

Abstract
In humans, obesity is associated with long QT, increased frequency of premature ventricular complexes, and sudden cardiac death. The mechanisms of the pro-arrhythmic electrophysiologic remodeling of obesity are poorly understood. We tested the hypothesis that there is decreased expression of voltage-gated potassium channels in the obese heart, leading to long QT. Using implanted telemeters, we found that diet-induced obese (DIO) wild-type mice have impaired cardiac repolarization, demonstrated by long QT, as well as more frequent ventricular ectopy, similar to obese humans. DIO mice have reduced protein and mRNA levels of the potassium channel Kv1.5 caused by a reduction of the transcription factor cyclic AMP response element binding protein (CREB) in DIO hearts. We found that CREB knock-down by siRNA reduces Kv1.5, CREB binds to the Kv1.5 promoter in the heart, and CREB increases transcription of mouse and human Kv1.5 promoters. The reduction in CREB protein during lipotoxicity can be rescued by inhibiting protein kinase D (PKD). Our results identify a mechanism for obesity-induced electrophysiologic remodeling in the heart, namely PKD-induced reduction of CREB, which in turn decreases expression of the potassium channel Kv1.5.
Keywords: obesity, arrhythmia, cyclic AMP response element binding protein, Kv1.5, protein kinase D
1. Introduction
The rising prevalence of diabetes and obesity, which often lead to heart failure and arrhythmias, is a concerning public-health epidemic. After adjusting for other cardiovascular risk factors, obesity is associated with altered cardiomyocyte electrical properties, including impaired repolarization, manifested by prolongation of the QT interval on the electrocardiogram [1, 2]. Obesity is also associated with arrhythmias. Both the Framingham and Atherosclerosis Risk in Communities Study (ARIC) studies found a significant association between obesity and atrial fibrillation [3, 4]. Obese humans have an increased frequency of premature ventricular complexes (PVCs) compared to control subjects, which appears to be unrelated to hypertension or ventricular hypertrophy [5, 6]. More importantly, obesity also confers an increased risk of sudden cardiac death, which is generally caused by arrhythmias [7–9]. Obesity induced cardiomyopathy is a common cause of non-ischemic sudden cardiac death [10]. The mechanisms leading to arrhythmia and sudden death in obesity are unresolved but there are several lines of evidence that abnormal cardiomyocyte lipid deposition has a role in the pathophysiology. Increased cardiomyocyte lipid stores are observed in obese patients and this has been termed lipotoxic cardiomyopathy, or “fatty heart” [11].
Arrhythmias are frequently due to abnormal cardiac ion channel expression or function. During the cardiomyocyte action potential, voltage-gated potassium (Kv) channels are responsible for repolarization. Reduced potassium channel protein levels or function cause prolonged repolarization and long QT, as in the genetic syndromes long-QT 1 and 2, as well as drug-induced long-QT. Reduced potassium channel expression or function has been implicated in other disease states such as heart failure and hypertrophy [12, 13].
There is compelling evidence that intracellular lipid content can regulate ion channels. We have previously demonstrated an arrhythmic phenotype in a transgenic model of lipotoxic cardiomyopathy, the peroxisome proliferator-activated receptor gamma (PPARγ) cardiac-overexpression mouse, which has increased cardiomyocyte lipid content. This model has impaired repolarization from a decrease in potassium channel protein levels, causing ventricular tachycardia and sudden cardiac death [14]. Other transgenic lipotoxic models, such as the PPARα overexpression mouse and MHC-FATP mouse, also have reduced Kv currents [15, 16]. Although transgenic mice are powerful tools, overexpressing a single gene during the entire life of a mouse may not cause the same pathophysiology as gradual onset diet-induced obesity. Thus the diet-induced obese (DIO) mouse is more physiologically relevant model for the molecular mechanisms causing cardiac abnormalities.
The specific transcription factors involved in potassium channel gene expression in the heart are not well established, but it has been appreciated for some time that cyclic AMP response elements (CREs) are present in the promoters of some Kv channels and other ion channels [17, 18]. The CRE binding protein (CREB), a transcription factor, is known to be expressed in human heart tissue [19]. In canine models, a decrease in CREB can cause repolarization abnormalities after artificial pacing [20–22]. We hypothesized that a reduction in cardiac CREB could cause the prolonged QT of obesity. Here, we show that protein kinase D (PKD) is activated in obese hearts, leading to a reduction in CREB protein which in turn causes decreased expression of Kv channels. Reduced CREB is a novel mechanism causing pro-arrhythmic electrophysiologic remodeling in obesity.
2. Materials and Methods
2.1 Animal Care and Echocardiography
Animal protocols were approved by the Columbia University Institutional Animal Care and Use Committee and were carried out in accordance with the NIH guidelines for the care and use of laboratory animals. Male C57BL/6J DIO and age-matched control mice were purchased from Jackson Labs. DIO mice were fed a high-fat diet (60% kcal from fat) starting at the age of 6 weeks; the control diet contained 10% of kcal from fat. DIO and age-matched controls were 18–20 weeks old at the time of telemetry or sacrifice. PPARγ mice were back-crossed into the C57BL6 background for more than ten generations [14]. Transthoracic echocardiography was performed using a high-resolution imaging system with a 30-MHz imaging transducer (Vevo 770; VisualSonics, Toronto, ON, Canada). Mice were sedated with isoflurane during echocardiography. Care was taken to minimize sedation by monitoring the heart rate during the procedure.
