

Abstract
Diabetes and obesity are both associated with lipotoxic cardiomyopathy exclusive of coronary artery disease and hypertension. Lipotoxicities have become a public health concern and are responsible for a significant portion of clinical cardiac disease. These abnormalities may be the result of a toxic metabolic shift to more fatty acid and less glucose oxidation with concomitant accumulation of toxic lipids. Lipids can directly alter cellular structures and activate downstream pathways leading to toxicity. Recent data have implicated fatty acids and fatty acyl coenzyme A, diacylglycerol and ceramide in cellular lipotoxicity, which may be caused by apoptosis, defective insulin signaling, endoplasmic reticulum stress, activation of protein kinase C, MAPK activation or modulation of PPARs.
Keywords: heart failure, lipotoxicity, fatty acid oxidation, diacylglycerol, ceramide, apoptosis, insulin signaling, endoplasmic reticulum stress, protein kinase C, MAPK, PPAR
INTRODUCTION
The purpose of this review is to discuss how lipids, which are important for cardiac function, can also be detrimental when their amount or distribution is in excess and may lead to abnormal cardiac structure and function. The heart, under normal conditions, is primarily dependent on fatty acids (FAs). On the other hand, the preferred substrate of cardiac energy metabolism switches from FAs to glucose for ATP production in pathological situations such as ischemia, advanced hypertrophy and heart failure. Although this “flexibility” is considered as an adaptive process, several studies have demonstrated that both inhibition and profound elevation of cardiac FA oxidation, which is regulated by multiple factors (Figure 1) can be pathogenic. Defective cardiac lipid uptake1 impairs optimal heart function. This finding may be explained, in part, by the limitation of the normal hearts for substituting glucose for lipids. In contrast, excessive FA oxidation is also harmful2, 3. Whether this is due to lipid toxicity with inappropriate regulation of lipid uptake or overwhelming FA oxidation alone will be discussed in this review.
Figure 1.
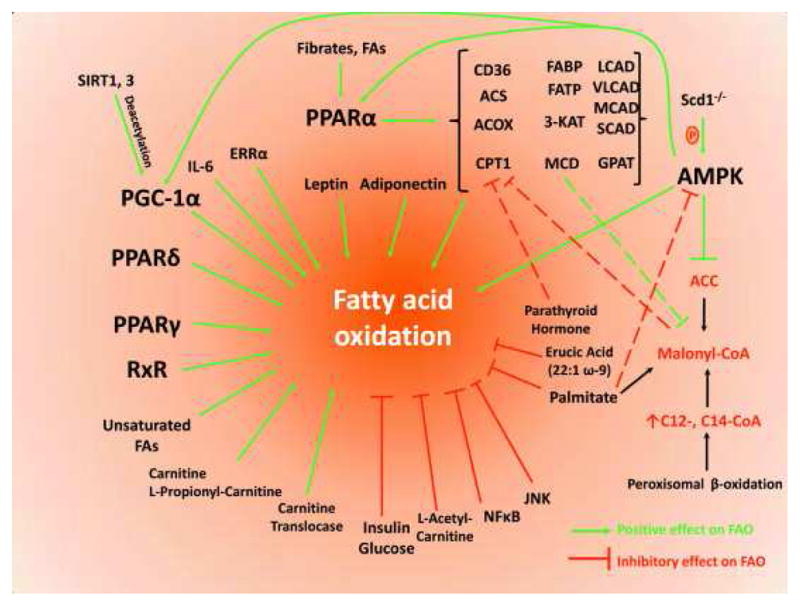
Proteins and transcriptional factors that modulate fatty acid oxidation
The delivery and uptake of lipids to the heart
FAs are the primary energetic lipids metabolically used by the heart. In fact, FAs are estimated to supply approximately 70% of the heart’s energy requirements2. Moreover, studies from more than four decades ago suggest that esterified FAs are the major source of cardiac lipids in humans4. FA supplies for the heart are derived from dietary fat, as well as by hepatic FAs synthesized from carbohydrates, and FAs released following adipose tissue lipolysis. In the circulation, most FAs are present either esterified to glycerol as a component of lipoprotein triglycerides (TG) and phospholipids, or unesterified, free fatty acids (FFAs) that circulate bound to albumin. Some FAs are acquired by tissues, including the heart, as a component of either whole lipoprotein particles or smaller lipid vesicles that dissociate from larger lipoproteins during lipolysis. Studies in humans demonstrated extraction of esterified i.e. lipoprotein-associated FAs by the heart4. In isolated hearts, most triglyceride uptake requires extracellular lipolysis5. TGs are the primary source of energy-producing FAs and circulate in the bloodstream within chylomicrons and very low density lipoproteins (VLDL). Lipoprotein-derived FAs are acquired by the heart following TG hydrolysis, which is mediated by endothelial-bound lipoprotein lipase (LpL). Intracellular esterified lipids stored in the form of triglycerides are converted to FFAs via the intracellular enzymes hormone sensitive lipase (HSL) and adipose triglyceride lipase (ATGL)6, 7, which seems to be a major circuit for providing FAs in the ATP production machinery8, 9.
Lipoprotein-derived FFAs are created when plasma lipoproteins interact with endothelial cell-associated LpL10. The heart is a particularly rich source of this enzyme. Of note, mice with expression of LpL solely in cardiomyocytes have normal plasma triglyceride levels11. In the neonatal and presumable prenatal period, cardiac LpL expression is very low as the heart primarily consumes glucose. Shortly after birth and with the consumption of high- fat milk, LpL is rapidly induced in cardiac tissue12–14. Fasting reduces adipose tissue LpL activity, but increases cardiac LpL15. This is thought to be a response to the increased circulating fraction of TG and FFA under starvation. Active cardiac LpL increases in the setting of defective glucose utilization and hypoinsulinemia16, 17. In contrast, insulin is needed to maintain LpL expression in isolated cardiomyocytes18. Hypertension reduces cardiac LpL as the heart shifts to greater glucose utilization19. Cardiac-specific loss of LpL converts the heart’s “metabolic preference” towards greater glucose uptake and oxidation20, while constitutive cardiomyocyte-specific expression of glycosylphosphatidylinositol (GPI)-anchored LPL leads to cardiomyopathy21.
FFA uptake occurs via cell surface receptors, the most well characterized being CD3622, and via biophysical non-receptor transport, also known as “flip-flop”23. CD36 knockout (CD36−/−) mice have reduced FFA uptake into muscle and heart22. Humans with a genetic defect in CD36 also have defective cardiac long chain FA uptake and an approximately 3-fold increase in glucose uptake24. Although CD36-deficient humans tend to have more hypertriglyceridemia and insulin resistance, they have not been reported to have cardiomyopathy25. Cardiac function in CD36−/− mice is also normal. One group reported that CD36−/− hearts respond normally to ischemia reperfusion26, an observation leading the investigators to conclude that glucose uptake compensates for defective FA uptake. However, other investigators reported that CD36 deficiency led to impaired cardiac contraction during ischemia, which was corrected by inclusion of caprylic acid, a medium chain FA, to the perfusate27. Differences in the conditions for these two studies likely contributed to the opposing results.
CD36 is required downstream of LpL for the uptake of at least a part of lipoprotein-derived FAs, as well as for non-esterified FFAs28. It is possible that a transporter other than CD36 functions in normal or CD36−/− mice. Most likely, this transporter is a member of the fatty acid transport protein (FATP) gene family. Consistent with this hypothesis, FATP1 expression is increased in CD36−/− hearts suggesting a compensatory regulation of gene expression29. However, FATP1 knockout mice30 do not have a cardiac phenotype. FATP6 has been reported to be the FATP most robustly expressed in the heart31. Further, intracellular accumulation of FAs requires trapping and esterification to CoA which also involves fatty acid transport proteins (FATPs) and long chain acylCoA synthase (ACS).
Figure 2 depicts the major proteins that are involved in fatty acid uptake and delivery to the mitochondria for oxidation and ATP production.
Figure 2. Lipid delivery to the cardiomyocyte.
