monocyte chemotactic protein-1 (MCP-1; or CC-chemokine ligand-2 [CCL2]) which then binds to its receptors (CCR2) on microglia and stimulates their migration towards PVN preautonomic neurons. In addition, it has been shown that AT1R are expressed on microglia and angiotensin receptor blockers reduce LPS-induced inflammation in vitro (Miyoshi et al., 2008). Conversely, the recent generation of a transgenic AT1R reporter mouse indicates that, under normotensive in vivo conditions, brain AT1R expression is almost exclusively neuronal (Gonzalez et al., 2012). However, it is possible that during hypertension, the expression pattern of AT1R changes and that microglia begin to express the receptor. Then again, it is also possible that microglia sense neuronal AT1R stimulation, and that this triggers their activation in response to Ang-II.
Figure 1. Interactions between microglia, astrocytes and preautonomic RVLM-projecting PVN neurons.
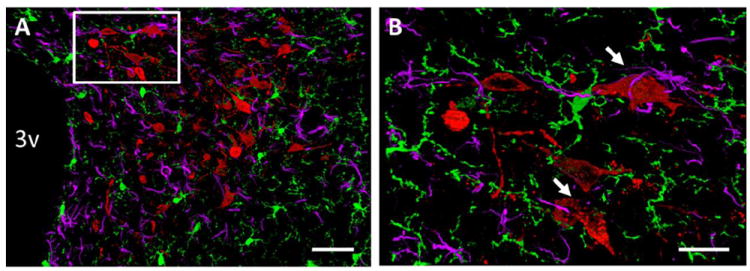
(A) 20× and (B) 63× images of microglia (green), astrocytes (purple) and preautonomic RVLM-projecting neurons within an adult mouse PVN. Adult mice were injected bilaterally into the RVLM with the retrograde tract tracer, fluorogold. Seven days later mice were perfused and brains were processed for triple-label immunohistochemistry for Iba-1 (a microglial marker), GFAP (an astrocytic marker), and fluorogold (preautonomic neurons) using previously published protocols (Ulrich-Lai et al., 2011; Thaler et al., 2012). 3v = third cerebral ventricle. Arrows highlight interactions between microglia, astrocytes and RVLM-projecting neurons. Scale bars = 50 μm (A) and 20 μm (B).
Astrocytes
Astrocytes also participate in innate immune reactions within the nervous system. They express TLRs and produce proinflammatory factors that can act on cells within the brain, as well as anti-inflammatory cytokines and neurotrophic factors, such as CNTF, which promote the repair of the injured CNS (Van Der Voorn et al., 1999; Panenka et al., 2001; McKimmie and Graham, 2010; Allaman et al., 2011; Gorina et al., 2011; Kang et al., 2012; Werry et al., 2012). Astrocytes regulate neuronal function via several mechanisms including the release of glutamate, ATP and other signaling molecules. Astrocytes buffer CNS potassium, remove excess cytotoxic glutamate and modulate blood flow (Gurden et al., 2006; Belanger et al., 2011; Ransohoff and Brown, 2012). Astrocytes also express the GABA transporter 3, which removes GABA and thereby contributes to GABA tone in various neural circuits, including those that regulate sympathetic outflow and blood pressure (Park et al., 2009). Moreover, astrocytes and microglia interact to regulate one another’s function and the function of neurons (Kloss et al., 1997; Bezzi et al., 2001a; Bezzi et al., 2001b; Belanger et al., 2011; Liu et al., 2011). They secrete factors that modulate the activity of microglia and they are impacted by factors secreted by microglia (DeWitt et al., 1998; Yang et al., 1998; Bezzi et al., 2001a). Importantly, astrocytes, like microglia, lie in close proximity to preautonomic PVN neurons (Figure 1) and are capable of sensing and responding to hypertensive and obesigenic stimuli (Buckman et al., 2012; Gerber and Bale, 2012; Thaler et al., 2012). Consequently, it is conceivable that astrocyte-neuron and/or astrocyte-microglia interactions contribute to blood pressure regulation.
Another mechanism by which astrocytes participate in the neuroimmune regulation of blood pressure depends on the fact that they represent a key cellular compartment for RAS actions within the brain. Angiotensinogen is primarily expressed within astrocytes (Stornetta et al., 1988; Intebi et al., 1990 & Deschepper, 1990; McKinley et al., 2003; Sherrod et al., 2005) and astrocyte AGT is critical for the integrity of the BBB (Kakinuma et al., 1998). Astrocytes also express AT1R and Ang-II can elicit immune responses via their activation (Kandalam and Clark, 2010; Finsen and Owens, 2011; Fuchtbauer et al., 2011). Furthermore, there is some evidence linking the protective effects of the ACE2-Ang-(1-7)-Mas axis’s on cardiovascular function to astrocytes, as these cells likely mediate Ang-(1-7)’s cardiovascular regulatory actions in the RVLM (Guo et al., 2010). On the other hand, although it is clear that AT1R are present on astrocytes and that activation of astrocytic AT1R induces inflammation, at present, the Ang-II-AT1R pathway within astrocytes has not been directly linked to the regulation of cardiovascular function.
4.4. Recruitment of Immune Cells to Specific Brain Nuclei
Many disturbances to homeostasis, including hypertension and obesity are associated with increased numbers of immune cells in distinct brain nuclei; however whether these cells are of central and/or peripheral origin is not always clear. Resident microglia may migrate within the brain and accumulate in damaged regions, while exogenous microglial progenitors, produced and mobilized from bone marrow, penetrate the brain and migrate to injured areas upon insult (Lawson et al., 1992; Imai et al., 1999; Malm et al., 2005; Stalder et al., 2005; Simard et al., 2006; Imai et al., 2007). Accumulating evidence suggests that the SNS regulates mobilization of microglial progenitors (Katayama et al., 2006). To this end, interference with the SNS, via either the administration of a β-blocker or the disruption of catecholaminergic neurons, reduces the number of HSPCs mobilized by granulocyte colony-stimulating factor (Katayama et al., 2006).
Once mobilized, resident microglia and microglial progenitors are directed to infiltrate specific injured brain areas, at least in part, by chemokine signaling. Chemokines and their receptors are vital for the recruitment of immune cells to specific sites of inflammation throughout the body and for development of inflammatory responses. For example, within the brain, MCP-1/CCL-2 stimulates its receptor, CCR2 to regulate the communication between neurons, astrocytes and microglia, thereby mediating the recruitment of both resident and peripheral immune cells to sites of injury within the CNS (Old and Malcangio, 2012). CCR2 is expressed on monocytes, and within the brain, predominantly on microglia. MCP-1/CCL2 is produced by glial cells (Glabinski et al., 1996; Gourmala et al., 1997; Van Der Voorn et al., 1999) and is released from neuronal synaptic vesicles (Thacker et al., 2009; Van Steenwinckel et al., 2011) to mediate the recruitment of monocytes to injured brain tissue. Importantly, this system is critical for recruiting additional microglial progenitors to the brain parenchyma. Using green fluorescent protein transgenic bone marrow chimeric mice, Schilling and colleagues were able to differentiate between resident and infiltrating immune cells and determine that mice deficient in this pathway have reduced infiltrating immune cells, rather than resident microglia following transient cerebral ischemia (Schilling et al., 2009a; Schilling et al., 2009b; Schuette-Nuetgen et al., 2012).
In addition to the recruitment of microglial progenitors to the CNS, peripheral immune cells are trafficked to the brain during certain pathologies, such as cerebral inflammatory diseases and peripheral organ inflammation (D’Mello et al., 2009; Conductier et al., 2010). For example, elevated peripheral TNFα signaling during hepatic inflammation enhances CCL2 production in microglia, leading to the recruitment of CCR2-expressing monocytes into the brain (D’Mello et al., 2009).