2.2 Telemetry and ECG analysis
Telemetry devices (Data Sciences International, model EA-F20) were implanted in mice under inhaled isoflurane anesthesia. The two subcutaneous leads were positioned to approximate ECG limb lead II. The mice recovered for one week after surgery before initiating recordings. Six hours of recording were obtained on two consecutive days from each mouse. ECG intervals were measured manually using Ponemah 3 software from recordings with minimal artifact at approximately the same time of day. Intervals were averaged from 4 consecutive beats when the heart rate was between 540–560 beats per minute and QTc was calculated by the formula QTc=QT/(RR/100) [23]. PVC and arrhythmia counts were tallied manually by a blinded reader.
2.3 Immunoblots
Cardiac protein lysates were made from rapidly harvested tissue by homogenizing ventricular tissue in buffer containing 1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10 mM EDTA, 10 mM EGTA, phosphatase inhibitor cocktail, and protease inhibitors (complete mini-tablet, calpain I and 11 inhibitors, Roche) using a Mini-BeadBeater (Glen Mills, Inc). Lysates were used for PAGE and then transferred to nitrocellulose membranes for immunoblots. The following antibodies were used: anti-Cx43 (Invitrogen), anti-phospho ERK (Thr202/Tyr204, Cell Signaling) and anti-total ERK (Santa Cruz), anti-Kv1.4 anti-Kv1.5 and anti-Kv2.1 (Alomone), anti-Kv4.2 and anti-CREB (Thermo-Scientific Pierce), anti-phospo-PKD (Ser916) and anti-total PKD (Cell Signaling), and anti-tubulin and anti-lamin A/C (Santa Cruz Biotechnology). Nuclear extracts were obtained using the CelLytic NuCLEAR kit from Sigma. Chemiluminescence signal was obtained using a Kodak Image Station 400R Pro digital camera with Kodak Molecular Imaging Software v4.5.1. Signal intensity was quantified using ImageJ software (NIH). Blots for tubulin or lamin were performed to normalize loading of lanes, using the same membrane.
2.4 Real-time PCR
Ventricular tissue was homogenized with a Mini-BeadBeater and RNA was then purified using a Qiagen RNeasy kit (#74104). cDNA was synthesized using the Applied Biosystems high capacity RNA to cDNA kit (#4387406) and diluted to 10 ng/μL for use as a template (20 ng template was used for each 20 μL reaction). Real-time PCR was performed using an Applied Biosystems StepOne Plus Real-Time PCR system with StepOne Software v2.0 and inventoried primers from Applied Biosystems. PCR was performed for 40 cycles with automated detection of crossing threshold; the ΔΔCT method was used for relative quantification. PCR reactions were performed with duplicate wells and with actin as a reference.
2.5 Chromatin Immunoprecipitation
Mouse ventricular muscle was homogenized and sonicated to shear DNA and then immunoprecipitation was performed with an anti-CREB antibody and the Magna ChIP kit (Millipore #17–600). PCR primers were designed to amplify 260–360 bp regions of the promoter that contained predicted CRE; primer sequences are listed in the online supplement.
2.6 Promoter-Luciferase Vectors and Mutagenesis
Proximal promoters (1.5–2.2 kb regions upstream from the start of transcription) were cloned by PCR from BAC templates (bacpac.chori.org) and ligated into the pGL3 basic vector (Promega). Constructs were verified by DNA sequencing. Restriction enzymes and ligase were from New England Biolabs. Mutagenesis to eliminate CREs was performed with the Agilent kit (#210515) following the manufacturer’s protocol. Mutagenesis primers were designed to substitute two base pairs in the middle of CREs (which are 6 bp in length) to disrupt the binding site sequence.
2.7 Cell Culture, Transfection, Chemical and siRNA Treatments
Luciferase constructs were co-transfected into rat cardiomyocyte H9c2 cells (ATCC) with a beta-gal reporter vector as a transfection control using Lipofectamine (Invitrogen). The rat CREB1 expression vector was created by Marc Montminy (Salk Institute) and purchased from Addgene (#22395). Standard 6-well tissue culture plates were used. The anti-CREB siRNA and scramble siRNA control constructs were purchased from Santa Cruz Biotechnology (SC-72030-V) and used as per the manufacturer’s protocol. Sodium palmitate was purchased from Sigma, and was dissolved to make a l0mM solution in sterile water with 10% fatty-acid free BSA (Sigma) and then diluted to final concentration 0.2 mM in media. CID755673 was purchased from Torcris, and PD98059 from Cell Signaling. Cells were pretreated with chemical inhibitors or vehicle control (DMSO) for 30 minute prior to the addition of palmitate.
2.8 Immunohistochemistry
Heart tissue was fixed with 4% paraformaldehyde, embedded in paraffin wax, and then sectioned. Sections were stained with hematoxylin and eosin. Cross sectional area (CSA) was measured from short-axis sections of left ventricle using ImageJ software. Oil red-O staining was performed on flash-frozen ventricular tissue as previously described [24].