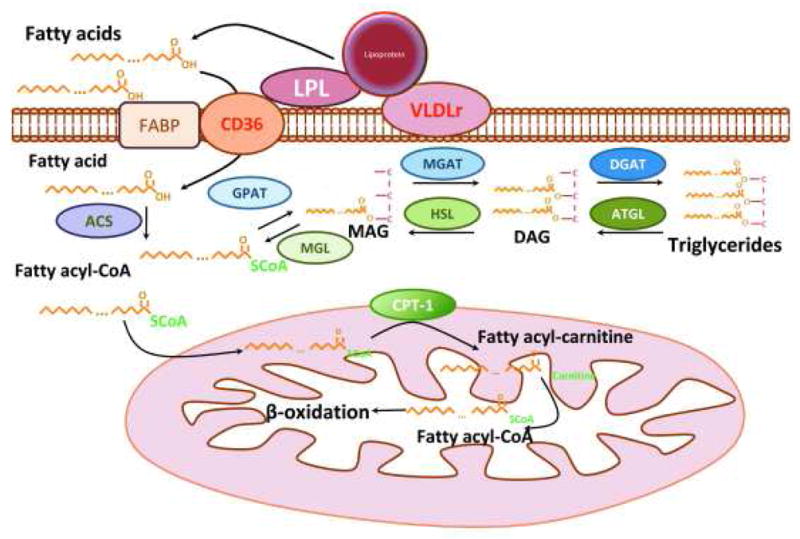
Fatty acids derived from triglyceride-rich lipoproteins, chylomicrons, and VLDL are hydrolyzed by lipoprotein lipase. Lipoprotein-derived fatty acids or albumin-bound free fatty acids are internalized by the cells via membrane receptors such as CD36 or other transporters, or via non-receptor diffusion through the membrane, known as “flip-flop”. Internalization of whole lipoproteins or lipoprotein remnants via lipoprotein receptors is also possible. Upon release into cardiomyocytes fatty acids are converted to fatty acyl-CoAs and can then gradually incorporated to a glycerol backbone forming mono-, di- and tri-acyl-glycerols (triglycerides). Fatty acyl-CoAs can be released via triglyceride lipolysis that is mediated by ATGL, HSL and MGL and with the contribution of Cpt-1 they enter mitochondria for β-oxidation and production of ATP.
Toxic lipids and lipid intermediates
Not all lipids are toxic. Specifically, the status of TG as a toxic molecule is unresolved. Despite the fact that increased TG levels correlate with insulin resistance in the setting of obesity and the metabolic syndrome, genetic experiments suggest that TG is by itself solely a marker for general lipid overload by the cells. Saturated LCFAs, particularly palmitic acid (PA, 16:0), are considered to be a more potent cause of lipotoxicity than unsaturated LCFA, such as oleic acid (OA, 18:1). This has been correlated with increased intracellular ceramide levels due to distinct enzyme specificity; PA leads to increased ceramide formation compared to OA32–34. Cardiolipotoxic animal models are characterized by increased myocardial lipid content in the absence of obesity. Transgenic mice expressing a GPI-anchored form of human LpL driven by the cardiac muscle–specific α-myosin heavy chain (αMHC) promoter (αMHC-LpLGPI) have normal plasma lipid levels but increased lipid uptake from circulating lipoproteins and intracellular cardiomyocyte lipid accumulation. Ceramide accounts, at least partially, for the aggravating consequences of the increased cardiac lipid accumulation of the MHC-LpLGPI mice35. Treatment of these mice with myriocin, a de novo ceramide synthesis inhibitor, normalizes intramyocardial ceramide levels and corrects cardiac hypertrophy35. However, this treatment improved survival only slightly, indicating that additional mechanisms, which are triggered by lipids other than ceramide may also mediate cardiac lipotoxicity.
Di-Acyl Glycerol (DAG) is another lipid that mediates FA-induced toxicity and is implicated as a cause of insulin resistance in skeletal muscle and liver36–38. In TG synthesis, DAG acyl transference (DGAT) adds the final FA onto DAG and converts the toxic lipid intermediate DAG to TG. Two non-homologous DGAT genes have been cloned, DGAT139 and DGAT240. Overexpression of DGAT1 in hearts of lipotoxic models, such as the ACS-overexpressing mice prevents from cardiac dysfunction although increased lipid accumulation still occurs41. This suggests a role for this enzyme in clearing toxic lipid intermediates into the stable and non-toxic storage for TG. On the other hand, DGAT1 deficient mice are resistant to weight gain when fed a western-style high fat diet42, 43. This condition is associated with increased total energy expenditure due to increased physical activity in these animals. Thus, based on these data, ceramide and diacylglycerol rather than TG appear to be toxic and contributing to deteriorating cardiac function.
Models of lipid toxicity
Two human conditions are thought to be caused by excess cardiac lipid accumulation: obesity-related cardiomyopathy, a dilated cardiomyopathy associated with normal coronary arteries and sudden cardiac death44, and diabetic cardiomyopathy with decreased cardiac function disproportional to coronary artery disease45. In studies of pathological specimens46, cardiac lipid uptake and oxidation47, and MR cardiac TG analysis48, 49 establish that cardiac dysfunction is associated with defects in cardiac lipid metabolism and lack of intracellular TG-derived FAs mobilization that leads to TG accumulation.
It is not very clear whether cardiomyopathy is due to excess FA oxidation or accumulation of toxic lipids or both. Oxidation of long chain FAs such as palmitate requires the transfer of FA-CoA into the mitochondria by carnitine palmitoyltransferase (Cpt)-1. Cpt-1-mediated FA consumption is critical as shown in Cpt-1β+/− mice that have aggravated pressure overload-induced cardiac hypertrophy caused by lipotoxicity50. Cpt-1 function is inhibited through a negative feed-back loop by malonyl-CoA, a product of acetyl-CoA carboxylase (ACC). Malonyl-CoA was reduced and cardiac function was improved in mice with cardiomyocyte-specific ACC2 ablation that underwent pressure overload via transverse aortic constriction51. ACC2−/− have increased FAO and this led to reduced left ventricular mass52. Pharmacological inhibitors of Cpt-1, such as etomoxir, ethyl-2-tetradecyl glycidate and oxfenicine switch energy metabolism from FA to glucose oxidation53. It has been proposed that such metabolic change from oxidation of FA to the energetically less O2-consuming glucose or lactate oxidation is beneficial in the post-ischemic heart failure54. On the other hand prolonged inhibition of Cpt-1 by etomoxir, in combination with high-fat diet, resulted in excess triglyceride accumulation and lipotoxicity in skeletal muscle of rats55.
A number of spontaneous and targeted rodent mutations have reproduced dilated cardiomyopathy with excess lipid accumulation. Rodents such as the Zucker rat56, 57 and the db/db mouse58 that have genetic defects in leptin signaling pathways demonstrate reduced cardiac glucose oxidation, increased FA oxidation, lipid accumulation, and cardiac dysfunction. More recently, studies have shown that decreased cardiac function linked to aging is blunted in CD36−/− mice59.
A number of genetically modified mice develop dilated cardiomyopathy associated with lipid accumulation. Several models have transgenic expression of proteins that increase lipid uptake by the heart. Cardiomyocyte-specific expression of long chain acyl CoA synthase 1 (ACS1)60, the enzyme that traps FA intracellularly by esterification to CoA, leads to both systolic and diastolic heart dysfunction. Fatty acid transport protein 1 (FATP1) overexpression also causes lipotoxic cardiomyopathy61; this protein is postulated to modulate FA trapping and uptake. Cardiomyocyte-anchored LpL is associated with greater uptake of plasma lipids and dilated cardiomyopathy21. All three genetically modified mice in the studies cited have increased cardiac lipid content, but also greater FA oxidation.
Additional models were created by overexpression of the FA metabolic transcription factors PPARα and PPARγ. Cardiomyocyte-specific PPARα transgenic mice have an increase in FA oxidation genes, greater FA oxidation, and less glucose oxidation and GLUT4 expression62. Cardiomyocyte specific PPARα transgenic mice fed with a long-chain-fatty-acid-containing diet exerted a severe lipotoxic cardiomyopathic phenotype in diabetic background, whereas the lipotoxic effect was ameliorated when diet was switched to a medium-chain triglyceride-enriched diet63. Reduction of lipid uptake in the PPARα transgenic model using either a whole body deletion of CD3629 or a cardiac specific-deletion of LpL64 corrected the cardiomyopathy. PPARγ transgenic mice have a similar increase in FA metabolic genes, but no decrease in GLUT465. Although it seems paradoxical that these mice develop lipotoxicity rather than reduced cardiac lipid stores, lipid uptake pathways involving either CD36 or LpL are upregulated by PPARs and increased lipid uptake must exceed oxidation. Interestingly, cardiomyocyte-specific overexpression of PPARγ on a PPARα−/− genetic background led to improvement in fatty acid oxidation, cardiac function and survival rates despite similar cardiac TG and toxic lipids, DAG and ceramide, accumulation, as compared to PPARγ66. Notably, acylcarnitine content was decreased and so apoptosis, ROS levels, and endoplasmic reticulum stress marker were. Although these models suggest that lipid accumulation is the culprit, the well-known toxic effects of excess lipid oxidation in perfused heart models67, especially in the setting of ischemia, does not rule out toxicity due to excess lipid oxidation.
