

Abstract
Annexins, found in most eukaryotic species, are cytosolic proteins that are able to bind negatively-charged phospholipids in a calcium-dependent manner. Annexin A4 (AnxA4) has been implicated in diverse cellular processes, including the regulation of exocytosis and ion-transport; however, its precise mechanistic role is not fully understood. AnxA4 has been shown to aggregate on lipid layers upon Ca2+ binding in vitro, a characteristic that may be critical for its function. We have utilized advanced fluorescence microscopy to discern details on the mobility and self-assembly of AnxA4 after Ca2+ influx at the plasma membrane in living cells. Total internal reflection microscopy in combination with Förster resonance energy transfer reveals that there is a delay between initial plasma membrane binding and the beginning of self-assembly and this process continues after the cytoplasmic pool has completely relocated. Number-and-brightness analysis suggests that the predominant membrane bound mobile form of the protein is trimeric. There also exists a pool of AnxA4 that forms highly immobile aggregates at the membrane. Fluorescence recovery after photobleaching suggests that the relative proportion of these two forms varies and is correlated with membrane morphology.
Introduction
Annexins make up a pervasive, structurally related class of proteins found in most eukaryotic species. There are 12 annexin family members expressed in mammalian cells (1); these are involved in a panoply of cellular functions such endo- and exocytosis, actin configuration, signaling, and plasma membrane repair (2). Although their precise mechanistic roles in many of these processes have yet to be completely elucidated, a shared trait of almost all annexins is their ability to bind negatively-charged phospholipids in a calcium-dependent manner (3–5). The mammalian annexin family member A4 (AnxA4) is found primarily in epithelial cells (6) and has been implicated in a wide range of cellular processes, including membrane aggregation (7), synaptic exocytosis (8), and the downregulation of the transcription factor NF-κB (9). Additionally, alterations in AnxA4 expression have been associated with certain types of cancer and seem to contribute to increased invasiveness and resistance to chemotherapy, perhaps by enhancing drug efflux (10,11). AnxA4 also negatively regulates the Ca2+-activated Cl− conductance by preventing the phosphorylation of the ion channel by the calmodulin-dependent protein kinase II (12,13). AnxA4 is thought to self-associate upon Ca2+-induced membrane binding, restricting the mobility of phospholipids and membrane-bound components (7,14,15). This characteristic may be a key factor in this protein’s ability to affect membrane architecture in ways that lead to tethering and hindrance of transport channels and signaling components. However, the molecular details of this process have thus far only been observed in vitro, using purified proteins assembled on artificial lipid layers (7,16–19). X-ray crystallography, electron microscopy, and atomic force microscopy have shown that AnxA4 crystallizes as a trimer on phospholipid layers when bound to Ca2+ and may, under certain conditions, associate in larger order arrays on synthetic membranes. Although a detailed model for AnxA4 self-assembly has been constructed based on these in vitro studies, it remains to be determined whether this in vitro model applies to the situation in living cells. To get a better biophysical understanding of this protein’s role in cell biology, we have utilized a range of fluorescent microscopy techniques to determine whether AnxA4 assembles in a way that enables it to scaffold membrane and signaling components. Here, we describe our findings regarding the self-association and mobility of AnxA4 at the plasma membrane.
Materials and Methods
Constructs
The annexinA4-eYFP construct was a kind gift from Carsten Schultz (14). The AgeI and BsrGI sites were used to swap eYFP for mCherry (a kind gift of Roger Tsien (20)), superGFP2 (sGFP2), superYFP2 (sYFP2), and mTurquoise (mTq) (21). The tandem sGFP2 was constructed by PCR-based cloning of the sGFP2 with the forward primer
and the reverse primer
This introduced a recognition site for the restriction endonuclease Pfc23II, which produces BsrGI compatible overhangs. This latter site was used to clone the sGFP2 PCR product into the sGFP2_C1 plasmid. All constructs were confirmed by sequencing.
Cell culture and transfection
HeLa cells were obtained from the American Tissue Culture Collection and N1E-115 cells were obtained from the European Collection of Cell Cultures. Cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with GlutaMAX and 10% fetal bovine serum (Invitrogen, Bleiswijk, NL) and grown for at least two days in phenol-free media before imaging. Cells were plated on 24-mm No. 1 round cover glasses (Menzel-Gläser, Braunschweig, Germany) in six-well plates. Transfections were done with Lipofectamine 2000 reagent according to the manufacturer’s protocol (Invitrogen). Twenty-four hours after transfection, cells were imaged live in microscopy medium (137 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 20 mM d-glucose, 20 mM HEPES). Elevation of intracellular Ca2+ levels was achieved by adding ionomycin (Sigma, St. Louis, MO) at a final concentration of 10 μM to the imagining medium.