Because of the critical role of MCP-1/CCL-2 in mobilization of immune cells in CNS disease states, it is possible that this system is important for immune activation and sympathoexcitation at the level of the PVN during hypertension. Under normal conditions, MCP-1/CCL2 is densely expressed in the PVN and other hypothalamic nuclei (Banisadr et al., 2005). In addition, CCR2 is upregulated in the PVN during restraint stress, a challenge that causes elevations in blood pressure (Reyes et al., 2003). These findings are consistent with a role for CCL2/CCR2 signaling in the recruitment of immune cells to the PVN during hypertension, and our preliminary data provide direct support for this idea. Specifically, we have demonstrated that central (intracerebroventricular) infusion of Ang II at a level that causes hypertension also increases MCP-1/CCL-2 expression in the PVN (Shi et al., 2011). Furthermore, elevations in PVN MCP-1/CCL-2 expression are also observed in SHR (Shi et al.), animals that display enhanced Ang-II/AT1R signaling in the PVN. In summary, the data available thus far support the idea that a critical component of sympathoexcitation and neurogenic hypertension is activation of and recruitment of immune cells within and to the PVN, and MCP-1/CCL-2 may play a major role.
5. Roles of the sympathetic nervous, renin-angiotensin and immune systems in obesity-related hypertension
One important strategy to treat or prevent hypertension is to develop therapeutics that target predisposing conditions. In this regard, obesity is a principal risk factor for the development of hypertension and these ailments are very often co-morbid (Garrison et al., 1987; Cassano et al., 1990; Brown et al., 2000; Timpson et al., 2009; Weiss et al., 2009; Armitage et al., 2012). Importantly, both pathologies are independently associated with activation of the SNS and RAS (Grassi et al., 1995; Alonso-Galicia et al., 1996; Vaz et al., 1997; Cooper et al., 1998; Rumantir et al., 1999; Grassi et al., 2003; de Kloet et al., 2010; Armitage et al., 2012) and when obese individuals reduce sympathetic drive or RAS activity either pharmacologically or by losing weight, their blood pressure is decreased (Grassi et al., 1998; Wofford et al., 2001; Straznicky et al., 2005). Obesity modulates the neural circuitry that controls sympathetic outflow and blood pressure and several mechanisms for the enhanced sympathetic drive during obesity have been proposed (Haynes, 2005; Stocker et al., 2006; Purkayastha et al., 2011; Hilzendeger et al., 2012; Yiannikouris et al., 2012a). Of importance, many of the factors that are thought to contribute to increased blood pressure during obesity are also associated with increased recruitment and activation of immune cells within the CNS.
It is widely-accepted that diet-induced and genetic models of obesity are accompanied by elevated inflammation in peripheral tissues and the brain (Xu et al., 2003; Shoelson et al., 2006; Cani et al., 2007; Shoelson and Goldfine, 2009; Thaler and Schwartz, 2010; Mathis and Shoelson, 2011; Thaler et al., 2012). In regards to the brain, high-fat diet consumption increases the expression of microglial and astrocytic markers within several brain regions (Pistell et al., 2010). In the mediobasal hypothalamus (including the ARC and median eminence), high-fat feeding elevates inflammation, microglial activation and astrogliosis in as little as 1-3 days (Thaler et al., 2012) and this inflammatory milieu persists after long-term high-fat diet feeding (Thaler et al., 2012), implying that perhaps just one meal of high-fat diet or long-term high-fat diet feeding can have a deleterious impact on the CNS.
The enhanced inflammation within mediobasal hypothalamus is then thought to contribute to alterations in metabolism (Zhang et al., 2008), glucose homeostasis (Posey et al., 2009; Milanski et al., 2012), and, more recently, to cardiovascular regulation (Purkayastha et al., 2011). Consistent with this, Purkayastha et al. determined that hypothalamic Iκκ-β and NFκB represents a key mechanism linking the consumption of high-fat diet to the dysregulation of energy, glucose and cardiovascular homeostasis. Of relevance, adenoviral activation of NFκB within the mediobasal hypothalamus leads to elevated blood pressure, while its inactivation prevents obesity-associated increases in blood pressure. Further, inflammation specifically within proopiomelanocortin neurons, which are particularly important for the regulation of energy balance, may contribute to blood pressure regulation, as TNFα causes increases in blood pressure and Iκκ-β phosphorylation in proopiomelanocortin neurons, while Iκκ-β deletion in proopiomelanocortin neurons prevents TNFα and obesity-induced increases in blood pressure.
An unanswered question, however, is whether or not high-fat diet consumption leads to inflammation in other cardiovascular control regions of the brain, thereby contributing to obesity-related hypertension. There is some recent evidence that genetic and diet-induced obesity in mice leads to astrogliosis within the PVN (Buckman et al., 2012). Moreover, our preliminary studies indicate that like chronic Ang-II administration, high-fat diet causes inflammation within the PVN.
Obesity is associated with adipose tissue dysfunction, characterized by hypertrophied adipocytes, increased adipose inflammation, infiltration of macrophages and by altered expression and secretion of various adiposity factors, collectively termed ‘adipokines,’ that may then act in the brain to control SNS activity. Of particular interest for this review, Ang-II can be considered an adipokine as it is produced by adipose tissue and its levels are positively-correlated with body mass in both humans and rodents (de Kloet et al., 2010). Considering that Ang-II is a potent activator of the SNS, one potential mechanism for the augmented sympathetic drive during obesity is this increased level of Ang-II. AT1R blockade reduces SNS activity in obese hypertensive humans (Grassi et al., 2003) and genetically obese rats have a greater magnitude in the decrease in blood pressure that follows AT1R blockade (Alonso-Galicia et al., 1996). Overexpression of AGT specifically within adipose tissue leads to elevated blood pressure (Massiéra et al., 2001; Kalupahana et al., 2011), while adipose tissue-specific deletion of AGT prevents obesity-related hypertension (Yiannikouris et al., 2012a).
Based on these findings and the observation that Ang-II induces hypothalamic inflammation in hypertensive models, it is intriguing to hypothesize that this peptide contributes to recruitment and activation of immune cells within the CNS during obesity which then facilitates sympathetic outflow and leads to obesity-related hypertension. Although several indirect lines of evidence suggest this may be the case, this hypothesis has not directly been tested. Moreover, this phenomenon is likely complex and several mechanisms contributing to obesity-related neuroimmune communication in hypertension have been proposed. For example, it is well known that other adipokines such as leptin and insulin can impact the SNS and increase immune factors in specific brain regions. Leptin acts at it receptors in cardiovascular control centers of the brain to increase sympathetic outflow to several tissues (Haynes et al., 1999; Haynes, 2005; Mark et al., 2009), to impair baroreflex control of heart rate (Arnold et al., 2009) and to increase blood pressure Correia et al. (2001). Importantly, although a key mechanism for obesity is resistance to the appetite suppressing effects of leptin, the sympathoexcitatory effects of leptin are preserved during obesity (Mark et al., 1999) and it is possible that leptin is a critical mediator of obesity-related hypertension. It is also apparent that, similar to Ang-II, leptin activates its receptor not only on neurons, but also on microglia and astrocytes, and it is possible that leptin’s actions at these cell-types may contribute to its sympathoexcitatory effects (Pinteaux et al., 2007; Tang et al., 2007; Lafrance et al., 2010). Lastly, there are substantial interactions between adiposity factors, such as insulin and leptin, and Ang-II, suggestive of additional avenues by which the RAS can influence blood pressure during obesity (Cassis et al., 2001; Hilzendeger et al., 2012).
6. A model for immune/central nervous system interactions during hypertension
Based upon the compelling evidence from the reviewed literature, it is tempting to speculate that an immune/central nervous system reciprocal interaction is critical for the regulation of cardiovascular and metabolic function. Hypertensive stimuli, such as obesity and chronic stress, cause increases in circulating factors, a key factor being Ang-II. These factors are then sensed by CVOs such as the SFO which then transmit these signals to the preautonomic region of PVN. This pathway then directly increases sympathetic outflow and blood pressure by stimulating spinally and RVLM-projecting neurons. During neurogenic hypertension, it is well known that there is also an enhancement of the direct neuronal actions of Ang-II via AT1R in the PVN, resulting in an over-stimulation of preautonomic neurons. Although the precise mechanism by which Ang-II becomes present in the brain is a subject of debate, there is substantial evidence that this peptide is synthesized by the brain-specific RAS (Lenkei et al., 1997; Morimoto et al., 2002; McKinley et al., 2003; Grobe et al., 2008; Cuadra et al., 2010; Grobe et al., 2010; de Kloet et al., 2011; Yamazato et al., 2011). Here we are proposing that the enhanced actions of Ang-II in neurogenic hypertension involve direct effects at microglia and possibly astroglia and sustained induction of both the central and peripheral immune systems. In terms of microglia, we suggest that Ang-II directly activates these cells, and indirectly (via MCP-1/CCL-2) released from neurons) causes their migration towards preautonomic neurons (Figure 2). It is possible that a similar scenario occurs with astroglia. This proinflammatory microenvironment within the PVN then stimulates the brain to signal via the SNS to mobilize the peripheral immune system and to the bone marrow to mobilize microglial progenitors that are recruited to the PVN, likely via a CCL2/CCR2-dependent mechanism. This increase in microglial progenitors within the PVN then contributes to the population of innate immune cells within the PVN, then feeding-forward to enhance and sustain elevations in blood pressure by further influencing the activity of preautonomic PVN neurons. The mechanism(s) by which microglia interact with preautonomic neurons to influence their activity has not yet been elucidated and is therefore an important avenue for future studies.