If lipid accumulation occurs in the setting of reduced FA oxidation, it confirms that excess lipids are at least one cause of toxicity. Two lines of genetically modified mice that develop cardiac dysfunction and lipid accumulation have been created. Using a line of mice in which LpL expression was driven by a fragment of its natural promoter, a line of mice expressing LpL specifically in cardiac muscle was created11. Lacking any other mutations, these mice appear normal. However, when FA oxidation was reduced by crossing onto the PPARα knockout background, cardiac dysfunction ensued68. Further, lipid oxidation decreased, lipid uptake was presumably not affected, and cardiomyocyte lipid accumulation occurred. Similarly, when FA oxidation was reduced by a tissue-specific knockout of PPARδ, lipid accumulation and cardiomyopathy occurred69. On the other hand, constitutive cardiomyocyte-specific expression of PPARδ induced the expression of FAO-associated genes and did not lead to lipid accumulation and cardiac dysfunction70. Besides elevated FAO, the prevention of cardiac lipid accumulation and organ dysfunction in the αMHC-PPARδ mice may be attributed to increased expression of Angiopoetin-like 471, which is an inhibitor of LpL and therefore may minimize cardiac lipid uptake. Accordingly, ATGL−/− mice show reduced FAO and massive cardiac lipid accumulation that are associated with severe cardiac dysfunction. Cardiac dysfunction in these mice is corrected upon pharmacological activation of PPARα, also indicating that FA-mediated PPARα activation relies on intracellular TG lipolysis72. Consistently, cardiomyocyte-ATGL overexpression seems to be beneficial for mice that undergo pressure overload9. However, this benefit cannot be attributed to increased FAO, which is surprisingly reduced, possibly as a secondary event to the increased glucose catabolism. In mice with total gene deletion of the other important cardiac lipase, HSL, cardiac TG lipase activity was decreased, but cardiac TG was not dramatically changed and there was no overt cardiac phenotype73. These data implicate lipid accumulation, rather than excess oxidation, as a cause of heart dysfunction.
Signaling effects of lipids linked to cardiac dysfunction
Several lipids have been implicated in pathological processes that might contribute to lipotoxic cardiomyopathy: FA/fatty acyl CoA, acylcarnitine, unesterified cholesterol, lysolecithin, ceramide, and DAG. These lipids are thought to cause apoptosis, inflammation, mitochondrial dysfunction, and/or defective intracellular signaling (Figure 3).
Figure 3. Metabolic pathways triggered by palmitic acid leading to apoptosis and cardiomyopathy.

Several mechanisms may account for the toxic effects of saturated fatty acids: 1. Palmitic Acid is used by Serine Palmitoyl Transferase to generate cytoplasmic ceramide, which activates JNK1/2 that interacts with Bax in the mitochondrial membrane and results in the release of cytochrome C due to reduction of cardiolipin (ceramide-dependent mechanism). 2. Palmitic acid can inhibit AMPK, which increases malonyl-CoA levels, inhibits CPT-1, and causes accumulation of fatty acids and lipotoxicity. 3. Palmitic acid contributes to the increase of DAG that induces the upregulation of PKC. PKC inhibits the insulin-signaling pathway by blocking IRS-1. 4. The insulin-signaling pathway can be inhibited by ceramide-mediated increased PP2A, which dephosphorylates (inactivates) the anti-apoptotic PKB/Akt; inhibition of PKB/Akt may induce apoptosis. Dephosphorylated Akt reduces AMPK activity that can lead to lipotoxicity (see above). 5. Palmitic acid may be incorporated into phospholipid and TG species in microsomal membranes, resulting in compromised ER membrane integrity and redistribution of protein-folding chaperones to the cytosol (ER stress). 6. Esterification of palmitate can also cause ER fission directly. Intensive ER stress may result in apoptosis.
Apoptosis pathways
Myocardial dysfunction has been attributed, among other factors, to apoptosis induced by abnormal conditions such as obesity, diabetes, and aging74. In most of these abnormalities, cardiac lipid overload is associated with and thought to contribute to the initiation of the apoptotic cascade. Studies, primarily in isolated cardiomyocytes, have implicated saturated FAs as a primary cause of apoptosis. Treatment of isolated neonatal rat ventricular myocytes with palmitic acid alters mitochondrial physiology and leads to apoptosis associated with cardiolipin loss, cytochrome c release, mitochondrial swelling, and DNA laddering75, 76. These pathways are summarized in Figure 3.
Saturated FA-induced apoptosis has been attributed to changes in cellular lipid content and/or excess lipid oxidation. Ceramide, one lipid class proposed to be toxic, is synthesized via two major pathways. In the first pathway, increased cellular palmitate drives de novo ceramide synthesis. Palmitate is initially converted to palmitoyl-CoA. Then, serine palmitoyltransferase (SPT), the rate limiting enzyme of ceramide de novo synthesis, catalyzes the condensation of palmitoyl-CoA and serine, producing 3-keto-sphinganine77. Subsequent reactions lead to the sequential synthesis of sphinganine, dihydroceramide, and ceramide78, 79. In the second pathway, ceramide is released from sphingomyelin after it is hydrolyzed by sphingomyelinase80.
Support for a non-toxic effect of greater FA oxidation in the non-ischemic heart is provided by studies associated with modulation of 5′-AMP-activated protein kinase (AMPK). Activation of AMPK with 5-aminoimidazole-4-carboxyamide-1-beta-D-ribofuranoside (AICAR) attenuated the apoptotic effect of palmitate on cardiomyocytes75. This specific effect of AMPK was attributed to reduced malonyl-CoA levels, increased FA transport and oxidation in mitochondria, and, presumably, decreased intracellular levels of toxic lipids75. Nevertheless, AMPK is a “master regulator” of mechanisms relevant to cardiac energy production through FA or glucose catabolism. AMPK is activated by an increase in intracellular AMP:ATP ratio81 and has been correlated with both cardiac FA uptake and oxidation. Activation of AMPK with AICAR increases protein levels of CD36 in cardiac myocytes and, subsequently, long chain FA transport in the heart82. AMPK-mediated increase of long chain FA uptake by mouse cardiomyocytes correlates with CD36 expression83. AMPK-mediated increase of FA uptake by cardiac cells also takes place by recruiting LpL to the coronary lumen, an event that increases LpL activity84.
ROS generation has also been implicated in palmitate-induced programmed cell death85,86. In CHO cells, reduction of palmitate-generated ROS by two ROS scavengers prevents apoptosis. In contrast, in neonatal rat cardiomyocytes, palmitate-induced apoptosis was neither associated with increased ROS nor rescued by antioxidants. In cultured aortic endothelial cells, FFAs increased ROS production87, especially in the setting of hyperglycemia.
Lipid-induced defective insulin signaling
There may be several mechanisms by which insulin signaling is cardioprotective and anti-apoptotic: 1) augmenting glucose oxidation, especially during ischemia, 2) direct activation of survival pathways downstream of Akt, 3) changes in cardiac perfusion due to eNOS activation. Insulin is an important regulator of myocardial substrate metabolism, but it also exerts regulatory effects on intracellular Ca2+ handling and cell survival. Via either of these pathways, defective insulin signaling might exacerbate lipotoxic cardiomyopathy. Insulin resistance is one of the earliest observed cardiac defects found in mice given a high-fat diet88.
Lipid accumulation in the heart can lead to the development of cardiomyocyte insulin resistance characterized by predominant utilization of FA for cardiac energy, decreased glucose uptake, defective contractile response to insulin, and decreased cardiac efficiency caused by oxygen waste for noncontractile purposes89–93. In contrast, mice with a cardiac-specific deletion of insulin receptors demonstrate increased glucose uptake and oxidation, and develop smaller hearts94.