2.9 Bioinformatics
DNA sequence alignment was performed using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and VISTA using the AVID alignment program (http://genome.lbl.gov/vista/mvista/about.shtml). Transcription factor binding sites were predicted using Matinspector (http://www.genomatix.de/cgi-bin/matinspector).
2.10 Statistical Analysis
Results are presented as mean ±SEM. The unpaired t-test was used for comparisons of means. A 2-tailed value of P < 0.05 was considered statistically significant but was reduced by the Bonferroni correction for multiple comparisons when appropriate. ANOVA was used to compare multiple groups with Bonferroni post-hoc test using Prism software, v5.
3. Results
3.1 Diet-induced obese (DIO) mice have long QT, increased ventricular ectopy, and reduced cardiac Kv protein levels
Wild type (WT) mice become obese if fed a high-fat diet for 3 months (Sup Fig 1A)[25]. This causes lipid overload of ventricular myocytes as demonstrated by oil red-O staining (Sup Fig 2). To determine if obesity causes heart rhythm abnormalities in mice, DIO mice were implanted with telemeters. ECG intervals showed a significant increase in QT interval in DIO mice, 15% longer than controls (Table 1). DIO mice also have increased frequency of ventricular ectopy, with approximately ten times as many PVCs per hour as age-matched non-obese controls (Fig 1). Thus, DIO mice demonstrate pro-arrhythmic electrophysiologic abnormalities similar to obese humans. Two of six DIO mice had spontaneous non-sustained ventricular tachycardia, which was never observed in control mice. Echocardiography demonstrated normal systolic function for DIO mice at this age (Sup Fig 1B), indicating that the pro-arrhythmic abnormalities are not secondary to heart failure. As reported previously, DIO mice had mild left ventricular hypertrophy (Sup Fig 1D)[26].
Figure 1. DIO mice have long QT and more frequent ventricular ectopy.

A. Representative ECGs from age-matched control and DIO mice at comparable heart rates, brackets indicate QT intervals. B. PVC counts per hour, N=6 each, mean + SEM, *= p<0.05 by t-test C. Normal sinus rhythm followed by a non-sustained ectopic ventricular rhythm in a DIO mouse.
Figure 2. DIO mice have decreased Kv1.5 protein and mRNA.
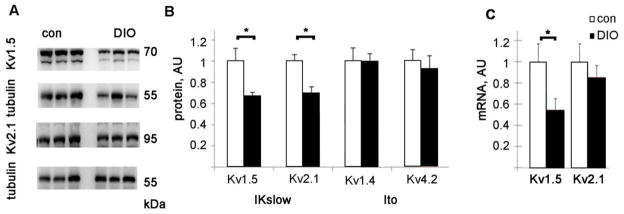
A. Representative immunoblots of Kv protein. B. Graph of Kv protein levels normalized to control levels after adjusting for tubulin loading, AU= arbitrary units. C. Kv1.5 and Kv2.1 mRNA levels, normalized to control levels. AU= arbitrary units. N=8 each, mean + SEM, *= p<0.05 by t-test.
Table 1.
ECG parameters in DIO and age-matched control mice
| RR | PR | QRS | QT | QTc | |
|---|---|---|---|---|---|
| Control | 110.5±2.4 | 34.6±1.1 | 12.7±0.5 | 45.2±1.2 | 43.2±1.0 |
| DIO | 106.8±2.4 | 35.2±0.3 | 12.4±0.4 | 51.2±1.4* | 49.6±1.7* |
Means in milliseconds ± SEM, N=6 for each group,
indicates p < 0.05 by t-test
Repolarization of the mouse ventricular myocyte is dependent on several voltage-gated potassium channels [27]. The two main repolarizing currents are the transient outward current, termed Ito, and the slower inactivating IKslow. To evaluate the cause of the impaired repolarization in DIO mice, immunoblots of the Kv proteins involved in repolarization were performed. There was a significant decrease in the protein levels of Kv1.5 and Kv2.1, the Kv channels responsible for Ikslow, in the DIO heart (Fig 2). In contrast, Kv1.4 and Kv4.2, which are responsible for the Ito current, were not significantly different from controls.
To determine if the cause of the decrease in Kv channel proteins could be due to transcriptional downregulation, we performed quantitative real time (qRT) PCR. qRT-PCR showed a significant decrease in the mRNA levels of Kv1.5 (Kcna5), approximately half that of controls. This is consistent with transcriptional down-regulation of Kv1.5 in obese hearts (Fig 2C). The mRNA for Kv2.1 (Kcnb1) had a non-significant decrease in DIO hearts compared to controls.
3.2 Lipotoxic mice have reduced cardiac CREB protein levels and CREB upregulates Kv1.5 transcription
Since the mRNA levels of Kv1.5 were decreased, we hypothesized that there is transcriptional downregulation of Kv1.5 in the obese heart. We examined the promoters of the Kv genes in silico and found that several possible cAMP response elements (CRE) are found in the mouse and human promoters of the corresponding genes Kcna5 and KCNA5. (http://www.genomatix.de/cgi-bin/matinspector).