Figure 2. Proposed model for immune/central nervous system interactions during hypertension.
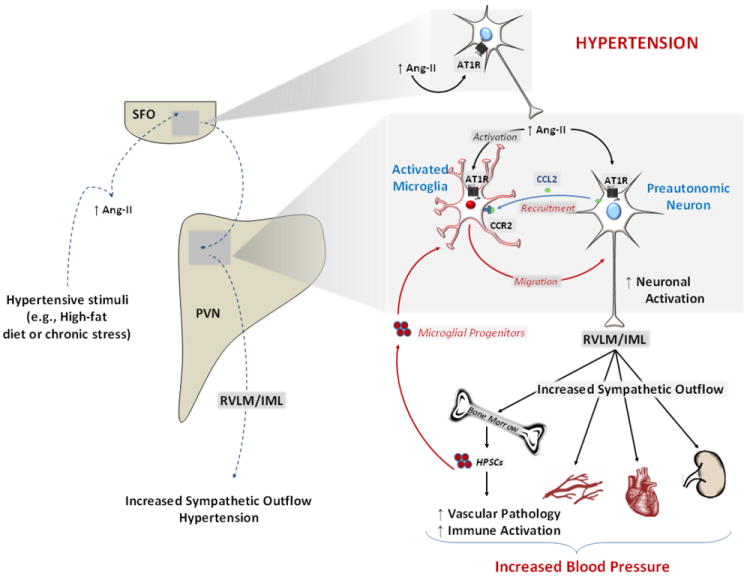
Hypertensive stimuli (e.g., obesity and chronic stress), cause increases in circulating factors (e.g., Ang-II) that are sensed by CVOs, such as the SFO. The SFO then transmits these signals to the preautonomic region of PVN, leading to stimulation of intermediolateral cell column (IML) and rostralventrolateral medulla (RVLM)-projecting neurons and resulting in increased sympathetic outflow and blood pressure. Furthermore, during neurogenic hypertension, the enhanced direct neuronal actions of Ang-II at AT1R in the PVN leads to an over-stimulation of preautonomic neurons. Direct effects of Ang-II at microglia leads to sustained induction of both the central and peripheral immune systems. We propose that initially Ang-II directly activates microglia, and indirectly (via MCP-1/CCL-2 released from neurons) causes their migration towards preautonomic neurons. This proinflammatory microenvironment within the PVN then stimulates the brain to signal via the SNS to mobilize the peripheral immune system and to the bone marrow to mobilize microglial progenitors that are recruited to the PVN, likely via a CCL2/CCR2-dependent mechanism. This increase in microglial progenitors within the PVN then contributes to the population of innate immune cells within this nucleus, feeding-forward to activate preautonomic PVN neurons thereby augmenting and sustaining the elevations in blood pressure.
We also propose that hypertensive and obesigenic stimuli threaten the integrity of the BBB in the key cardiovascular and metabolic control regions, the PVN and ARC and that this is sensed by the innate immune cells of the brain, further exacerbating their responses, and contributing to the microglial activation and astrogliosis within these brain regions. It is possible that this astrogliosis and microglial activation initially occurs in attempt to protect the neurons from peripheral factors, but ends up being detrimental.
It is clear that during many conditions that pre-dispose to hypertension there are elevated levels of the peripheral RAS activity that is sensed by the brain. Although the focus here is on Ang-II, other factors, such as leptin, proinflammatory cytokines and fatty acids may also activate this pathway during obesity to exacerbate cardiovascular dysfunction. We believe that these collective neuroimmune interactions facilitate the progression of neurogenic hypertension.
7. Consideration of RAS/Neuroimmune System interactions as novel therapeutic targets in hypertension
Importantly, the reviewed studies reveal a promising new avenue for the development of therapeutics for patients with neurogenic hypertension. As discussed throughout this review, chronic immune activation, increases in SNS activity and enhanced RAS activity are common features of hypertension and reciprocal communication between these systems likely contributes to increased blood pressure. Therapeutics that target the RAS or SNS are already widely-utilized to reduce blood pressure in hypertensive patients. The concept that has only recently been appreciated is that the connection between the RAS/SNS and the immune system may serve as another opportunity for the development of anti-hypertensive therapeutics. In this regard, it has been hypothesized that AT1R blockade may have utility for the treatment of inflammatory brain disorders (Benicky et al., 2009; Benicky et al., 2011). Furthermore, numerous clinically-approved pharmacological therapeutics that target inflammatory cascades and are currently prescribed for other pathologies, may perhaps also be exploited to treat hypertension. For example, Etanercept, which inhibits TNFα actions, is widely utilized to treat autoimmune disorders and reduces blood pressure in rodent studies (Tran et al., 2009; Venegas-Pont et al., 2010)
Additional support for the probable therapeutic utility of targeting this pathway is reflected by the fact that this model may not only underlie hypertension, but it may also causally link hypertension to its pre-disposing conditions. While there are several pre-disposing conditions that may activate the proposed pathway, in this review, particular emphasis was placed on the applicability of the proposed model to obesity-related hypertension. Therefore, when evaluating the therapeutic potential of targeting RAS/neuroimmune system interactions during obesity-related hypertension, it is also important to consider, that the SNS, RAS and immune system are also involved in other related pathologies, such as obesity itself and diabetes (Hotamisligil, 2003, 2006; de Kloet et al., 2010; Thaler and Schwartz, 2010). Developing a clear understanding of the overlap and distinction between the roles of the systems in the regulation of energy balance and glucose homeostasis versus their roles in the regulation of cardiovascular function may, therefore, also open avenues for the development of novel therapeutics to treat or prevent several aspects of the metabolic syndrome.
Collectively, the reviewed studies provide support for a key RAS/neuroimmune interactions in the regulation of blood pressure are reveal a promising new avenue for the development of therapeutics for patients with neurogenic hypertension.
Acknowledgments
This work was supported by an American Heart Association Postdoctoral Fellowship (12POST11550013), by an NIH T32 Training Grant (HL-083810), and by NIH grants HL-076803, HL-093186 and HL-33610.