The FA-induced impairment of the insulin/IRS1/PI3K/Akt pathway has been suggested to be a causative factor in diabetic cardiomyopathy. The mechanism that underlies the effect of the accumulated myocardial FA on insulin signaling has not been elucidated; however, several mechanisms have been proposed based on findings in cells besides cardiomyocytes. ROS play a major role in the development of insulin resistance, as was shown by the amelioration of insulin resistance following ROS attenuation95. Both ceramide and DAG have also been implicated in defective insulin signaling and reduced glucose uptake in muscle.
Ceramide-mediated insulin resistance occurs in saturated fat feeding96. Although the mechanism is not completely clear, ceramide prevents insulin-mediated activation of Akt/PKB97–99. Ceramide may do this by blocking phosphorylation of Akt/PKB and/or by stimulating protein phosphatase 2A, thereby leading to the dephosphorylation of Akt/PKB100. In C2C12 myotubes, overexpression of acid ceramidase, which catalyzes the conversion of ceramide to sphingosine i.e. the reduction of intracellular ceramide levels, attenuated the inhibitory effects of saturated FFAs on insulin signaling101.
DAG blocks upstream signaling events by promoting the serine phosphorylation of IRS-1, resulting in its deactivation102–104. This process, at least in skeletal muscle, may be mediated by activation of PKCθ105 or other PKCs106. Systemic insulin resistance in patients with heart failure was accompanied by increased toxic lipid intermediates, DAG and ceramide. Mechanical unloading after left ventricular assist device implantation was shown to decrease myocardial levels of DAG and ceramide and improved insulin/phosphatidylinositol-3 kinase/Akt pathway activation107.
It may be that the insulin-resistance associated with lipotoxicity is an adaptive process. Insulin signaling may exacerbate lipid accumulation in the heart by increasing FA uptake. Besides its well-known function in glucose uptake, regulation, and catabolism, insulin redistributes CD36 from intracellular stores to cell membranes of rat cardiac myocytes108, a process that might increase intracellular FA stores. This process occurs via the actions of the forkhead transcription factor FoxO1 in muscle cells109. Moreover, following insulin stimulation, long chain FAs that are taken up by cardiomyocytes are less likely to be oxidized, thereby leading to the increased storage of toxic lipids.
PKC activation
Several lipids are associated with activation of protein kinase C (PKC). PKCs are a family of 12 serine/threonine protein kinases that are capable of modifying the activity of many cellular proteins. Several PKCs are highly expressed in adult myocardium and have been implicated in the regulation of contractility, gene expression, and growth110. A number of studies have demonstrated that PKC activation relies on binding of DAG36–38 or ceramide111–121 and translocation to the membrane. Overexpression of PKCβ, specifically in the myocardium of transgenic mice, leads to a cardiomyopathy associated with myocardial necrosis and thickened left and right ventricular walls resulting in an increase in the number of cardiomyocytes and the size of the interstitial extracellular matrix122. A later study showed that male Sprague-Dawley rats that were assigned to a high-fat diet (saturated fat from coconut oil) had increased activation of PKCβ2, as evidenced by greater membrane translocation and cardiac hypertrophy123. Thus, lipid-mediated activation of PKC could exacerbate lipotoxic cardiomyopathy.
Several PKC isoforms are activated in failing hearts124. PKCα and PKCε confer negative inotropic effects in cardiomyocytes125, 126. PKCβ impairs Ca2+ handling, increases cardiomyocyte necrosis and ventricular wall thickening122, 123, 127, 128. Genetic128–130 and pharmacologic128, 130, 131 inhibition of PKCs improves cardiac responsiveness to catecholamines and heart function in cardiomyopathic mice. Of note, myocardial tissue from heart failure patients107, cardiolipotoxic mice32 and a palmitate treated-human cardiomyocyte cell line32 have increased PKCα and PKCδ activation. In addition cardiac lipotoxicity animal models and palmitate-treated cells show compromised β-adrenergic receptor responsiveness, which is corrected by inhibition of the PKC pathway32. Thus, PKC signaling is activated by toxic lipids and is associated with heart failure.
MAP Kinases
Mitogen Activated Protein Kinases (MAPKs) constitute a major group of kinases participating in critical intracellular signal transduction and regulation pathways. Members of the MAPK family, including Erk1, Erk2, JNK1, JNK2, and p38, have been associated with the control of survival signaling in cardiomyocytes following oxidative stress and are known to modulate insulin signaling132, 133. Erk and JNK activation also occurs in a genetic heart failure animal model (mutated lamin A/C gene) and treatment of the animals with Erk and JNK inhibitors prevented left ventricular end-systolic dilatation, increased ejection fraction, and decreased myocardial fibrosis134. Although MAPKs have been implicated in cardiac development and disease, as well as in cardiomyocyte apoptosis135, 136, they may also play a role in FA-induced toxicity. Palmitate treatment of primary neonatal rat ventricular myocytes was found to activate Erk1/2, p38, and JNK137. However, a MEK1/2 inhibitor or a p38 kinase inhibitor had no effect on baseline or palmitate-induced apoptosis137. Activated JNK1 has been implicated in the induction of apoptosis in rat cardiomyocytes that undergo ischemia/reoxygenation stress138. The same study showed that the apoptotic effect of ceramide in rat cardiomyocytes can be mediated by activation of JNK and can be attenuated by administration of antisense JNK1 or JNK2. JNK interacts with proapoptotic Bax on the mitochondrial membrane136. Treatment of the same cells with a low concentration of oleate along with palmitate inhibited both palmitate-induced JNK activation and apoptotic events137. Inhibition of JNK has also been associated with improved FAO in the heart139 and adipose tissue140. These data suggest that MAPKs may be involved in lipid-mediated apoptosis or suppression of FAO and thus may, at least partially, account for compromised cardiac function.
ER stress
Accumulation of FAs has been linked to the induction of endoplasmic reticulum (ER) stress. Specifically, palmitate-induced ER stress has been considered to be a secondary event that follows oxidative stress and generation of ROS, and results in an induction of eukaryotic elongation factor (eEF) 1A-1, which interferes with the integrity of the cytoskeleton and causes cellular death141. A more direct mechanism of palmitate-induced ER stress has also been proposed by the same study. This mechanism involves the incorporation of palmitate in phospholipid and TG species in microsomal membranes such that ER membrane integrity is compromised and protein-folding chaperones are redistributed to the cytosol142. Another study has reported that the esterification of palmitate can directly cause ER fission143. Myocardial ER-stress markers were elevated in a rat heart failure model (left anterior descending coronary arteries ligation) and their expression was alleviated by treatment of the rats with atorvastatin, which improved left ventricular function and reduced cardiac fibrosis144. Thus, ER-stress driven by cardiac lipid accumulation may contribute in the development of heart failure.
PPARs
Nuclear receptors, particularly PPARs, have a major role in the control of fatty acid oxidation. The PPAR family consists of three members, PPARα, PPARβ/δ, usually reported as PPARδ, and PPARγ. PPARα has been tightly associated with increased fatty acid oxidation in the heart62 and skeletal muscle145. Similarly, PPARδ has been shown to activate fatty acid oxidation in the heart146. PPARγ, besides its well-known role as a major regulator of lipogenesis65, 147, contributes also in fatty acid oxidation in cardiac66 and skeletal muscle148. PPARα-mediated fatty acid oxidation in the heart and other tissues relies on the activation of peroxisomal and mitochondrial enzymes such as, acyl-CoA oxidase (AOX) and carnitine palmitoyl-transferase I (CptI). A major cγ-activator of PPARα-mediated fatty acid oxidation, at the transcriptional level, is the PPARγ-coactivator-1 (PGC-1)149. Heart failure150, as well as myocardial infarction151, hypoxia152, 153, inflammatory markers such as IL-1β154, IL-6154, NF-κB155, and reactive oxygen species155 downregulate PPARα expression. PPARα gene expression levels and subsequent fatty acid oxidation are upregulated by estrogen related receptor (ERR)α, which acts in conjunction with PGC-1α and binds directly to the PPARα promoter156. In addition, ERRα gene expression is induced by PGC-1α156, 157 at the transcriptional level indicating a positive feedback loop in the coordination of PGC-1α and ERRα towards an increase of PPARα gene expression.