We hypothesized that a decrease in cardiac CREB protein could cause a reduction in Kv1.5 transcription in obesity. Immunoblots confirmed that DIO mice have a significant decrease in CREB protein levels in ventricular tissue compared to controls (Fig 3A,B). CREB mRNA levels, assessed by qRT-PCR, were not significantly reduced, implying that downregulation is not at the level of transcription but probably occurs at the protein level (Sup Fig 3). Since CREB is a transcription factor we assessed the levels of CREB in the nucleus. Consistent with our hypothesis, immunoblots using nuclear extracts of ventricular tissue from DIO and age-matched control mice showed a significant reduction in CREB nuclear content (Fig 3C,D). Since the nucleus is the site of action for a transcription factor this supports a decrease in CREB-mediated transcription in the DIO heart.
Figure 3. DIO mice have decreased cardiac CREB protein.
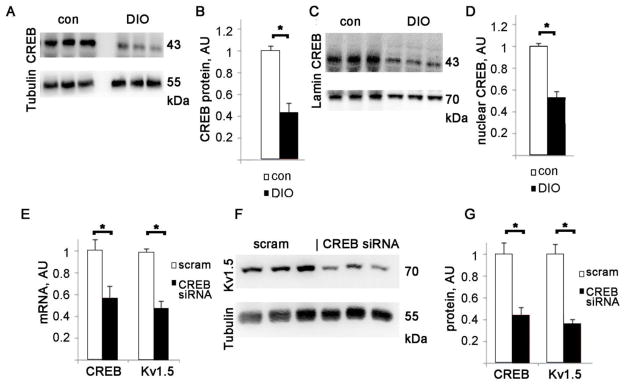
A. Representative CREB immunoblots from whole heart lysates. B. CREB protein levels in DIO mice and age-matched controls, normalized to control levels after adjusting for tubulin loading, AU= arbitrary units, error bars are SEM. N=8 each for DIO and matched controls, * = p<0.01 by t-test. C. CREB immunoblots from nuclear extracts, with lamin A/C as a loading control. D. Nuclear CREB protein levels in DIO mice and age-matched controls, normalized to control levels after adjusting for lamin A/C loading, arbitrary units, error bars are SEM. N=4 each for DIO and matched controls, * = p<0.05 by t-test. E. Graph of CREB and Kv1.5 mRNA levels in H9c2 after treatment with anti-CREB siRNA or control_scramble siRNA, normalized to control levels, AU= arbitrary units. F. Representative immunoblots of Kv1.5 after anti-CREB siRNA or control treatment, G. Graph of CREB and Kv1.5 protein levels after anti-CREB siRNA, arbitrary units, normalized to control levels after adjusting for tubulin loading, AU= arbitrary units. N=6 each, mean + SEM, *= p<0.05 by t-test.
To determine if reduced CREB is a general feature of lipotoxic cardiomyopathy, we performed immunoblots of heart lysates from PPARγ cardiac-overexpression mice, a transgenic model of lipotoxicity. We have previously shown that the PPARγ mouse has decreased Kv1.5 levels, long QT, and frequent ventricular ectopy [14]. PPARγ mice also have a significant reduction in CREB compared to wild-type littermates (Sup Fig 4), demonstrating that a reduction of CREB is a common feature of at least two lipotoxic models.
Figure 4. CREB increases the transcription of mouse and human potassium channels.

A. Luciferase constructs show that CREB increases the transcriptional activity of the mouse Kv1.5 promoter. Y-axis shows luciferase signal normalized to control. Representative experiment done with triplicates, mean + SEM, * = p<0.05 by t-test. B. Kv1.5 promoter-luciferase construct shows a dose-response relationship with CREB. Numbers indicate nanograms of CREB vector, which progressively increases the transcriptional activity of the mouse Kv1.5 promoter. The means of the groups are significantly different by ANOVA (p = 0.008). C. PCR products of CREB ChIP from mouse heart tissue using Kv1.5 promoter primers. MWM = molecular weight marker, the lowest 3 bands are 100, 200, and 300 bp. Fos is a positive control. Schematic shows the relative locations of the CRE in the luciferase construct. D. Luciferase experiments showing that mutating the CREs identified by ChIP, alone or combined as a double mutant, reduces CREB-induced expression. * = p<0.01 compared to negative control, #= p<0.01 compared to WT promoter (Bonferroni correction for multiple comparisons). E. Human Kv1.5 promoter-luciferase construct shows a dose-response relationship with CREB. The means of the groups are significantly different by ANOVA (p=0.0005). F. Mutating 2 CREs in the human Kv1.5 promoter, alone or combined as a double mutant, significantly reduces CREB-induced expression (symbols same as D) G. The HERG promoter is also upregulated by CREB. * p<0.05 by t-test. For all experiments, luciferase activity was adjusted to the internal β-gal as a transfection control for each well.
To better understand the molecular mechanisms of downregulation of Kv1.5 we used the H9c2 cell line, derived from rat ventricular myocytes [28]. H9c2 cells express CREB and several ion channels including Kv1.5. To evaluate the role of CREB in Kv1.5 transcription we transfected H9c2 with CREB siRNA. This caused a significant decrease in CREB mRNA and protein (Fig 3E,G). Kv1.5 mRNA and protein levels were also significantly reduced, to about half of scramble siRNA control (Fig 3E,F,G). This shows that CREB protein level is an important determinant of Kv1.5 transcription under baseline conditions.