TIR FLIM-FRET and ratiometric-FRET
To measure the self-assembly of AnxA4 at the plasma membrane, we utilized fluorescence lifetime imaging microscopy (FLIM), which is an intensity-independent technique that is much less sensitive to artifacts that can potentially plague intensity-based Förster resonance energy transfer (FRET) measurements. By combining this method with a total internal reflection (TIR) modality, we were able to confine our analysis to regions within ∼80 nm of the cover-glass interface (22). The theory behind FLIM has been described in detail previously in Gadella et al. (23). Our TIR-FLIM instrumental setup is outlined in Gadella (24). Briefly, an Axiovert 200M inverted microscope with a α-Plan Fluar 100× 1.45 NA oil-immersion objective (Zeiss, Jena, Germany) was utilized. By off-axis coupling of the laser input into the microscope, the excitation light has an offset with respect to the back focal-plane and is totally reflected at the glass/water interface at the cover glass (22). Samples were excited using a 514-nm Argon laser (CVI Melles Griot, Carlsbad, CA) modulated at 75.1 MHz by an acousto-optic modulator; fluorescence was selected with a BP 530–560 emission filter (Chroma Technology, Bellows Falls, VT). FLIM stacks of 12 phase steps were acquired in permutated recording order to reduce artifacts due to photobleaching (25). Software for acquisition, processing, and analysis of the data was written in MATLAB Ver. 6.1 (The MathWorks, Natick, MA). Figures and movies were generated using a custom-written macro for the software IMAGEJ (National Institutes of Health, Bethesda, MD).
To gain the time-resolution necessary to track the kinetics of AnxA4 self-association, we performed ratio-FRET experiments with a setup that allowed us to acquire donor and acceptor images simultaneously. This imaging was performed on an inverted Eclipse Ti microscope (Nikon, Melville, NY) equipped with a total internal reflection fluorescence (TIRF) system using a 60× ApoTIRF 1.49 NA oil objective. Samples were excited with a continuous-wave DPSS 445-ST-I (450 nm, 500 mW; Sintec Optronics Technology, Singapore). Excitation light was directed through a multi-pass 440/514/561 nm dichroic mirror (Chroma Technology). The emission light passes through an Opto Split II image splitter (Cairn Research, Edinburgh, Scotland) equipped with a 506-nm beam splitter and 479/40 and 545/50 emission filters (Chroma Technology) to project dual images (mTurquoise (mTq) channel and superYFP2 (sYFP2) channel) onto the chip of an iXon 897 EMCCD (Andor Scientific, South Windsor, CT). Ratio-FRET intensity curves were calculated from average intensity values from split wavelength images. The values of the yellow channel were corrected for bleedthrough of the mTq emission by subtracting 40% of the values of the blue channel (percentage was acquired by a mTq-only labeled sample). Average intensities were subsequently divided with the average of eight frames before stimulation for normalization to 1. FRET ratios were calculated by division of the normalized average intensities of the corrected yellow channel by those of the cyan channel. The membrane localization curve was obtained by the summation of the uncorrected average intensities of the cyan and yellow channel.
Number-and-brightness analysis
To determine the stoichiometry of the AnxA4 self-association, we made use of number-and-brightness analysis, a fluctuation-based method that, unlike FRET, can provide information on the number of proteins that are physically associated in a complex (26). Images were acquired on an Olympus IX81 equipped with a FluorView FV1000 scan and confocal detection head (Olympus, Melville, NY) coupled to a custom-made detection unit containing MPD avalanche photodiodes (PicoQuant, Westfield, MA). The 488-nm excitation light from an CW Argon laser (CVI Melles Griot) was focused with a 60× UPLS Apochromat, 1.2 NA water immersion objective into the sample. Fluorescence passed through a 525DF45 band-pass filter (Chroma Technology) placed just before the detector. The laser power was typically set at ∼5 μW. For a typical measurement, images of 128 × 128 pixels were acquired, having a pixel dimension of either 207 or 414 nm. The pixel dwell time was set at 100 μs. These parameters were established by determining what was necessary to detect an approximate twofold difference between a cytosolic monomeric sGFP2 and a cytosolic tandem-dimer sGFP2 expressed in HeLa cells (Table 2 and see Table S1 in the Supporting Material).
Table 2.
Molecular brightness analysis of controls and AnxA4-sGFP2
| Sample | Brightness (kHz/bin) | Mean ± SE | Fold | n |
|---|---|---|---|---|
| sGFP2 (control) | 0.035 | 0.003 | 1.00 | 6 |
| Tandem-sGFP2 (control) | 0.062 | 0.005 | 1.77 | 6 |
| AnxA4-sGFP2 (cytoplasm) | 0.032 | 0.004 | 0.91 | 7 |
| AnxA4-sGFP2 + ionomycin (plasma membrane) | 0.108 | 0.005 | 3.09 | 7 |
AnxA4-sGFP2 values were measured before and after ionomycin-induced membrane binding. Fold values are normalized to monomeric sGFP2.