Fatty acid oxidation can also be triggered by nuclear receptors other than PPARα, such as PPARγ and PPARδ. Mice with constitutive PPARγ expression specifically in the hearts of PPARα−/− mice (αMHC-PPARγ/PPARα−/−) have elevated fatty acid oxidation levels, as well as improved cardiac function and survival as compared to the cardiolipotoxic αMHC-PPARγ mice although lipids still accumulate in the heart 66. These observations indicated that overexpression of PPARγ can substitute for PPARα suppression or even deletion. PPARγ agonists have also increased fatty acid oxidation in type 2 diabetic human muscle cells148. Co-activation of PPARδ and PGC-1α can induce hepatic fatty acid oxidation. Thus, one method to overcome a marked reduction in PPARα, which occurs in heart failure, might be via overexpression of other members of the PPAR gene family.
Both PPARα and PGC-1α gene expression levels are increased by AMPK158–160, which has itself been correlated with elevated cardiac transporter-mediated fatty acid uptake82 and oxidation161 levels. Mice that express a dominant negative form of AMPK cannot increase mitochondrial biogenesis in response to energy starvation162. Similarly, mice that express an inactive AMPK show impaired fasting-induced expression of lipid oxidative genes163. On the other hand mice carrying a constitutively active AMPK have elevated expression of fatty acid oxidation genes163–165.
CONCLUSIONS
Despite its preference for lipids, the heart is also vulnerable to the pathological effects of lipid overload. Surprisingly, pathways required for acquisition of cardiac lipids are relatively poorly understood. FA uptake requires CD36 and LpL acting either in series or in parallel. The heart, like other tissues, can obtain more lipid than can be oxidized or stored in a non-toxic manner. It is unknown which specific lipid or array of different lipids activate processes that cause cardiac dysfunction. Pharmacologic and dietary studies that include genetic manipulation have implicated ceramide, DAG, and FA/fatty acyl CoAs in apoptosis, defective insulin signaling, and mitochondrial dysfunction. In vivo, one process or a combination of dysfunctional processes might be needed for clinical pathology.
As studies proceed in this area, coincident investigations in other organs need to be observed in order to determine whether the process that leads to lipotoxic cardiomyopathy is common or heart-specific.
Acknowledgments
Funding
Dr. Konstantinos Drosatos is supported by a National Heart, Lung, and Blood Institute (NHLBI) K99/R00 award (1K99HL112853-01). Dr. P. Christian Schulze is supported by a NHLBI K23 award (K23HL24534).
Footnotes
Disclosure
Konstantinos Drosatos declares that he has no conflict of interest.
P. Christian Schulze declares that he has no conflict of interest.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Augustus AS, Buchanan J, Park TS, Hirata K, Noh HL, Sun J, Homma S, D’Armiento J, Abel ED, Goldberg IJ. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J Biol Chem. 2006;281:8716–8723. doi: 10.1074/jbc.M509890200. [DOI] [PubMed] [Google Scholar]
- 2.Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: Part i: General concepts. Circulation. 2002;105:1727–1733. doi: 10.1161/01.cir.0000012466.50373.e8. [DOI] [PubMed] [Google Scholar]
- 3.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 4.Ballard FB, Danforth WH, Naegle S, Bing RJ. Myocardial metabolism of fatty acids. J Clin Invest. 1960;39:717–723. doi: 10.1172/JCI104088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamboli A, O’Looney P, Vander Maten M, Vahouny GV. Comparative metabolism of free and esterified fatty acids by the perfused rat heart and rat cardiac myocytes. Biochim Biophys Acta. 1983;750:404–410. doi: 10.1016/0005-2760(83)90046-2. [DOI] [PubMed] [Google Scholar]
- 6.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 7.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem. 2004;279:47066–47075. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- 8•.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor pparalpha. Circulation research. 2010;107:233–241. doi: 10.1161/CIRCRESAHA.110.221713. This is a paper that sheds light on an important aspect of cardiac metabolism, which pertains to whether fatty acids that are used for cardiac ATP production are obtained from intracellular store of triglycerides or not. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kienesberger PC, Pulinilkunnil T, Sung MM, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR. Myocardial atgl overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol. 2012;32:740–750. doi: 10.1128/MCB.06470-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg IJ. Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. J Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- 11.Levak-Frank S, Hofmann W, Weinstock PH, Radner H, Sattler W, Breslow JL, Zechner R. Induced mutant mouse lines that express lipoprotein lipase in cardiac muscle, but not in skeletal muscle and adipose tissue, have normal plasma triglyceride and high-density lipoprotein-cholesterol levels. Proc Natl Acad Sci U S A. 1999;96:3165–3170. doi: 10.1073/pnas.96.6.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chajek T, Stein O, Stein Y. Pre- and post-natal development of lipoprotein lipase and hepatic triglyceride hydrolase activity in rat tissues. Atherosclerosis. 1977;26:549–561. doi: 10.1016/0021-9150(77)90122-8. [DOI] [PubMed] [Google Scholar]
- 13.Singh-Bist A, Komaromy MC, Kraemer FB. Transcriptional regulation of lipoprotein lipase in the heart during development in the rat. Biochem Biophys Res Commun. 1994;202:838–843. doi: 10.1006/bbrc.1994.2006. [DOI] [PubMed] [Google Scholar]
- 14.Semenkovich CF, Chen SH, Wims M, Luo CC, Li WH, Chan L. Lipoprotein lipase and hepatic lipase mrna tissue specific expression, developmental regulation, and evolution. J Lipid Res. 1989;30:423–431. [PubMed] [Google Scholar]
- 15.Ruge T, Wu G, Olivecrona T, Olivecrona G. Nutritional regulation of lipoprotein lipase in mice. Int J Biochem Cell Biol. 2004;36:320–329. doi: 10.1016/s1357-2725(03)00256-5. [DOI] [PubMed] [Google Scholar]
- 16.Pulinilkunnil T, Abrahani A, Varghese J, Chan N, Tang I, Ghosh S, Kulpa J, Allard M, Brownsey R, Rodrigues B. Evidence for rapid “metabolic switching” through lipoprotein lipase occupation of endothelial-binding sites. J Mol Cell Cardiol. 2003;35:1093–1103. doi: 10.1016/s0022-2828(03)00205-0. [DOI] [PubMed] [Google Scholar]
- 17.Sambandam N, Abrahani MA, Craig S, Al-Atar O, Jeon E, Rodrigues B. Metabolism of vldl is increased in streptozotocin-induced diabetic rat hearts. Am J Physiol Heart Circ Physiol. 2000;278:H1874–H1882. doi: 10.1152/ajpheart.2000.278.6.H1874. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Severson DL. Regulation of myocardial lipoprotein lipase activity by diabetes and thyroid hormones. Can J Physiol Pharmacol. 1994;72:1259–1264. doi: 10.1139/y94-180. [DOI] [PubMed] [Google Scholar]
- 19.Masuzaki H, Jingami H, Matsuoka N, Nakagawa O, Ogawa Y, Mizuno M, Yoshimasa Y, Yamamoto T, Nakao K. Regulation of very-low-density lipoprotein receptor in hypertrophic rat heart. Circ Res. 1996;78:8–14. doi: 10.1161/01.res.78.1.8. [DOI] [PubMed] [Google Scholar]
- 20.Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R, Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. 2004;279:25050–25057. doi: 10.1074/jbc.M401028200. [DOI] [PubMed] [Google Scholar]
- 21.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (lpl) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of cd36 knockout mice. J Biol Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton JA. Fatty acid transport: Difficult or easy? J Lipid Res. 1998;39:467–481. [PubMed] [Google Scholar]
- 24.Fukuchi K, Nozaki S, Yoshizumi T, Hasegawa S, Uehara T, Nakagawa T, Kobayashi T, Tomiyama Y, Yamashita S, Matsuzawa Y, Nishimura T. Enhanced myocardial glucose use in patients with a deficiency in long-chain fatty acid transport (cd36 deficiency) J Nucl Med. 1999;40:239–243. [PubMed] [Google Scholar]
- 25.Yamashita S, Hirano K, Kuwasako T, Janabi M, Toyama Y, Ishigami M, Sakai N. Physiological and pathological roles of a multi-ligand receptor cd36 in atherogenesis; insights from cd36-deficient patients. Mol Cell Biochem. 2007;299:19–22. doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
- 26.Kuang M, Febbraio M, Wagg C, Lopaschuk GD, Dyck JR. Fatty acid translocase/cd36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation. 2004;109:1550–1557. doi: 10.1161/01.CIR.0000121730.41801.12. [DOI] [PubMed] [Google Scholar]