To verify that CREB can regulate Kv expression, we cloned 1.5–2 kb of the 5′ flanking regions, containing the proximal promoters with predicted CRE, for mouse Kv1.5 (gene Kcna5) and Kv4.2 (gene Kcnd2) into the luciferase reporter vector pGL3. The H9c2 rat ventricular myocyte cell line was used since cardiac-specific cofactors may be required for CREB-mediated transcription. The promoter-luciferase vectors were cotransfected with a CREB expression vector or the pcDNA3 vector without insert as a negative control. CREB transfection consistently caused a significant increase in the transcriptional activity of the Kv1.5 proximal promoter, but not the Kv4.2 promoter (Fig 4A). Further, the Kv1.5 proximal promoter shows a dose-response relationship when progressively greater amounts of CREB vector are transfected (Fig 4B). This suggests a specific interaction between CREB and DNA sequences in the core promoter of Kv1.5.
3.3 Chromatin Immunoprecipitation shows that CREB binds to the Kv1.5 promoter in the heart
To determine if CREB binds to the Kv1.5 promoter in the heart, mouse ventricular tissue was homogenized and chromatin immunoprecipitation (ChIP) was performed with an anti-CREB antibody. We designed primer pairs encompassing three different predicted CRE sequences in the Kv1.5 core promoter (Fig 4C). Two of the primer pairs amplified bands of predicted size, corresponding to CREs located about 1180 and 800 bp upstream of the Kv1.5 start codon, which we refer to as CRE A and B for convenience. The third, CRE C, closer to the start codon, gave a negative result by ChIP. Identical results were obtained from lysates from two different WT mice. This demonstrates that CREB binds to at least two sites in the Kv1.5 proximal promoter in the mouse heart. To further establish the specificity of the CREB interaction at these sites in the promoter, the mouse Kv1.5 promoter-luciferase vector was mutated at the two CREs that showed binding by ChIP, substituting two base pairs in each CRE to eliminate the binding sequence for CREB. Single mutant promoters were created and also a double mutant promoter, by sequential mutagenesis. Mutating either of these CREs reduced CREB-mediated expression significantly, and mutating both abolished the increase of transcription of the Kv1.5 core promoter by CREB (Fig 4D).
3.4 CREB regulates the human Kv1.5 and HERG promoters
We next compared the mouse and human Kv1.5 promoters in silico to determine if regulation was likely to be conserved between species. Although non-coding DNA is generally less conserved during evolution than protein-coding DNA, the promoter regions upstream from the mouse and human KCNA5 genes show significant similarity. Comparing the human and mouse promoters using BLAST (up to 3 kb upstream of the start codon), there is a 600 bp stretch that is 74% identical between mouse and human, including a CRE consensus site. A VISTA alignment of the core promoters of human, mouse, and rat KCNA5 is shown in supplemental figure 4. The degree of similarity suggests conserved elements that are important for transcriptional regulation, such as transcription factor binding sites. We cloned the proximal promoter of human Kv1.5 into the luciferase reporter vector and found that co-transfection with CREB caused an increase in expression similar to its mouse counterpart (Fig 4E). There are two predicted CREs in the human proximal promoter of Kv1.5. We performed mutagenesis on these two CREs, singly and in combination to form a double mutant. Mutating the predicted CREs reduces CREB-mediated expression significantly (Fig 4F). In the human heart, Kv1.5 is a relatively atrial-specific channel, and HERG (aka KCNH2) is more important for ventricular repolarization at resting heart rates. To examine if human ventricular repolarization could be regulated by CREB, we cloned the HERG core promoter into the luciferase reporter vector. Cotransfection experiments using H9c2 cells showed that the HERG proximal promoter also had a significant response to CREB, approximately 3.5x baseline activity (Fig 4G). This indicates that the mechanism of CREB transcriptional regulation may be conserved in several mammalian Kv channels, modulating ventricular repolarization.
3.5 Extracellular signal-regulated kinase (ERK) and protein kinase D (PKD) are activated in the DIO heart
There are several potential mechanisms of CREB protein reduction in the DIO heart. In our efforts to identify the cause of decreased CREB protein levels, we examined ERK and PKD activation since both have been implicated in increasing the degradation of CREB protein [29]. ERK phosphorylation correlates with its activity [30]. Immunoblots of ventricular lysates with a phospho-specific antibody demonstrated that ERK was significantly more phosphorylated in the DIO heart, about 2.5x age-matched non-obese controls (Fig 5). PKD is a serine/threonine kinase in the calcium/calmodulin-dependent protein kinase family that has been implicated in hypertrophy and regulating myofilament calcium sensitivity [31, 32]. PKD was also significantly more phosphorylated, and thus more active, in the DIO heart (Fig 5).
Figure 5. ERK and PKD are activated in DIO hearts.