Abbreviations
- ACE
angiotensin-converting enzyme
- ACE2
angiotensin-converting enzyme 2
- AGT
angiotensinogen
- Ang-II
angiotensin-II
- Ang-(1-7)
angiotensin (1-7)
- AT1R
angiotensin type-1 receptor
- AT2R
angiotensin type-2 receptor
- ARC
arcuate nucleus of the hypothalamus
- BBB
blood-brain barrier
- CCL2
C-C chemokine ligand type-2
- CCR2
C-C chemokine receptor type-2
- CVO
circumventricular organ
- GFAP
glial fibrillary acidic protein
- IL-10
interleukin-10
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- IML
intermediolateral cell column
- Iba-1
ionized calcium binding adaptor molecule 1
- MIF
macrophage migration inhibitory factor
- MCP-1
monocyte chemotactic protein-1
- NTS
nucleus of the solitary tract
- PVN
paraventricular nucleus of the hypothalamus
- PRR
pro-renin receptor
- RAS
renin-angiotensin system
- RVLM
rostral ventral lateral medulla
- SFO
subfornical organ
- SHR
spontaneously hypertensive rat
- SNS
sympathetic nervous system
- TPOR
thiol-protein oxidoreductase
- TNFα
tumor necrosis factor-α
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abboud FM. Effects of sodium, angiotensin, and steroids on vascular reactivity in man. Fed Proc. 1974;33:143–149. [PubMed] [Google Scholar]
- Adams JM, McCarthy JJ, Stocker SD. Excess dietary salt alters angiotensinergic regulation of neurons in the rostral ventrolateral medulla. Hypertension. 2008;52:932–937. doi: 10.1161/HYPERTENSIONAHA.108.118935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal D, Welsch MA, Keller JN, Francis J. Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res Cardiol. 2011;106:1069–1085. doi: 10.1007/s00395-011-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Alonso-Galicia M, Brands MW, Zappe DH, Hall JE. Hypertension in obese Zucker rats. Role of angiotensin II and adrenergic activity. Hypertension. 1996;28:1047–1054. doi: 10.1161/01.hyp.28.6.1047. [DOI] [PubMed] [Google Scholar]
- Armitage JA, Burke SL, Prior LJ, Barzel B, Eikelis N, Lim K, et al. Rapid onset of renal sympathetic nerve activation in rabbits fed a high-fat diet. Hypertension. 2012;60:163–171. doi: 10.1161/HYPERTENSIONAHA.111.190413. [DOI] [PubMed] [Google Scholar]
- Arnold AC, Shaltout HA, Gallagher PE, Diz DI. Leptin Impairs Cardiovagal Baroreflex Function at the Level of the Solitary Tract Nucleus. Hypertension. 2009;54:1001–1008. doi: 10.1161/HYPERTENSIONAHA.109.138065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M, Ganten D. It’s renin in the brain: transgenic animals elucidate the brain renin angiotensin system. Circ Res. 2002;90:8–10. [PubMed] [Google Scholar]
- Badoer E. Microglia: activation in acute and chronic inflammatory states and in response to cardiovascular dysfunction. Int J Biochem Cell Biol. 2010;42:1580–1585. doi: 10.1016/j.biocel.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res. 1992;599:223–229. doi: 10.1016/0006-8993(92)90395-p. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin R-D, Mechighel P, Kitabgi P, Rostène W, Parsadaniantz SM. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: Evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol. 2005;489:275–292. doi: 10.1002/cne.20598. [DOI] [PubMed] [Google Scholar]
- Banks WA. Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des. 2005;11:973–984. doi: 10.2174/1381612053381684. [DOI] [PubMed] [Google Scholar]
- Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Song CK. Brain-adipose tissue neural crosstalk. Physiol Behav. 2007;91:343–351. doi: 10.1016/j.physbeh.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benicky J, Sanchez-Lemus E, Pavel J, Saavedra JM. Anti-inflammatory effects of angiotensin receptor blockers in the brain and the periphery. Cell Mol Neurobiol. 2009;29:781–792. doi: 10.1007/s10571-009-9368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B, Larson-Meyer DE, Ravussin E, Volaufova J, Soros A, Cefalu WT, et al. Impaired Insulin Sensitivity and Elevated Ectopic Fat in Healthy Obese vs. Nonobese Prepubertal Children. Obesity. 2012;20:371–375. doi: 10.1038/oby.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55:233–238. doi: 10.1002/glia.20459. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001a;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Vesce S, Volterra A. Neuron-astrocyte cross-talk during synaptic transmission: physiological and neuropathological implications. Prog Brain Res. 2001b;132:255–265. doi: 10.1016/S0079-6123(01)32081-2. [DOI] [PubMed] [Google Scholar]
- Bosnyak S, Welungoda IK, Hallberg A, Alterman M, Widdop RE, Jones ES. Stimulation of angiotensin AT2 receptors by the non-peptide agonist, Compound 21, evokes vasodepressor effects in conscious spontaneously hypertensive rats. Br J Pharmacol. 2010;159:709–716. doi: 10.1111/j.1476-5381.2009.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa EA, Sved AF, Speth RC. Angiotensin modulation of rostral ventrolateral medulla (RVLM) in cardiovascular regulation. Mol Cell Endocrinol. 2009;302:167–175. doi: 10.1016/j.mce.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VA. Differential brain angiotensin-II type I receptor expression in hypertensive rats. J Vet Sci. 2011;12:291–293. doi: 10.4142/jvs.2011.12.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56:879–884. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CD, Higgins M, Donato KA, Rohde FC, Garrison R, Obarzanek E, et al. Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res. 2000;8:605–619. doi: 10.1038/oby.2000.79. [DOI] [PubMed] [Google Scholar]
- Buckman LB, Thompson MM, Moreno HN, Ellacott KLJ. Regional astrogliosis in the mouse hypothalamus in response to obesity. J Comp Neurol. 2012:n/a–n/a. doi: 10.1002/cne.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke GM, Sica DA, Frishman WH. Renal sympathetic denervation for the treatment of systemic hypertension. Cardiol Rev. 2012;20:274–278. doi: 10.1097/CRD.0b013e3182651f91. [DOI] [PubMed] [Google Scholar]
- Busche S, Gallinat S, Fleegal MA, Raizada MK, Sumners C. Novel role of macrophage migration inhibitory factor in angiotensin II regulation of neuromodulation in rat brain. Endocrinology. 2001;142:4623–4630. doi: 10.1210/endo.142.11.8502. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cao X, Peterson JR, Wang G, Anrather J, Young CN, Guruju MR, et al. Angiotensin II-dependent hypertension requires cyclooxygenase 1-derived prostaglandin E2 and EP1 receptor signaling in the subfornical organ of the brain. Hypertension. 2012;59:869–876. doi: 10.1161/HYPERTENSIONAHA.111.182071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-kappaBin the paraventricular nucleus. Hypertension. 2012;59:113–121. doi: 10.1161/HYPERTENSIONAHA.111.182154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar-Bauguil S, Cousin B, Bour S, Casteilla L, Penicaud L, Carpene C. Adipose tissue lymphocytes: types and roles. J Physiol Biochem. 2009;65:423–436. doi: 10.1007/BF03185938. [DOI] [PubMed] [Google Scholar]
- Cassano PA, Segal MR, Vokonas PS, Weiss ST. Body fat distribution, blood pressure, and hypertension. A prospective cohort study of men in the normative aging study. Ann Epidemiol. 1990;1:33–48. doi: 10.1016/1047-2797(90)90017-m. [DOI] [PubMed] [Google Scholar]
- Cassis LA. Role Of Angiotensin-Ii In Brown Adipose Thermogenesis During Cold-Acclimation. American Journal Of Physiology. 1993;265:E860–E865. doi: 10.1152/ajpendo.1993.265.6.E860. [DOI] [PubMed] [Google Scholar]
- Cassis LA, Dwoskin LP. Presynaptic modulation of neurotransmitter release by endogenous angiotensin II in brown adipose tissue. J Neural Transm Suppl. 1991;34:129–137. doi: 10.1007/978-3-7091-9175-0_17. [DOI] [PubMed] [Google Scholar]
- Cassis LA, English V, Helton M. Angiotensin II regulates leptin secretion: A potential link between obesity and hypertension. Faseb Journal. 2001;15:A78–A78. [Google Scholar]
- Cechetto DF, Saper CB. Neurochemical organization of the hypothalamic projection to the spinal cord in the rat. J Comp Neurol. 1988;272:579–604. doi: 10.1002/cne.902720410. [DOI] [PubMed] [Google Scholar]
- Conductier G, Blondeau N, Guyon A, Nahon J-L, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Cooper R, Forrester T, Ogunbiyi O, Muffinda J. Angiotensinogen levels and obesity in four black populations. ICSHIB Investigators. J Hypertens. 1998;16:571–575. doi: 10.1097/00004872-199816050-00003. [DOI] [PubMed] [Google Scholar]