- 27.Irie H, Krukenkamp IB, Brinkmann JF, Gaudette GR, Saltman AE, Jou W, Glatz JF, Abumrad NA, Ibrahimi A. Myocardial recovery from ischemia is impaired in cd36-null mice and restored by myocyte cd36 expression or medium-chain fatty acids. Proc Natl Acad Sci U S A. 2003;100:6819–6824. doi: 10.1073/pnas.1132094100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS, Goldberg IJ. Chylomicron- and vldl-derived lipids enter the heart through different pathways: In vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. 2010;285:37976–37986. doi: 10.1074/jbc.M110.174458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. Cd36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 30.Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, Mozell RL, Tan G, Stricker-Krongrad A, Hirsch DJ, Fillmore JJ, Liu ZX, Dong J, Cline G, Stahl A, Lodish HF, Shulman GI. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gimeno RE, Ortegon AM, Patel S, Punreddy S, Ge P, Sun Y, Lodish HF, Stahl A. Characterization of a heart-specific fatty acid transport protein. J Biol Chem. 2003;278:16039–16044. doi: 10.1074/jbc.M211412200. [DOI] [PubMed] [Google Scholar]
- 32.Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, Agarwal R, Yu S, Jiang H, Steinberg SF, Blaner WS, Koch WJ, Goldberg IJ. Cardiomyocyte lipids impair beta-adrenergic receptor function via pkc activation. Am J Physiol Endocrinol Metab. 2011;300:E489–499. doi: 10.1152/ajpendo.00569.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Sharov V, Sabbah HN, Tserng KY, Hoit BD, Ernsberger P, Young ME, Stanley WC. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. Am J Physiol Heart Circ Physiol. 2006;291:H38–44. doi: 10.1152/ajpheart.01295.2005. [DOI] [PubMed] [Google Scholar]
- 35.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase c isoform beta ii and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newton AC, Johnson JE. Protein kinase c: A paradigm for regulation of protein function by two membrane-targeting modules. Biochim Biophys Acta. 1998;1376:155–172. doi: 10.1016/s0304-4157(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 38.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase c. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 39.Cheng D, Meegalla RL, He B, Cromley DA, Billheimer JT, Young PR. Human acyl-coa:Diacylglycerol acyltransferase is a tetrameric protein. Biochem J. 2001;359:707–714. doi: 10.1042/0264-6021:3590707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, Farese RV., Jr Cloning of dgat2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem. 2001;276:38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. Dgat1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–36323. doi: 10.1074/jbc.M109.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buhman KK, Smith SJ, Stone SJ, Repa JJ, Wong JS, Knapp FF, Jr, Burri BJ, Hamilton RL, Abumrad NA, Farese RV., Jr Dgat1 is not essential for intestinal triacylglycerol absorption or chylomicron synthesis. J Biol Chem. 2002;277:25474–25479. doi: 10.1074/jbc.M202013200. [DOI] [PubMed] [Google Scholar]
- 43.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking dgat. Nat Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 44.Alpert MA. Obesity cardiomyopathy: Pathophysiology and evolution of the clinical syndrome. Am J Med Sci. 2001;321:225–236. doi: 10.1097/00000441-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 46.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 47.Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, Gropler RJ. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47:598–604. doi: 10.1016/j.jacc.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 48.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: A 1h-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 49.O’Donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol. 2008;44:315–322. doi: 10.1016/j.yjmcc.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, Yang Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation. 2012;126:1705–1716. doi: 10.1161/CIRCULATIONAHA.111.075978. This paper shows the importance of fatty acid oxidation in preventing cardiac hypertrophy driven by severe pressure overload and indicates caution that the clinical use of fatty acid oxidation inhibitors should be applied with. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Kolwicz SC, Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl coa carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728–738. doi: 10.1161/CIRCRESAHA.112.268128. This paper shows that ablation of the ACC2 enzyme that regulates the formation of a fatty acid oxidation inhibitor, malonyl-CoA, improves fatty acid oxidation in pressure-overload hearts and leads to attenuation of cardiac hypertrophy with a significant reduction in fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Essop MF, Camp HS, Choi CS, Sharma S, Fryer RM, Reinhart GA, Guthrie PH, Bentebibel A, Gu Z, Shulman GI, Taegtmeyer H, Wakil SJ, Abu-Elheiga L. Reduced heart size and increased myocardial fuel substrate oxidation in acc2 mutant mice. Am J Physiol Heart Circ Physiol. 2008;295:H256–265. doi: 10.1152/ajpheart.91489.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rupp H, Jacob R. Metabolically-modulated growth and phenotype of the rat heart. Eur Heart J. 1992;13 (Suppl D):56–61. doi: 10.1093/eurheartj/13.suppl_d.56. [DOI] [PubMed] [Google Scholar]
- 54.Bristow M. Etomoxir: A new approach to treatment of chronic heart failure. Lancet. 2000;356:1621–1622. doi: 10.1016/S0140-6736(00)03149-4. [DOI] [PubMed] [Google Scholar]
- 55.Dobbins RL, Szczepaniak LS, Bentley B, Esser V, Myhill J, McGarry JD. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 56.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2005;288:H2102–2110. doi: 10.1152/ajpheart.00935.2004. [DOI] [PubMed] [Google Scholar]
- 58.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 59.Koonen DP, Febbraio M, Bonnet S, Nagendran J, Young ME, Michelakis ED, Dyck JR. Cd36 expression contributes to age-induced cardiomyopathy in mice. Circulation. 2007;116:2139–2147. doi: 10.1161/CIRCULATIONAHA.107.712901. [DOI] [PubMed] [Google Scholar]
- 60.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 62.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by pparalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for pparalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ, Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-alpha activators. Circulation. 2010;121:426–435. doi: 10.1161/CIRCULATIONAHA.109.888735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of ppargamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ. Ppargamma-induced cardiolipotoxicity in mice is ameliorated by pparalpha deficiency despite increases in fatty acid oxidation. J Clin Invest. 2010;120:3443–3454. doi: 10.1172/JCI40905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kantor PF, Dyck JR, Lopaschuk GD. Fatty acid oxidation in the reperfused ischemic heart. Am J Med Sci. 1999;318:3–14. doi: 10.1097/00000441-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 68.Nohammer C, Brunner F, Wolkart G, Staber PB, Steyrer E, Gonzalez FJ, Zechner R, Hoefler G. Myocardial dysfunction and male mortality in peroxisome proliferator-activated receptor alpha knockout mice overexpressing lipoprotein lipase in muscle. Lab Invest. 2003;83:259–269. doi: 10.1097/01.lab.0000053916.61772.ca. [DOI] [PubMed] [Google Scholar]
- 69.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 70.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors pparbeta/delta and pparalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Georgiadi A, Lichtenstein L, Degenhardt T, Boekschoten MV, van Bilsen M, Desvergne B, Muller M, Kersten S. Induction of cardiac angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ Res. 2010;106:1712–1721. doi: 10.1161/CIRCRESAHA.110.217380. [DOI] [PubMed] [Google Scholar]
- 72••.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. Atgl-mediated fat catabolism regulates cardiac mitochondrial function via ppar-alpha and pgc-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. This paper shows that fatty acids that are released via ATGL-mediated lipolysis of cardiac triglycerides are essential for PPARα activation and fatty acid oxidation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci U S A. 2000;97:787–792. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32:1552–1562. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hickson-Bick DL, Buja ML, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol. 2000;32:511–519. doi: 10.1006/jmcc.1999.1098. [DOI] [PubMed] [Google Scholar]
- 76.Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2000;279:H2124–2132. doi: 10.1152/ajpheart.2000.279.5.H2124. [DOI] [PubMed] [Google Scholar]
- 77.Weiss B, Stoffel W. Human and murine serine-palmitoyl-coa transferase--cloning, expression and characterization of the key enzyme in sphingolipid synthesis. Eur J Biochem. 1997;249:239–247. doi: 10.1111/j.1432-1033.1997.00239.x. [DOI] [PubMed] [Google Scholar]