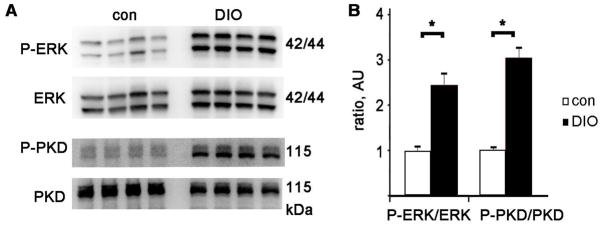
A Representative immunoblots of phosphorylated ERK 1/2, the same membrane, stripped and reprobed with total ERK antibody, and below P-PKD (Ser916) and total PKD. B. Graph of P-ERK divided by total ERK and P-PKD divided by total PKD, normalized to control, arbitrary units. N= 8 each group, mean + SEM, * = p<0.01 by t-test
3.6 PKD inhibition during lipid treatment normalizes CREB
To further analyze the molecular mechanisms involved in CREB reduction in obesity, we treated H9c2 cells with lipid to mimic the lipotoxic phenotype of obesity. H9c2 cells exposed to a non-toxic dose of palmitate (PA, a saturated fatty acid, 0.2 mM) for 24 hours showed a decrease in CREB protein similar to that found in DIO hearts. A time-course showed that the decrease in CREB was preceded by activation of ERK and PKD (Fig 6A,B). ERK activation occurs within one hour of PA treatment; PKD activation peaks several hours later. CID755673 is a specific and potent inhibitor of PKD. Inhibition of PKD with CID755673 at 20 μM (a concentration that has been shown to be selective in cell culture [33]) normalized CREB protein levels during palmitate treatment (CREB protein level 91% of control. Fig 6 C,D). Inhibition of ERK with PD98059, however, did not prevent the decrease in CREB protein. We therefore conclude that PKD activation plays an important role in the reduction of CREB protein during lipid overload (Fig 6E).
Figure 6. PKD inhibition normalizes CREB during lipid exposure.

A,B. Time course of phospho-ERK and phospho-PKD during palmitate treatment in H9c2, duplicate lanes at baseline and 3 time-points. C. Immunoblots of CREB and tubulin for H9c2 cells after overnight treatment with palmitate +/− PKD inhibitor. D. Graph of immunoblot results from H9c2, normalized to control levels after adjusting for tubulin loading, AU= arbitrary units, n=6 each, the means of the groups are significantly different by ANOVA (p=0.0001); *= p<0.01 compared to control (Bonferroni post-hoc); con= control, PA = palmitate, CID = CID755673 (PKD inhibitor, 20 μM) PD = PD98059 (ERK inhibitor, 25 μM). E. Schematic of proposed pathway. ROS = reactive oxygen species
4. Discussion
We report five major findings. First, WT diet-induced obese mice have long QT and increased ventricular ectopy, similar to obese humans. Second, DIO mice have reduced ventricular expression of the voltage-gated potassium channel Kv1.5. Third, DIO hearts have reduced levels of CREB protein, as does a cardiac cell line exposed to lipid. Fourth, CREB binds to and increases the transcription of the Kv1.5 promoter. Fifth, PKD is activated in the DIO heart as well as a cardiac cell line exposed to lipid, and a selective PKD inhibitor normalizes the CREB protein level in the cell line during lipid treatment.
It is well established that abnormal metabolism can affect cardiac repolarization but only a few studies have explored the molecular mechanisms. For example, hypo- or hyperthyroidism can alter the expression of several cardiac ion channels, including Kv1.5 [34]. The mechanistic links between metabolism and ion channel regulation are poorly understood.
The CREB transcription factor regulates a diverse group of genes by binding to short DNA sequences called cAMP response elements (CREs) in promoters, and is known to have a role in the regulation of cellular metabolism [35]. Earlier work had shown that cAMP can regulate Kv1.5 promoter reporter constructs, and that recombinant CREB protein can bind to DNA oligos containing a Kv1.5 CRE by means of electromobility gel shift assays [17]. Although this early work was suggestive, we have gone further by showing that CREB transfection increases the transcription of human and mouse Kv1.5 promoter reporter constructs, and that ChIP demonstrates binding of CREB to the Kv1.5 promoter in mouse heart. This is a significant advance since our experiments give stronger evidence that CREB upregulates Kv1.5 in the heart. It had also previously been demonstrated that a reduction in CREB downregulates Kv4.3 in the canine heart during artificial pacing [21]. Electrophysiologic abnormalities have been reported recently in cardiac-specific CREB KO mice [36] which is strong support of the regulation of cardiac repolarization by CREB. In rodent models of diabetes and obesity, vascular CREB protein levels are reduced [37]. Interestingly, CREB is activated in adipocytes in obese mice, demonstrating differential regulation of CREB in different tissues [38].
A limitation of using mouse models to study heart rhythm is that mice have different cardiac Kv channels than humans. Since we show that both the human and mouse promoters of Kv1.5 respond to CREB, this pathway may be involved in the increased risk of arrhythmia observed in obese humans. In humans, the KCNA5-encoded channel Kv1.5 is a relatively atrial-specific, and patients with atrial fibrillation have downregulated protein levels of Kv1.5 [39]. It may be that the downregulation of Kv1.5 in obese mice contributes to ventricular arrhythmias, and that downregulation of Kv1.5 in obese humans contributes more to atrial fibrillation, which is significantly more prevalent in obese humans [4]. Since the HERG promoter also shows a response to CREB, the increase in QT interval seen in obese humans may be the result of reduced cardiac CREB, causing a decrease in HERG expression and impaired repolarization in the ventricles.