- Correia ML, Morgan DA, Sivitz WI, Mark AL, Haynes WG. Leptin acts in the central nervous system to produce dose-dependent changes in arterial pressure. Hypertension. 2001;37:936–942. doi: 10.1161/01.hyp.37.3.936. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadra AE, Shan Z, Sumners C, Raizada MK. A current view of brain renin-angiotensin system: Is the (pro)renin receptor the missing link? Pharmacol Ther. 2010;125:27–38. doi: 10.1016/j.pharmthera.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampney R. Medullary pathways regulating sympathetic outflow: the need for more lateral thinking. Am J Physiol Regul Integr Comp Physiol. 2004;286:R446–448. doi: 10.1152/ajpregu.00696.2003. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Coleman MJ, Fontes MA, Hirooka Y, Horiuchi J, Li YW, et al. Central mechanisms underlying short- and long-term regulation of the cardiovascular system. Clin Exp Pharmacol Physiol. 2002;29:261–268. doi: 10.1046/j.1440-1681.2002.03640.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Monnot A, Neal AU, Martin AA, Horton JJ, Zheng W. The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood-brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiol Behav. 2012;107:26–33. doi: 10.1016/j.physbeh.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet AD, Krause EG, Scott KA, Foster MT, Herman JP, Sakai RR, et al. Central angiotensin-II has catabolic action at white and brown adipose tissue. Am J Physiol Endocrinol Metab. 2011 doi: 10.1152/ajpendo.00307.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet AD, Krause EG, Woods SC. The renin angiotensin system and the metabolic syndrome. Physiol Behav. 2010;100:525–534. doi: 10.1016/j.physbeh.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1136–1142. doi: 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011a;300:F734–742. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Lund H, Mattson DL. High dietary protein exacerbates hypertension and renal damage in Dahl SS rats by increasing infiltrating immune cells in the kidney. Hypertension. 2011b;57:269–274. doi: 10.1161/HYPERTENSIONAHA.110.154302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Seagard JL, Hopp FA, Kampine JP. Differential control of sympathetic activity to kidney and skeletal muscle by ventral medullary neurons. J Auton Nerv Syst. 1992;37:1–10. doi: 10.1016/0165-1838(92)90139-8. [DOI] [PubMed] [Google Scholar]
- Der Sarkissian S, Huentelman MJ, Stewart J, Katovich MJ, Raizada MK. ACE2: A novel therapeutic target for cardiovascular diseases. Prog Biophys Mol Biol. 2006;91:163–198. doi: 10.1016/j.pbiomolbio.2005.05.011. [DOI] [PubMed] [Google Scholar]
- DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol. 1998;149:329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- Diez-Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27:12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- Ding ZQ, Li YW, Wesselingh SL, Blessing WW. Transneuronal labelling of neurons in rabbit brain after injection of herpes simplex virus type 1 into the renal nerve. J Auton Nerv Syst. 1993;42:23–31. doi: 10.1016/0165-1838(93)90338-u. [DOI] [PubMed] [Google Scholar]
- Dobrenis K, Chang HY, Pina-Benabou MH, Woodroffe A, Lee SC, Rozental R, et al. Human and mouse microglia express connexin36, and functional gap junctions are formed between rodent microglia and neurons. J Neurosci Res. 2005;82:306–315. doi: 10.1002/jnr.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Dorresteijn JA, Visseren FL, Spiering W. Mechanisms linking obesity to hypertension. Obes Rev. 2012;13:17–26. doi: 10.1111/j.1467-789X.2011.00914.x. [DOI] [PubMed] [Google Scholar]
- Egan BM, Zhao Y, Axon R. US trends in prevalence, awareness, treatment, and control of hypertension, 1988-2008. JAMA: The Journal of the American Medical Association. 2010;303:2043–2050. doi: 10.1001/jama.2010.650. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed] [Google Scholar]
- Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Bohm M. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet. 2010;376:1903–1909. doi: 10.1016/S0140-6736(10)62039-9. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Eckardt D, Theis M, Willecke K, Bennett MV, Saez JC. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc Natl Acad Sci U S A. 2001;98:4190–4195. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsen B, Owens T. Innate immune responses in central nervous system inflammation. FEBS Lett. 2011;585:3806–3812. doi: 10.1016/j.febslet.2011.05.030. [DOI] [PubMed] [Google Scholar]
- Fisher JP, Fadel PJ. Therapeutic strategies for targeting excessive central sympathetic activation in human hypertension. Exp Physiol. 2010;95:572–580. doi: 10.1113/expphysiol.2009.047332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiria-Oliveira AH, Blanch GT, Li H, Colombari E, Colombari DS, Sumners C. Macrophage migration inhibitory factor in the nucleus of solitary tract decreases blood pressure in SHRs. Cardiovasc Res. 2012 doi: 10.1093/cvr/cvs297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchtbauer L, Groth-Rasmussen M, Holm TH, Lobner M, Toft-Hansen H, Khorooshi R, et al. Angiotensin II Type 1 receptor (AT1) signaling in astrocytes regulates synaptic degeneration-induced leukocyte entry to the central nervous system. Brain Behav Immun. 2011;25:897–904. doi: 10.1016/j.bbi.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, et al. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- Garrison RJ, Kannel WB, Stokes J, 3rd, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med. 1987;16:235–251. doi: 10.1016/0091-7435(87)90087-9. [DOI] [PubMed] [Google Scholar]
- Gerber AR, Bale TL. Antiinflammatory treatment ameliorates HPA stress axis dysfunction in a mouse model of stress sensitivity. Endocrinology. 2012;153:4830–4837. doi: 10.1210/en.2012-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, et al. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol. 1996;156:4363–4368. [PubMed] [Google Scholar]
- Goldstein DS. Plasma catecholamines and essential hypertension. An analytical review. Hypertension. 1983;5:86–99. doi: 10.1161/01.hyp.5.1.86. [DOI] [PubMed] [Google Scholar]
- Gonzalez AD, Wang G, Waters EM, Gonzales KL, Speth RC, Van Kempen TA, et al. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience. 2012;226:489–509. doi: 10.1016/j.neuroscience.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Gourmala NG, Buttini M, Limonta S, Sauter A, Boddeke HW. Differential and time-dependent expression of monocyte chemoattractant protein-1 mRNA by astrocytes and macrophages in rat brain: effects of ischemia and peripheral lipopolysaccharide administration. J Neuroimmunol. 1997;74:35–44. doi: 10.1016/s0165-5728(96)00203-2. [DOI] [PubMed] [Google Scholar]
- Grassi G. Role of the sympathetic nervous system in human hypertension. J Hypertens. 1998;16:1979–1987. doi: 10.1097/00004872-199816121-00019. [DOI] [PubMed] [Google Scholar]
- Grassi G. Sympathetic neural activity in hypertension and related diseases. Am J Hypertens. 2010;23:1052–1060. doi: 10.1038/ajh.2010.154. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Cattaneo BM, Bolla GB, Lanfranchi A, Colombo M, et al. Sympathetic activation in obese normotensive subjects. Hypertension. 1995;25:560–563. doi: 10.1161/01.hyp.25.4.560. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Colombo M, Bolla G, Cattaneo BM, Cavagnini F, et al. Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation. 1998;97:2037–2042. doi: 10.1161/01.cir.97.20.2037. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Dell’Oro R, Trevano FQ, Bombelli M, Scopelliti F, et al. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertens. 2003;21:1761–1769. doi: 10.1097/00004872-200309000-00027. [DOI] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Quarti-Trevano F. The ‘neuroadrenergic hypothesis’ in hypertension: current evidence. Exp Physiol. 2010;95:581–586. doi: 10.1113/expphysiol.2009.047381. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, et al. The brain Renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab. 2010;12:431–442. doi: 10.1016/j.cmet.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 2008;23:187–193. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Liu B, Tang F, Lane S, Souslova EA, Chudakov DM, et al. Astroglia are a possible cellular substrate of angiotensin(1-7) effects in the rostral ventrolateral medulla. Cardiovasc Res. 2010;87:578–584. doi: 10.1093/cvr/cvq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurden H, Uchida N, Mainen ZF. Sensory-evoked intrinsic optical signals in the olfactory bulb are coupled to glutamate release and uptake. Neuron. 2006;52:335–345. doi: 10.1016/j.neuron.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007a;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Marvar PJ, Czesnikiewicz-Guzik M, Korbut R. Perivascular adipose tissue as a messenger of the brain-vessel axis: role in vascular inflammation and dysfunction. J Physiol Pharmacol. 2007b;58:591–610. [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Goronzy J, Weyand C. Is hypertension an immunologic disease? Curr Cardiol Rep. 2008;10:464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. doi: 10.3389/fphys.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–1197. doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WG. Role of leptin in obesity-related hypertension. Exp Physiol. 2005;90:683–688. doi: 10.1113/expphysiol.2005.031237. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33:542–547. doi: 10.1161/01.hyp.33.1.542. [DOI] [PubMed] [Google Scholar]