Image time-stacks of at least 100 image scans were collected at time intervals of 1.95 s per frame. The PicoQuant .pt3 data-files containing photon arrival times were subsequently converted to intensity image-time Tiff stacks using an in-house developed program that was written in C++ (Qt 4.7.0; http://qt.digia.com/Product/). The stacks were subsequently analyzed using a custom-written number-and-brightness macro in the software IMAGEJ (National Institutes of Health), in which the equations from Digman et al. (26) were implemented. The apparent brightness Bx,y for each pixel at position (x, y) was calculated using
| (1) |
where
denotes the estimated time-averaged intensity at position (x,y) and
denotes the estimated variance of the intensity at position (x, y). Because we made use of true photon-counting avalanche photodiode detectors, with offset = 0 and S-factor, S = 1, the corrections for detector offset and S-factor (27) that are necessary for analog photomultiplier tubes were not required. Therefore, the true molecular brightness, at position (x, y), can be directly estimated from
| (2) |
Photobleaching and minor cell movement will lead to spurious increase of the variance. Therefore, a detrending filter was applied based on the time course of averaged images using the software IMAGEJ plugin written by Jay Unruh (http://research.stowers-institute.org/imagejplugins/). Despite the application of this filter, regions of more marked cell movement still showed artifacts due to increased variance; therefore, cells or regions of cells that displayed obvious movement artifacts were excluded from the analysis. Average molecular brightness values and corresponding standard errors of the mean were obtained from manually selected regions of interest (ROIs) in each cell, hence .
Confocal microscopy and FRAP
We made use of the quantitatively rigorous strategy of determining the spatial characteristics of the photobleaching and recovery (28) to discern, biologically, import parameters on plasma membrane-bound AnxA4, such as diffusion speed, proportionality of mobile/immobile fraction, and association kinetics (binding/unbinding). Confocal imaging was performed on an inverted Eclipse Ti microscope (Nikon) equipped with a 60× ApoTIRF 1.49 oil-immersion objective and a model No. CSU X-1 spinning disk (Yokagawa, Tokyo, Japan). HeLa cells expressing AnxA4-sGFP2 were imaged every 5 s for up to 30 min during which period 10 μM of ionomycin was added to the imaging media (see above) to raise intracellular Ca2+ levels and induce AnxA4 membrane binding. To take advantage of the Olympus tornado-scanning feature, fluorescence recovery after photobleaching (FRAP) experiments were performed on an inverted IX81 with FluorView FV1000 scan and confocal detection head using a 60× UPLS Apochromat, 1.2 NA water immersion objective (Olympus, Melville, NY). Lasers and filters used were essentially identical to those in the number-and-brightness analysis experiments. The upper plasma membrane was imaged (128 × 128 pixels with xy dimensions of 207 nm) at a scan speed of 10 μs/pixel (0.331 s/frame). A circular region with a 20-pixel radius (∼4.14 μm) was selected and bleached at 100% laser-power (∼500 μW) in tornado-scanning mode (bleaching from the center outwards) for a total bleach duration of 4 s. In total, 800 images were collected including 50 prebleach frames. Because the laser did not fully return to the lower laser power that was used for imaging just after the bleach, the first frame after bleaching was omitted from the analysis. A background intensity trace, Ib(t), was obtained from a ROI outside of the cell and a reference trace, Iref (t), was obtained from a ROI at a position distant from the bleached region. The raw fluorescence intensity I(x,y,t) was subsequently normalized using (29)
The center of the bleach area was estimated by averaging fitted (x0, y0) values that were obtained by fitting the following function to eight post-bleach frames:
| (3) |
with radius
Radial intensity profiles In(r,t), ranging from r = 0 up to r ∼8.28 μm were calculated by averaging the fluorescent intensity in 20 evenly spaced concentric rings with center and ring-width 0.414 μm. For each concentric ring also the standard error of the mean (mean ± SE), was calculated. The radial profiles were spatially normalized with the time-averaged radial profile of the pre-bleach frames, such that the prebleach fluorescent intensity profiles are normalized to unity. Assuming that the system is at steady state and the distribution of molecules (including binding sites) is approximately uniform at the macroscopic level, the fluorescence recovery due to lateral diffusion and lateral binding to and unbinding from immobile structures at the membrane can be approximated by the following two differential equations (28,30,31):
| (4a) |
| (4b) |