- 78.Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- 79.Merrill AH., Jr De novo sphingolipid biosynthesis: A necessary, but dangerous, pathway. J Biol Chem. 2002;277:25843–25846. doi: 10.1074/jbc.R200009200. [DOI] [PubMed] [Google Scholar]
- 80.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hardie DG, Carling D, Carlson M. The amp-activated/snf1 protein kinase subfamily: Metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 82.Chabowski A, Momken I, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Bonen A. Prolonged ampk activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol Cell Biochem. 2006;288:201–212. doi: 10.1007/s11010-006-9140-8. [DOI] [PubMed] [Google Scholar]
- 83.Habets DD, Coumans WA, Voshol PJ, den Boer MA, Febbraio M, Bonen A, Glatz JF, Luiken JJ. Ampk-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal cd36. Biochem Biophys Res Commun. 2007;355:204–210. doi: 10.1016/j.bbrc.2007.01.141. [DOI] [PubMed] [Google Scholar]
- 84.Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, Lu S, Jiang XC. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoe-deficient mice. J Biol Chem. 2005;280:10284–10289. doi: 10.1074/jbc.M412348200. [DOI] [PubMed] [Google Scholar]
- 85.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 86.Hickson-Bick DL, Sparagna GC, Buja LM, McMillin JB. Palmitate-induced apoptosis in neonatal cardiomyocytes is not dependent on the generation of ros. Am J Physiol Heart Circ Physiol. 2002;282:H656–664. doi: 10.1152/ajpheart.00726.2001. [DOI] [PubMed] [Google Scholar]
- 87.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and enos activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in c57bl/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 89.Iozzo P, Chareonthaitawee P, Dutka D, Betteridge DJ, Ferrannini E, Camici PG. Independent association of type 2 diabetes and coronary artery disease with myocardial insulin resistance. Diabetes. 2002;51:3020–3024. doi: 10.2337/diabetes.51.10.3020. [DOI] [PubMed] [Google Scholar]
- 90.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 91.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 92.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. 2006;55:466–473. doi: 10.2337/diabetes.55.02.06.db05-1164. [DOI] [PubMed] [Google Scholar]
- 93.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 94.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 96.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 97.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter glut4 translocation and akt kinase activity by ceramide. Mol Cell Biol. 1998;18:5457–5464. doi: 10.1128/mcb.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hajduch E, Balendran A, Batty IH, Litherland GJ, Blair AS, Downes CP, Hundal HS. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase b leading to a loss in downstream signalling in l6 skeletal muscle cells. Diabetologia. 2001;44:173–183. doi: 10.1007/s001250051596. [DOI] [PubMed] [Google Scholar]
- 99.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining akt in an inactive dephosphorylated state. Diabetes. 2001;50:2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 100.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of akt/protein kinase b. J Biol Chem. 2004;279:36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 101.Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280:20148–20153. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- 102.Kellerer M, Mushack J, Seffer E, Mischak H, Ullrich A, Haring HU. Protein kinase c isoforms alpha, delta and theta require insulin receptor substrate-1 to inhibit the tyrosine kinase activity of the insulin receptor in human kidney embryonic cells (hek 293 cells) Diabetologia. 1998;41:833–838. doi: 10.1007/s001250050995. [DOI] [PubMed] [Google Scholar]
- 103.Cortright RN, Azevedo JL, Jr, Zhou Q, Sinha M, Pories WJ, Itani SI, Dohm GL. Protein kinase c modulates insulin action in human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;278:E553–562. doi: 10.1152/ajpendo.2000.278.3.E553. [DOI] [PubMed] [Google Scholar]
- 104.Motley ED, Kabir SM, Eguchi K, Hicks AL, Gardner CD, Reynolds CM, Frank GD, Eguchi S. Protein kinase c inhibits insulin-induced akt activation in vascular smooth muscle cells. Cell Mol Biol (Noisy-le-grand) 2001;47:1059–1062. [PubMed] [Google Scholar]
- 105.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (irs-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 106.Farese RV, Sajan MP, Yang H, Li P, Mastorides S, Gower WR, Jr, Nimal S, Choi CS, Kim S, Shulman GI, Kahn CR, Braun U, Leitges M. Muscle-specific knockout of pkc-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest. 2007;117:2289–2301. doi: 10.1172/JCI31408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–2853. doi: 10.1161/CIRCULATIONAHA.111.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, Tandon NN, Van Der Vusse GJ, Bonen A, Glatz JF. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of fat/cd36. Diabetes. 2002;51:3113–3119. doi: 10.2337/diabetes.51.10.3113. [DOI] [PubMed] [Google Scholar]
- 109.Bastie CC, Nahle Z, McLoughlin T, Esser K, Zhang W, Unterman T, Abumrad NA. Foxo1 stimulates fatty acid uptake and oxidation in muscle cells through cd36-dependent and -independent mechanisms. J Biol Chem. 2005;280:14222–14229. doi: 10.1074/jbc.M413625200. [DOI] [PubMed] [Google Scholar]
- 110.Nishizuka Y. Protein kinase c and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 111.Aschrafi A, Franzen R, Shabahang S, Fabbro D, Pfeilschifter J, Huwiler A. Ceramide induces translocation of protein kinase c-alpha to the golgi compartment of human embryonic kidney cells by interacting with the c2 domain. Biochim Biophys Acta. 2003;1634:30–39. doi: 10.1016/j.bbalip.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 112.Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase c zeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- 113.Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M. Ceramide recruits and activates protein kinase c zeta (pkc zeta) within structured membrane microdomains. J Biol Chem. 2007;282:12450–12457. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- 114.Galve-Roperh I, Haro A, Diaz-Laviada I. Ceramide-induced translocation of protein kinase c zeta in primary cultures of astrocytes. FEBS Lett. 1997;415:271–274. doi: 10.1016/s0014-5793(97)00985-x. [DOI] [PubMed] [Google Scholar]
- 115.Huang HW, Goldberg EM, Zidovetzki R. Ceramides modulate protein kinase c activity and perturb the structure of phosphatidylcholine/phosphatidylserine bilayers. Biophys J. 1999;77:1489–1497. doi: 10.1016/S0006-3495(99)76996-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huwiler A, Fabbro D, Pfeilschifter J. Selective ceramide binding to protein kinase c-alpha and -delta isoenzymes in renal mesangial cells. Biochemistry. 1998;37:14556–14562. doi: 10.1021/bi981401i. [DOI] [PubMed] [Google Scholar]
- 117.Kajimoto T, Ohmori S, Shirai Y, Sakai N, Saito N. Subtype-specific translocation of the delta subtype of protein kinase c and its activation by tyrosine phosphorylation induced by ceramide in hela cells. Mol Cell Biol. 2001;21:1769–1783. doi: 10.1128/MCB.21.5.1769-1783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kajimoto T, Shirai Y, Sakai N, Yamamoto T, Matsuzaki H, Kikkawa U, Saito N. Ceramide-induced apoptosis by translocation, phosphorylation, and activation of protein kinase cdelta in the golgi complex. J Biol Chem. 2004;279:12668–12676. doi: 10.1074/jbc.M312350200. [DOI] [PubMed] [Google Scholar]
- 119.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase b (pkb)/akt by a pkczeta-dependent mechanism. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang G, Krishnamurthy K, Umapathy NS, Verin AD, Bieberich E. The carboxyl-terminal domain of atypical protein kinase czeta binds to ceramide and regulates junction formation in epithelial cells. J Biol Chem. 2009;284:14469–14475. doi: 10.1074/jbc.M808909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang G, Silva J, Krishnamurthy K, Tran E, Condie BG, Bieberich E. Direct binding to ceramide activates protein kinase czeta before the formation of a pro-apoptotic complex with par-4 in differentiating stem cells. J Biol Chem. 2005;280:26415–26424. doi: 10.1074/jbc.M501492200. [DOI] [PubMed] [Google Scholar]
- 122.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, King GL. Targeted overexpression of protein kinase c beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A. 1997;94:9320–9325. doi: 10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jalili T, Manning J, Kim S. Increased translocation of cardiac protein kinase c beta2 accompanies mild cardiac hypertrophy in rats fed saturated fat. J Nutr. 2003;133:358–361. doi: 10.1093/jn/133.2.358. [DOI] [PubMed] [Google Scholar]
- 124.Wang J, Liu X, Sentex E, Takeda N, Dhalla NS. Increased expression of protein kinase c isoforms in heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H2277–2287. doi: 10.1152/ajpheart.00142.2002. [DOI] [PubMed] [Google Scholar]