- Hendel MD, Collister JP. Contribution of the subfornical organ to angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2005;288:H680–685. doi: 10.1152/ajpheart.00823.2004. [DOI] [PubMed] [Google Scholar]
- Hilzendeger AM, Morgan DA, Brooks L, Dellsperger D, Liu X, Grobe JL, et al. A brain leptin-renin angiotensin system interaction in the regulation of sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2012;303:H197–206. doi: 10.1152/ajpheart.00974.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S53–55. doi: 10.1038/sj.ijo.0802502. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Huang XC, Richards EM, Sumners C. Mitogen-activated protein kinases in rat brain neuronal cultures are activated by angiotensin II type 1 receptors and inhibited by angiotensin II type 2 receptors. J Biol Chem. 1996;271:15635–15641. doi: 10.1074/jbc.271.26.15635. [DOI] [PubMed] [Google Scholar]
- Imai F, Sawada M, Suzuki H, Zlokovic BV, Kojima J, Kuno S, et al. Exogenous microglia enter the brain and migrate into ischaemic hippocampal lesions. Neurosci Lett. 1999;272:127–130. doi: 10.1016/s0304-3940(99)00592-3. [DOI] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, et al. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Intebi AD, Flaxman MS, Ganong WF, Deschepper CF. Angiotensinogen production by rat astroglial cells in vitro and in vivo. Neuroscience. 1990;34:545–554. doi: 10.1016/0306-4522(90)90163-x. [DOI] [PubMed] [Google Scholar]
- Jankord R, Zhang R, Flak JN, Solomon MB, Albertz J, Herman JP. Stress activation of IL-6 neurons in the hypothalamus. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2010;299:R343–R351. doi: 10.1152/ajpregu.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Shi P, Li H, Lu S, Braseth L, Cuadra AE, et al. Phosphate-activated glutaminase-containing neurons in the rat paraventricular nucleus express angiotensin type 1 receptors. Hypertension. 2009;54:845–851. doi: 10.1161/HYPERTENSIONAHA.109.134684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ, Charkoudian N, Wallin BG. A sympathetic view of the sympathetic nervous system and human blood pressure regulation. Exp Physiol. 2008;93:715–724. doi: 10.1113/expphysiol.2007.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JY, Zubcevic J, Qi Y, Afzal A, Carvajal JM, Thinschmidt JS, et al. Brain-Mediated Dysregulation of the Bone Marrow Activity in Angiotensin II-Induced Hypertension. Hypertension. 2012;60:1316–1323. doi: 10.1161/HYPERTENSIONAHA.112.199547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma Y, Hama H, Sugiyama F, Yagami K, Goto K, Murakami K, et al. Impaired blood-brain barrier function in angiotensinogen-deficient mice. Nat Med. 1998;4:1078–1080. doi: 10.1038/2070. [DOI] [PubMed] [Google Scholar]
- Kalupahana NS, Massiera F, Quignard-Boulange A, Ailhaud G, Voy BH, Wasserman DH, et al. Overproduction of Angiotensinogen From Adipose Tissue Induces Adipose Inflammation, Glucose Intolerance, and Insulin Resistance. Obesity (Silver Spring) 2011 doi: 10.1038/oby.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandalam U, Clark MA. Angiotensin II activates JAK2/STAT3 pathway and induces interleukin-6 production in cultured rat brainstem astrocytes. Regul Pept. 2010;159:110–116. doi: 10.1016/j.regpep.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Kang SS, Keasey MP, Cai J, Hagg T. Loss of neuron-astroglial interaction rapidly induces protective CNTF expression after stroke in mice. J Neurosci. 2012;32:9277–9287. doi: 10.1523/JNEUROSCI.1746-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, et al. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res. 2009;82:503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan H, Tanaka Y, Kunitake T, Ueta Y, Hayashida Y, Yamashita H. Activation of sympathetic outflow by recombinant human interleukin-1 beta in conscious rats. Am J Physiol. 1996;270:R479–485. doi: 10.1152/ajpregu.1996.270.2.R479. [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J Alzheimers Dis. 2010;21:207–219. doi: 10.3233/JAD-2010-091414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the Sympathetic Nervous System Regulate Hematopoietic Stem Cell Egress from Bone Marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Katovich MJ, Grobe JL, Huentelman M, Raizada MK. Angiotensin-converting enzyme 2 as a novel target for gene therapy for hypertension. Exp Physiol. 2005;90:299–305. doi: 10.1113/expphysiol.2004.028522. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kleiber AC, Zheng H, Sharma NM, Patel KP. Chronic AT1 receptor blockade normalizes NMDA-mediated changes in renal sympathetic nerve activity and NR1 expression within the PVN in rats with heart failure. American Journal of Physiology - Heart and Circulatory Physiology. 2010;298:H1546–H1555. doi: 10.1152/ajpheart.01006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CU, Kreutzberg GW, Raivich G. Proliferation of ramified microglia on an astrocyte monolayer: characterization of stimulatory and inhibitory cytokines. J Neurosci Res. 1997;49:248–254. [PubMed] [Google Scholar]
- Krause EG, de Kloet AD, Scott KA, Flak JN, Jones K, Smeltzer MD, et al. Blood-borne angiotensin II acts in the brain to influence behavioral and endocrine responses to psychogenic stress. J Neurosci. 2011;31:15009–15015. doi: 10.1523/JNEUROSCI.0892-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause EG, Melhorn SJ, Davis JF, Scott KA, Ma LY, de Kloet AD, et al. Angiotensin type 1 receptors in the subfornical organ mediate the drinking and hypothalamic-pituitary-adrenal response to systemic isoproterenol. Endocrinology. 2008;149:6416–6424. doi: 10.1210/en.2008-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet. 2009;373:1275–1281. doi: 10.1016/S0140-6736(09)60566-3. [DOI] [PubMed] [Google Scholar]
- Krum H, Sobotka P, Mahfoud F, Bohm M, Esler M, Schlaich M. Device-based antihypertensive therapy: therapeutic modulation of the autonomic nervous system. Circulation. 2011;123:209–215. doi: 10.1161/CIRCULATIONAHA.110.971580. [DOI] [PubMed] [Google Scholar]
- Lafrance V, Inoue W, Kan B, Luheshi GN. Leptin modulates cell morphology and cytokine release in microglia. Brain Behav Immun. 2010;24:358–365. doi: 10.1016/j.bbi.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Langhans W. Signals generating anorexia during acute illness. Proceedings of the Nutrition Society. 2007;66:321–330. doi: 10.1017/S0029665107005587. [DOI] [PubMed] [Google Scholar]
- Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Localization of renin expressing cells in the brain, by use of a REN-eGFP transgenic model. Physiol Genomics. 2004;16:240–246. doi: 10.1152/physiolgenomics.00131.2003. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48:405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- Lenkei Z, Palkovits M, Corvol P, Llorens-Cortès C. Expression of Angiotensin Type-1 (AT1) and Type-2 (AT2) Receptor mRNAs in the Adult Rat Brain: A Functional Neuroanatomical Review. Frontiers in Neuroendocrinology. 1997;18:383. doi: 10.1006/frne.1997.0155. [DOI] [PubMed] [Google Scholar]
- Levick SP, Murray DB, Janicki JS, Brower GL. Sympathetic Nervous System Modulation of Inflammation and Remodeling in the Hypertensive Heart. Hypertension. 2010;55:270–276. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D-P, Chen S-R, Pan H-L. Angiotensin II Stimulates Spinally Projecting Paraventricular Neurons through Presynaptic Disinhibition. J Neurosci. 2003;23:5041–5049. doi: 10.1523/JNEUROSCI.23-12-05041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D-P, Pan H-L. Angiotensin II Attenuates Synaptic GABA Release and Excites Paraventricular-Rostral Ventrolateral Medulla Output Neurons. J Pharmacol Exp Ther. 2005;313:1035–1045. doi: 10.1124/jpet.104.082495. [DOI] [PubMed] [Google Scholar]
- Li H, Gao Y, Freire CD, Raizada MK, Toney GM, Sumners C. Macrophage migration inhibitory factor in the PVN attenuates the central pressor and dipsogenic actions of angiotensin II. FASEB J. 2006;20:1748–1750. doi: 10.1096/fj.06-5836fje. [DOI] [PubMed] [Google Scholar]
- Li H, Gao Y, Qi Y, Katovich MJ, Jiang N, Braseth LN, et al. Macrophage migration inhibitory factor in hypothalamic paraventricular nucleus neurons decreases blood pressure in spontaneously hypertensive rats. Faseb J. 2008;22:3175–3185. doi: 10.1096/fj.08-108662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Ferguson AV. Subfornical organ efferents to paraventricular nucleus utilize angiotensin as a neurotransmitter. Am J Physiol Regul Integr Comp Physiol. 1993;265:R302–309. doi: 10.1152/ajpregu.1993.265.2.R302. [DOI] [PubMed] [Google Scholar]