PKD has been identified as a key mediator of cardiac hypertrophy [32], a condition which is associated with impaired repolarization [12]. PKD is activated by reactive oxygen species (ROS), and in neonatal rat cardiac myocytes it has been shown that ROS decrease CREB protein levels by PKD activation [29]. Cardiac ROS are known to be increased in diabetes and obesity, so oxidative stress is a plausible upstream cause of PKD activation in the obese heart [40](Fig 6G).
In summary, DIO mice have long QT and an increased frequency of PVCs. Our mechanistic studies support the contention that expression of the potassium channel Kv1.5 is reduced due to a decrease in the transcription factor CREB. PKD is activated in the obese heart and pharmacologic blockade of PKD normalizes CREB protein levels in a ventricular cell line treated with lipid. Reduced cardiac CREB by PKD activation is a novel mechanism causing pro-arrhythmic electrophysiologic remodeling in obesity.
Supplementary Material
Highlights.
Diet-induced obese mice have long QT, similar to obese humans.
Transcription of potassium channels is reduced because of lower cAMP response element binding protein (CREB) levels.
Reduced cardiac CREB is a novel mechanism causing electrophysiologic remodeling in obesity.
Acknowledgments
This work was supported by NIH grant 1K08HL105801, the Louis V. Gerstner Jr. Scholars Program, and the Lewis Katz Prize in Cardiovascular Research
Footnotes
Disclosures: none declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li W, Bai Y, Sun K, Xue H, Wang Y, Song X, et al. Patients with metabolic syndrome have prolonged corrected QT interval (QTc) Clin Cardiol. 2009 Dec;32(12):E93–9. doi: 10.1002/clc.20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramirez AH, Schildcrout JS, Blakemore DL, Masys DR, Pulley JM, Basford MA, et al. Modulators of normal electrocardiographic intervals identified in a large electronic medical record. Heart Rhythm. 2011 Feb;8(2):271–7. doi: 10.1016/j.hrthm.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huxley RR, Lopez FL, Folsom AR, Agarwal SK, Loehr LR, Soliman EZ, et al. Absolute and attributable risks of atrial fibrillation in relation to optimal and borderline risk factors: the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2011 Apr 12;123(14):1501–8. doi: 10.1161/CIRCULATIONAHA.110.009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang TJ, Parise H, Levy D, D’Agostino RB, Sr, Wolf PA, Vasan RS, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004 Nov 24;292(20):2471–7. doi: 10.1001/jama.292.20.2471. [DOI] [PubMed] [Google Scholar]
- 5.Messerli FH, Nunez BD, Ventura HO, Snyder DW. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch Intern Med. 1987 Oct;147(10):1725–8. doi: 10.1001/archinte.147.10.1725. [DOI] [PubMed] [Google Scholar]
- 6.Zemva A, Zemva Z. Ventricular ectopic activity, left ventricular mass, hyperinsulinemia, and intracellular magnesium in normotensive patients with obesity. Angiology. 2000 Feb;51(2):101–6. doi: 10.1177/000331970005100202. [DOI] [PubMed] [Google Scholar]
- 7.Filippi A, Sessa E, Jr, Mazzaglia G, Pecchioli S, Jr, Capocchi R, Jr, Caprari F, et al. Out of hospital sudden cardiac death in Italy: a population-based case-control study. J Cardiovasc Med (Hagerstown) 2008 Jun;9(6):595–600. doi: 10.2459/JCM.0b013e3282f2c9d0. [DOI] [PubMed] [Google Scholar]
- 8.Albert CM, Chae CU, Grodstein F, Rose LM, Rexrode KM, Ruskin JN, et al. Prospective study of sudden cardiac death among women in the United States. Circulation. 2003 Apr 29;107(16):2096–101. doi: 10.1161/01.CIR.0000065223.21530.11. [DOI] [PubMed] [Google Scholar]
- 9.Jouven X, Desnos M, Guerot C, Ducimetiere P. Predicting sudden death in the population: the Paris Prospective Study I. Circulation. 1999 Apr 20;99(15):1978–83. doi: 10.1161/01.cir.99.15.1978. [DOI] [PubMed] [Google Scholar]
- 10.Hookana E, Junttila MJ, Puurunen VP, Tikkanen JT, Kaikkonen KS, Kortelainen ML, et al. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm. 2011 Oct;8(10):1570–5. doi: 10.1016/j.hrthm.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 11.Szczepaniak LS, Victor RG, Orci L, Unger RH. Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res. 2007 Oct 12;101(8):759–67. doi: 10.1161/CIRCRESAHA.107.160457. [DOI] [PubMed] [Google Scholar]
- 12.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999 May;42(2):270–83. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 13.Aomine M, Yamato T. Electrophysiological properties of ventricular muscle obtained from spontaneously diabetic mice. Exp Anim. 2000 Jan;49(1):23–33. doi: 10.1538/expanim.49.23. [DOI] [PubMed] [Google Scholar]