- 125.Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase c-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 126.Narayan P, Valdivia HH, Mentzer RM, Jr, Lasley RD. Adenosine a1 receptor stimulation antagonizes the negative inotropic effects of the pkc activator dioctanoylglycerol. J Mol Cell Cardiol. 1998;30:913–921. doi: 10.1006/jmcc.1998.0648. [DOI] [PubMed] [Google Scholar]
- 127.Connelly KA, Kelly DJ, Zhang Y, Prior DL, Advani A, Cox AJ, Thai K, Krum H, Gilbert RE. Inhibition of protein kinase c-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ Heart Fail. 2009;2:129–137. doi: 10.1161/CIRCHEARTFAILURE.108.765750. [DOI] [PubMed] [Google Scholar]
- 128.Liu Q, Chen X, Macdonnell SM, Kranias EG, Lorenz JN, Leitges M, Houser SR, Molkentin JD. Protein kinase c{alpha}, but not pkc{beta} or pkc{gamma}, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Pkc-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 130.Hambleton M, Hahn H, Pleger ST, Kuhn MC, Klevitsky R, Carr AN, Kimball TF, Hewett TE, Dorn GW, 2nd, Koch WJ, Molkentin JD. Pharmacological- and gene therapy-based inhibition of protein kinase calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–582. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Boyle AJ, Kelly DJ, Zhang Y, Cox AJ, Gow RM, Way K, Itescu S, Krum H, Gilbert RE. Inhibition of protein kinase c reduces left ventricular fibrosis and dysfunction following myocardial infarction. J Mol Cell Cardiol. 2005;39:213–221. doi: 10.1016/j.yjmcc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 132.Kolter T, Uphues I, Eckel J. Molecular analysis of insulin resistance in isolated ventricular cardiomyocytes of obese zucker rats. Am J Physiol. 1997;273:E59–67. doi: 10.1152/ajpendo.1997.273.1.E59. [DOI] [PubMed] [Google Scholar]
- 133.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 134.Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin a/c gene. Circulation. 2011;123:53–61. doi: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-jun nh2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res. 2003;92:136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 136.Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, Izumo S. Direct activation of mitochondrial apoptosis machinery by c-jun n-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 137.Miller TA, LeBrasseur NK, Cote GM, Trucillo MP, Pimentel DR, Ido Y, Ruderman NB, Sawyer DB. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem Biophys Res Commun. 2005;336:309–315. doi: 10.1016/j.bbrc.2005.08.088. [DOI] [PubMed] [Google Scholar]
- 138.Hreniuk D, Garay M, Gaarde W, Monia BP, McKay RA, Cioffi CL. Inhibition of c-jun n-terminal kinase 1, but not c-jun n-terminal kinase 2, suppresses apoptosis induced by ischemia/reoxygenation in rat cardiac myocytes. Mol Pharmacol. 2001;59:867–874. doi: 10.1124/mol.59.4.867. [DOI] [PubMed] [Google Scholar]
- 139.Drosatos K, Drosatos-Tampakaki Z, Khan R, Homma S, Schulze PC, Zannis VI, Goldberg IJ. Inhibition of c-jun-n-terminal kinase increases cardiac ppar{alpha} expression and fatty acid oxidation and prevents lps-induced heart dysfunction. J Biol Chem. 2011;286:36331–36339. doi: 10.1074/jbc.M111.272146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu XX, Murray SF, Watts L, Booten SL, Tokorcheck J, Monia BP, Bhanot S. Reduction of jnk1 expression with antisense oligonucleotide improves adiposity in obese mice. Am J Physiol Endocrinol Metab. 2008;295:E436–445. doi: 10.1152/ajpendo.00629.2007. [DOI] [PubMed] [Google Scholar]
- 141.Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ET, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1a-1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 143.Turner MD. Fatty acyl coa-mediated inhibition of endoplasmic reticulum assembly. Biochim Biophys Acta. 2004;1693:1–4. doi: 10.1016/j.bbamcr.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 144.Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY, Yang P. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating er stress response. International journal of medical sciences. 2011;8:564–572. doi: 10.7150/ijms.8.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, Kelly DP. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005;1:133–144. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 146.Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE, Yang Q. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem Biophys Res Commun. 2004;313:277–286. doi: 10.1016/j.bbrc.2003.11.127. [DOI] [PubMed] [Google Scholar]
- 147.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by ppar gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 148.Cha BS, Ciaraldi TP, Park KS, Carter L, Mudaliar SR, Henry RR. Impaired fatty acid metabolism in type 2 diabetic skeletal muscle cells is reversed by ppargamma agonists. Am J Physiol Endocrinol Metab. 2005;289:E151–159. doi: 10.1152/ajpendo.00141.2004. [DOI] [PubMed] [Google Scholar]
- 149.Vega RB, Huss JM, Kelly DP. The coactivator pgc-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett. 2003;8:49–53. [PubMed] [Google Scholar]
- 151.Masamura K, Tanaka N, Yoshida M, Kato M, Kawai Y, Oida K, Miyamori I. Myocardial metabolic regulation through peroxisome proliferator-activated receptor alpha after myocardial infarction. Exp Clin Cardiol. 2003;8:61–66. [PMC free article] [PubMed] [Google Scholar]
- 152.Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001;166:7543–7548. doi: 10.4049/jimmunol.166.12.7543. [DOI] [PubMed] [Google Scholar]
- 153.Razeghi P, Young ME, Abbasi S, Taegtmeyer H. Hypoxia in vivo decreases peroxisome proliferator-activated receptor alpha-regulated gene expression in rat heart. Biochem Biophys Res Commun. 2001;287:5–10. doi: 10.1006/bbrc.2001.5541. [DOI] [PubMed] [Google Scholar]
- 154.Parmentier JH, Schohn H, Bronner M, Ferrari L, Batt AM, Dauca M, Kremers P. Regulation of cyp4a1 and peroxisome proliferator-activated receptor alpha expression by interleukin-1beta, interleukin-6, and dexamethasone in cultured fetal rat hepatocytes. Biochem Pharmacol. 1997;54:889–898. doi: 10.1016/s0006-2952(97)00256-6. [DOI] [PubMed] [Google Scholar]
- 155.Cabrero A, Alegret M, Sanchez RM, Adzet T, Laguna JC, Carrera MV. Increased reactive oxygen species production down-regulates peroxisome proliferator-activated alpha pathway in c2c12 skeletal muscle cells. J Biol Chem. 2002;277:10100–10107. doi: 10.1074/jbc.M110321200. [DOI] [PubMed] [Google Scholar]
- 156.Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator pgc-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (erralpha) J Biol Chem. 2003;278:9013–9018. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- 158.Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, Lee KU, Park JY. Ampk activation increases fatty acid oxidation in skeletal muscle by activating pparalpha and pgc-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 159.Meng RS, Pei ZH, Yin R, Zhang CX, Chen BL, Zhang Y, Liu D, Xu AL, Dong YG. Adenosine monophosphate-activated protein kinase inhibits cardiac hypertrophy through reactivating peroxisome proliferator-activated receptor-alpha signaling pathway. Eur J Pharmacol. 2009;620:63–70. doi: 10.1016/j.ejphar.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 160.Ravnskjaer K, Boergesen M, Dalgaard LT, Mandrup S. Glucose-induced repression of pparalpha gene expression in pancreatic beta-cells involves pp2a activation and ampk inactivation. J Mol Endocrinol. 2006;36:289–299. doi: 10.1677/jme.1.01965. [DOI] [PubMed] [Google Scholar]
- 161.Makinde AO, Gamble J, Lopaschuk GD. Upregulation of 5′-amp-activated protein kinase is responsible for the increase in myocardial fatty acid oxidation rates following birth in the newborn rabbit. Circ Res. 1997;80:482–489. doi: 10.1161/01.res.80.4.482. [DOI] [PubMed] [Google Scholar]
- 162.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. Amp kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Long YC, Barnes BR, Mahlapuu M, Steiler TL, Martinsson S, Leng Y, Wallberg-Henriksson H, Andersson L, Zierath JR. Role of amp-activated protein kinase in the coordinated expression of genes controlling glucose and lipid metabolism in mouse white skeletal muscle. Diabetologia. 2005;48:2354–2364. doi: 10.1007/s00125-005-1962-5. [DOI] [PubMed] [Google Scholar]
- 164.Barnes BR, Long YC, Steiler TL, Leng Y, Galuska D, Wojtaszewski JF, Andersson L, Zierath JR. Changes in exercise-induced gene expression in 5′-amp-activated protein kinase gamma3-null and gamma3 r225q transgenic mice. Diabetes. 2005;54:3484–3489. doi: 10.2337/diabetes.54.12.3484. [DOI] [PubMed] [Google Scholar]
- 165.Garcia-Roves PM, Osler ME, Holmstrom MH, Zierath JR. Gain-of-function r225q mutation in amp-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008;283:35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]