- Lind RW, Swanson LW, Ganten D. Angiotensin II immunoreactivity in the neural afferents and efferents of the subfornical organ of the rat. Brain Res. 1984;321:209–215. doi: 10.1016/0006-8993(84)90174-4. [DOI] [PubMed] [Google Scholar]
- Liu W, Tang Y, Feng J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011;89:141–146. doi: 10.1016/j.lfs.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. 276p following 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden CJ, Sved AF. Cardiovascular regulation after destruction of the C1 cell group of the rostral ventrolateral medulla in rats. Am J Physiol Heart Circ Physiol. 2003;285:H2734–2748. doi: 10.1152/ajpheart.00155.2003. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Mark AL, Agassandian K, Morgan DA, Liu X, Cassell MD, Rahmouni K. Leptin Signaling in the Nucleus Tractus Solitarii Increases Sympathetic Nerve Activity to the Kidney. Hypertension. 2009;53:375–380. doi: 10.1161/HYPERTENSIONAHA.108.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–1953. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- Marvar PJ, Harrison DG. Stress-dependent hypertension and the role of T lymphocytes. Exp Physiol. 2012 doi: 10.1113/expphysiol.2011.061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Vinh A, Thabet S, Lob HE, Geem D, Ressler KJ, et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry. 2012;71:774–782. doi: 10.1016/j.biopsych.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiéra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc J-M, et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001 doi: 10.1096/fj.01-0457fje. 01-0457fje. [DOI] [PubMed] [Google Scholar]
- Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nat Rev Immunol. 2011;11:81. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun. 2010;394:1006–1011. doi: 10.1016/j.bbrc.2010.03.111. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, et al. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35:901–918. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes. 2012;61:1455–1462. doi: 10.2337/db11-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T. Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide-induced stimulation of rat microglial cells by suppressing nuclear factor kappaB and activator protein-1 activation. Eur J Neurosci. 2008;27:343–351. doi: 10.1111/j.1460-9568.2007.06014.x. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Cassell MD, Sigmund CD. Glia- and neuron-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277:33235–33241. doi: 10.1074/jbc.M204309200. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001;130:169–175. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nunes FC, Braga VA. Chronic angiotensin II infusion modulates angiotensin II type I receptor expression in the subfornical organ and the rostral ventrolateral medulla in hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2011;12:440–445. doi: 10.1177/1470320310394891. [DOI] [PubMed] [Google Scholar]
- Old EA, Malcangio M. Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol. 2012;12:67–73. doi: 10.1016/j.coph.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Oparil S, Zaman MA, Calhoun DA. Pathogenesis of hypertension. Ann Intern Med. 2003;139:761–776. doi: 10.7326/0003-4819-139-9-200311040-00011. [DOI] [PubMed] [Google Scholar]
- Osborn JW, Hendel MD, Collister JP, Ariza-Guzman PA, Fink GD. The role of the subfornical organ in angiotensin II-salt hypertension in the rat. Exp Physiol. 2012;97:80–88. doi: 10.1113/expphysiol.2011.060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, et al. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci. 2001;21:7135–7142. doi: 10.1523/JNEUROSCI.21-18-07135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JB, Jo JY, Zheng H, Patel KP, Stern JE. Regulation of tonic GABA inhibitory function, presympathetic neuronal activity and sympathetic outflow from the paraventricular nucleus by astroglial GABA transporters. J Physiol. 2009;587:4645–4660. doi: 10.1113/jphysiol.2009.173435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Kasparov S. Differential effects of angiotensin II on cardiorespiratory reflexes mediated by nucleus tractus solitarii - a microinjection study in the rat. J Physiol. 1999;521(Pt 1):213–225. doi: 10.1111/j.1469-7793.1999.00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinteaux E, Inoue W, Schmidt L, Molina-Holgado F, Rothwell NJ, Luheshi GN. Leptin induces interleukin-1beta release from rat microglial cells through a caspase 1 independent mechanism. J Neurochem. 2007;102:826–833. doi: 10.1111/j.1471-4159.2007.04559.x. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson JW, Dampney RA, Boscan P, Pickering AE, Paton JF. Differential baroreflex control of sympathetic drive by angiotensin II in the nucleus tractus solitarii. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1954–1960. doi: 10.1152/ajpregu.00041.2007. [DOI] [PubMed] [Google Scholar]
- Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med. 2011;17:883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyner S, Coote JH. Identification of branching paraventricular neurons of the hypothalamus that project to the rostroventrolateral medulla and spinal cord. Neuroscience. 2000;100:549–556. doi: 10.1016/s0306-4522(00)00283-9. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes TM, Walker JR, DeCino C, Hogenesch JB, Sawchenko PE. Categorically Distinct Acute Stressors Elicit Dissimilar Transcriptional Profiles in the Paraventricular Nucleus of the Hypothalamus. The Journal of Neuroscience. 2003;23:5607–5616. doi: 10.1523/JNEUROSCI.23-13-05607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282:F191–201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- Rumantir MS, Vaz M, Jennings GL, Collier G, Kaye DM, Seals DR, et al. Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17:1125–1133. doi: 10.1097/00004872-199917080-00012. [DOI] [PubMed] [Google Scholar]
- Santos RA, Ferreira AJ, Simoes ESAC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol. 2008;93:519–527. doi: 10.1113/expphysiol.2008.042002. [DOI] [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Ringelstein EB, Kiefer R, Schabitz WR. Turnover of meningeal and perivascular macrophages in the brain of MCP-1-, CCR-2- or double knockout mice. Exp Neurol. 2009a;219:583–585. doi: 10.1016/j.expneurol.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009b;161:806–812. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Hering D, Sobotka P, Krum H, Lambert GW, Lambert E, et al. Effects of renal denervation on sympathetic activation, blood pressure, and glucose metabolism in patients with resistant hypertension. Front Physiol. 2012;3:10. doi: 10.3389/fphys.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med. 2009a;361:932–934. doi: 10.1056/NEJMc0904179. [DOI] [PubMed] [Google Scholar]
- Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD. Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension. 2009b;54:1195–1201. doi: 10.1161/HYPERTENSIONAHA.109.138610. [DOI] [PubMed] [Google Scholar]
- Schuette-Nuetgen K, Strecker JK, Minnerup J, Ringelstein EB, Schilling M. MCP-1/CCR-2-double-deficiency severely impairs the migration of hematogenous inflammatory cells following transient cerebral ischemia in mice. Exp Neurol. 2012;233:849–858. doi: 10.1016/j.expneurol.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. 2012;8:709–716. doi: 10.1038/nrendo.2012.114. [DOI] [PubMed] [Google Scholar]
- Shafton AD, Ryan A, Badoer E. Neurons in the hypothalamic paraventricular nucleus send collaterals to the spinal cord and to the rostral ventrolateral medulla in the rat. Brain Res. 1998;801:239–243. doi: 10.1016/s0006-8993(98)00587-3. [DOI] [PubMed] [Google Scholar]
- Sherrod M, Davis DR, Zhou X, Cassell MD, Sigmund CD. Glial-specific ablation of angiotensinogen lowers arterial pressure in renin and angiotensinogen transgenic mice. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1763–1769. doi: 10.1152/ajpregu.00435.2005. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Grobe JL, Desland FA, Raizada MK, Sumners C. Microglial activation by the brain renin-angiotensin system. FASEB J. 2011;25:661–662. [Google Scholar]