- 14.Morrow JP, Katchman A, Son NH, Trent CM, Khan R, Shiomi T, et al. Mice With Cardiac Overexpression of Peroxisome Proliferator-Activated Receptor gamma Have Impaired Repolarization and Spontaneous Fatal Ventricular Arrhythmias. Circulation. 2011 Dec 20;124(25):2812–21. doi: 10.1161/CIRCULATIONAHA.111.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marionneau C, Aimond F, Brunet S, Niwa N, Finck B, Kelly DP, et al. PPARalpha-mediated remodeling of repolarizing voltage-gated K+ (Kv) channels in a mouse model of metabolic cardiomyopathy. J Mol Cell Cardiol. 2008 Jun;44(6):1002–15. doi: 10.1016/j.yjmcc.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005 Feb 4;96(2):225–33. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 17.Mori Y, Matsubara H, Folco E, Siegel A, Koren G. The transcription of a mammalian voltage-gated potassium channel is regulated by cAMP in a cell-specific manner. J Biol Chem. 1993 Dec 15;268(35):26482–93. [PubMed] [Google Scholar]
- 18.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002 Aug 15;35(4):605–23. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 19.Muller FU, Boknik P, Horst A, Knapp J, Linck B, Schmitz W, et al. cAMP response element binding protein is expressed and phosphorylated in the human heart. Circulation. 1995 Oct 15;92(8):2041–3. doi: 10.1161/01.cir.92.8.2041. [DOI] [PubMed] [Google Scholar]
- 20.Patberg KW, Plotnikov AN, Quamina A, Gainullin RZ, Rybin A, Danilo P, Jr, et al. Cardiac memory is associated with decreased levels of the transcriptional factor CREB modulated by angiotensin II and calcium. Circ Res. 2003 Sep 5;93(5):472–8. doi: 10.1161/01.RES.0000088785.24381.2F. [DOI] [PubMed] [Google Scholar]
- 21.Patberg KW, Obreztchikova MN, Giardina SF, Symes AJ, Plotnikov AN, Qu J, et al. The cAMP response element binding protein modulates expression of the transient outward current: implications for cardiac memory. Cardiovasc Res. 2005 Nov 1;68(2):259–67. doi: 10.1016/j.cardiores.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Ozgen N, Lau DH, Shlapakova IN, Sherman W, Feinmark SJ, Danilo P, Jr, et al. Determinants of CREB degradation and KChIP2 gene transcription in cardiac memory. Heart Rhythm. 2010 Jul;7(7):964–70. doi: 10.1016/j.hrthm.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998 Mar;274(3 Pt 2):H747–51. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- 24.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007 Oct;117(10):2791–801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005 Dec;54(12):3530–40. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 26.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. Metabolic stress-induced activation of Fox01 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012 Mar 1;122(3):1109–18. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005 Oct;85(4):1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 28.Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991 Dec;69(6):1476–86. doi: 10.1161/01.res.69.6.1476. [DOI] [PubMed] [Google Scholar]
- 29.Ozgen N, Guo J, Gertsberg Z, Danilo P, Jr, Rosen MR, Steinberg SF. Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol Pharmacol. 2009 Oct;76(4):896–902. doi: 10.1124/mol.109.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000 Dec 1;19(23):6341–50. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuello F, Bardswell SC, Haworth RS, Yin X, Lutz S, Wieland T, et al. Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ Res. 2007 Mar 30;100(6):864–73. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- 32.Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A. 2008 Feb 26;105(8):3059–63. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, et al. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008 Nov 28;283(48):33516–26. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G, et al. Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: relation to cellular and integrated electrophysiology. Circ Res. 2003 Feb 7;92(2):234–42. doi: 10.1161/01.res.0000053185.75505.8e. [DOI] [PubMed] [Google Scholar]
- 35.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011 Mar;12(3):141–51. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulte JS, Seidl MD, Nunes F, Freese C, Schneider MD, Schmitz W, et al. CREB critically regulates action potential shape and duration in the adult mouse ventricle. Am J Physiol Heart Circ Physiol. 2012 Mar 16;15(302):H1998–2007. doi: 10.1152/ajpheart.00057.2011. [DOI] [PubMed] [Google Scholar]
- 37.Schauer IE, Knaub LA, Lloyd M, Watson PA, Gliwa C, Lewis KE, et al. CREB downregulation in vascular disease: a common response to cardiovascular risk. Arterioscler Thromb Vasc Biol. 2010 Apr;30(4):733–41. doi: 10.1161/ATVBAHA.109.199133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi L, Saberi M, Zmuda E, Wang Y, Altarejos J, Zhang X, et al. Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 2009 Mar;9(3):277–86. doi: 10.1016/j.cmet.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brundel BJ, Van Gelder IC, Henning RH, Tuinenburg AE, Wietses M, Grandjean JG, et al. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: differential regulation of protein and mRNA levels for K+ channels. Journal of the American College of Cardiology. 2001 Mar 1;37(3):926–32. doi: 10.1016/s0735-1097(00)01195-5. [DOI] [PubMed] [Google Scholar]
- 40.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007 Oct;56(10):2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.