- Shoelson SE, Goldfine AB. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med. 2009;15:373–374. doi: 10.1038/nm0409-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res. 2011;92:401–408. doi: 10.1093/cvr/cvr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, et al. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelings UM, Paulis L, Namsolleck P, Unger T. AT2 receptor agonists: hypertension and beyond. Curr Opin Nephrol Hypertens. 2012;21:142–146. doi: 10.1097/MNH.0b013e328350261b. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. Water deprivation activates a glutamatergic projection from the hypothalamic paraventricular nucleus to the rostral ventrolateral medulla. J Comp Neurol. 2006;494:673–685. doi: 10.1002/cne.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science. 1988;242:1444–1446. doi: 10.1126/science.3201232. [DOI] [PubMed] [Google Scholar]
- Straznicky NE, Lambert EA, Lambert GW, Masuo K, Esler MD, Nestel PJ. Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. J Clin Endocrinol Metab. 2005;90:5998–6005. doi: 10.1210/jc.2005-0961. [DOI] [PubMed] [Google Scholar]
- Sun C, Li H, Gao Y, Matsuura T, Upchurch PA, Raizada MK, et al. Lack of macrophage migration inhibitory factor regulation is linked to the increased chronotropic action of angiotensin II in SHR neurons. Hypertension. 2007;49:528–534. doi: 10.1161/01.HYP.0000257877.11495.cb. [DOI] [PubMed] [Google Scholar]
- Sun C, Li H, Leng L, Raizada MK, Bucala R, Sumners C. Macrophage migration inhibitory factor: an intracellular inhibitor of angiotensin II-induced increases in neuronal activity. J Neurosci. 2004;24:9944–9952. doi: 10.1523/JNEUROSCI.2856-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol. 1980;194:555–570. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- Tang CH, Lu DY, Yang RS, Tsai HY, Kao MC, Fu WM, et al. Leptin-induced IL-6 production is mediated by leptin receptor, insulin receptor substrate-1, phosphatidylinositol 3-kinase, Akt, NF-kappaB, and p300 pathway in microglia. J Immunol. 2007;179:1292–1302. doi: 10.4049/jimmunol.179.2.1292. [DOI] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, et al. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151:4109–4115. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpson NJ, Harbord R, Davey Smith G, Zacho J, Tybjaerg-Hansen A, Nordestgaard BG. Does greater adiposity increase blood pressure and hypertension risk?: Mendelian randomization using the FTO/MC4R genotype. Hypertension. 2009;54:84–90. doi: 10.1161/HYPERTENSIONAHA.109.130005. [DOI] [PubMed] [Google Scholar]
- Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330:219–228. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Jones KR, Ziegler DR, Cullinan WE, Herman JP. Forebrain origins of glutamatergic innervation to the rat paraventricular nucleus of the hypothalamus: differential inputs to the anterior versus posterior subregions. J Comp Neurol. 2011;519:1301–1319. doi: 10.1002/cne.22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Voorn P, Tekstra J, Beelen RHJ, Tensen CP, Van Der Valk P, De Groot CJA. Expression of MCP-1 by Reactive Astrocytes in Demyelinating Multiple Sclerosis Lesions. Am J Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steenwinckel J, Reaux-Le Goazigo A, Pommier B, Mauborgne A, Dansereau M-A, Kitabgi P, et al. CCL2 Released from Neuronal Synaptic Vesicles in the Spinal Cord Is a Major Mediator of Local Inflammation and Pain after Peripheral Nerve Injury. The Journal of Neuroscience. 2011;31:5865–5875. doi: 10.1523/JNEUROSCI.5986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. 1997;96:3423–3429. doi: 10.1161/01.cir.96.10.3423. [DOI] [PubMed] [Google Scholar]
- Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, et al. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56:643–649. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. The Journal of Neuroscience. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M, Paton JF. Gene expression profiles of major cytokines in the nucleus tractus solitarii of the spontaneously hypertensive rat. Auton Neurosci. 2008a;142:40–44. doi: 10.1016/j.autneu.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Waki H, Gouraud SS, Maeda M, Paton JF. Specific inflammatory condition in nucleus tractus solitarii of the SHR: novel insight for neurogenic hypertension? Auton Neurosci. 2008b;142:25–31. doi: 10.1016/j.autneu.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Waki H, Liu B, Miyake M, Katahira K, Murphy D, Kasparov S, et al. Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension. 2007;49:1321–1327. doi: 10.1161/HYPERTENSIONAHA.106.085589. [DOI] [PubMed] [Google Scholar]
- Wang Y-Y, Lin S-Y, Chuang Y-H, Chen C-J, Tung K-C, Sheu WH-H. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. American Journal of Physiology - Endocrinology And Metabolism. 2011;300:E155–E163. doi: 10.1152/ajpendo.00301.2010. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation. 2003;112:1796. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R, Shaw M, Savoye M, Caprio S. Obesity dynamics and cardiovascular risk factor stability in obese adolescents. Pediatr Diabetes. 2009;10:360–367. doi: 10.1111/j.1399-5448.2008.00504.x. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- Werry EL, Liu GJ, Lovelace MD, Nagarajah R, Bennett MR. Glutamate potentiates lipopolysaccharide-stimulated interleukin-10 release from neonatal rat spinal cord astrocytes. Neuroscience. 2012;207:12–24. doi: 10.1016/j.neuroscience.2012.01.039. [DOI] [PubMed] [Google Scholar]
- Wofford MR, Anderson DC, Jr, Brown CA, Jones DW, Miller ME, Hall JE. Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am J Hypertens. 2001;14:694–698. doi: 10.1016/s0895-7061(01)01293-6. [DOI] [PubMed] [Google Scholar]
- Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9:212. doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation. 2003;112:1821. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazato M, Ferreira AJ, Yamazato Y, Diez-Freire C, Yuan L, Gillies R, et al. Gene transfer of angiotensin-converting enzyme 2 in the nucleus tractus solitarius improves baroreceptor heart rate reflex in spontaneously hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2011;12:456–461. doi: 10.1177/1470320311412809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang L, Ju G. Evidence for hypothalamic paraventricular nucleus as an integrative center of neuroimmunomodulation. Neuroimmunomodulation. 1997;4:120–127. doi: 10.1159/000097330. [DOI] [PubMed] [Google Scholar]
- Yang L, Tanaka J, Zhang B, Sakanaka M, Maeda N. Astrocytes modulate nitric oxide production by microglial cells through secretion of serine and glycine. Biochem Biophys Res Commun. 1998;251:277–282. doi: 10.1006/bbrc.1998.9457. [DOI] [PubMed] [Google Scholar]
- Yao F, Sumners C, O’Rourke ST, Sun C. Angiotensin II increases GABAB receptor expression in nucleus tractus solitarii of rats. Am J Physiol Heart Circ Physiol. 2008;294:H2712–2720. doi: 10.1152/ajpheart.00729.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C-X, Al-Massadi O, Donelan E, Lehti M, Weber J, Ress C, et al. Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiology & Behavior. 2012a;106:485–490. doi: 10.1016/j.physbeh.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Yi CX, Tschop MH, Woods SC, Hofmann SM. High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech. 2012b doi: 10.1242/dmm.009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, et al. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension. 2012a;60:1524–1530. doi: 10.1161/HYPERTENSIONAHA.112.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, et al. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol. 2012b;302:R244–251. doi: 10.1152/ajpregu.00323.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, et al. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest. 2012;122:3960–3964. doi: 10.1172/JCI64583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhang ZH, Wei SG, Serrats J, Weiss RM, Felder RB. Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension. 2010;55:652–659. doi: 10.1161/HYPERTENSIONAHA.109.142836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–858. doi: 10.1016/j.neuroscience.2010.09.029. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler MG, Mills P, Dimsdale JE. Hypertensives’ pressor response to norepinephrine. Analysis by infusion rate and plasma levels. Am J Hypertens. 1991;4:586–591. [PubMed] [Google Scholar]
- Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, et al. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res. 2004a;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004b;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- Zubcevic J, Waki H, Raizada MK, Paton JF. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension. 2011;57:1026–1033. doi: 10.1161/HYPERTENSIONAHA.111.169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol. 2009;297:H1557–1566. doi: 10.1152/ajpheart.00073.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]