where f(r,t) and b(r,t) denote the laterally diffusing and bound fluorescent fractions, respectively; (s−1) denotes the off-rate of binding; (s−1) denotes the apparent on-rate of binding; and D (μm2·s−1) denotes the lateral diffusion coefficient. We assume that lateral diffusion, binding, and unbinding predominantly occur in the membrane and that there is no significant exchange between membrane and cytoplasm. The bound fraction represents larger immobile structures, with diffusion coefficient of zero. The model also allowed for an immobile fraction β that could not exchange molecules by binding and unbinding laterally in the membrane on short and intermediate recovery timescales. These represent molecules that are more deeply embedded in the larger structures and cannot shuttle between the membrane and the cytoplasm. The sum of all fractions, free f(r,t), bound b(r,t), and immobile s(r) just after bleaching, equals the initial fluorescent profile, , hence
The initial profile at t = 0 was estimated by smoothing the noisy raw profile by applying a Savitzky-Golay smoothing filter (32) twice—first on the temporal traces using 61 points and subsequently on the spatial profiles using 11 points, then selecting the smoothed profile at t = 0 as the initial profile. In both cases, the polynomial order was 3. The initial values of the free, bound, and immobile fraction profiles can be estimated using the following equations (28,30):
| (5a) |
| (5b) |
| (5c) |
The four model parameters, D, , , and β were estimated by fitting the model to the normalized radial profiles by minimizing the sum of weighted-squared errors,
| (6) |
where denotes the modeled normalized fluorescence intensity that was obtained with parameter vector
The data extraction, model, and optimization were implemented in MATLAB (The MathWorks). The differential equations were discretized with a finite volume scheme using polar coordinates and were solved numerically using the ode45 solver from MATLAB. The model system was extended to a maximum radius of 30.43 μm and the proper Dirichlet boundary conditions were applied to f(r,t) and b(r,t) to mimic bulk fractions. Parameter optimization was done with the nonlinear optimization function Lsqnonlin (trust region reflective algorithm) using box constraints (, , , and ).
Results
Timing of AnxA4 self-assembly at the plasma membrane tracked by FRET microscopy
To gain a more detailed understanding of the timing and nature of AnxA4 localization at the plasma membrane and the relation of this timing to self-association, we performed a series of experiments tracking this translocation via Förster resonance energy transfer (FRET) microscopy using total internal reflection fluorescence (TIRF) microscopy. We utilized two FRET methods—the quantitative fluorescence lifetime imaging microscopy (FLIM-FRET)-based measurement and the qualitative, but faster ratio-FRET method (33). AnxA4 fused to an optimized, monomeric variant of the green fluorescent protein, superGFP2 (sGFP2) (34), was transiently produced in HeLa cells. Consistent with previous studies (14,35), at low resting levels of Ca2+, AnxA4-sGFP2 is uniformly localized throughout the cytoplasm and the nucleus (Fig. 1 A). After inducing a sharp rise in intracellular Ca2+ levels by the addition of 10 μM ionomycin, AnxA4 relocalized to the plasma membrane (Fig. 1 B) and, after a delay that varied in duration, from the nucleoplasm to the nuclear membrane (Fig. 1 C) (see Movie S1 in the Supporting Material). Some in vitro studies on the closely-related annexin family members, AnxA5 and AnxB12, have suggested that the formation of trimers begins within milliseconds of membrane association and formation of two-dimensional crystals can occur at low protein concentrations, although the timing of this latter step has not been precisely determined (36–38). It was reported that AnxA5 membrane-bound monomers persist on in vitro lipid bilayers, even at high protein concentrations (39). Previously, FRET has been used to detect the self-association of AnxA4 in vitro (7) and in cells (14). The latter study did not attempt to discern the timing and stages of the self-association in relation to the relocation of AnxA4 to the plasma membrane, so the steps of self-assembly at the plasma membrane remain unclear. To determine the precise sequence of events of AnxA4 self-association in cells, we used the highly quantitative FRET method based on fluorescence lifetime measurements of the donor (FLIM-FRET). Donor and acceptor constructs of AnxA4 fused to the enhanced cyan fluorescent protein, mTurquoise (mTq) (21) and superYFP2 (sYFP2) (41), respectively, were coproduced in HeLa cells. The microscope was configured to make fluorescence lifetime measurements in TIRF mode (24). Lifetime measurements were taken in a time-series at ∼30-s intervals (the fastest iteration we could use and still collect enough phase-steps to get robust lifetime data). Although measurements were conducted in TIRF mode, the evanescent excitation wave extended far enough into the cytoplasm that we could make lifetime and intensity measurements of the cytoplasmic AnxA4-mTq protein before the addition of ionomycin. These measurements show a phase and modulation lifetime for mTq of ∼3.7 and 3.8 ns, respectively (Table 1), which is equivalent to what has been measured in cells producing AnxA4-mTq only (data not shown). As noted above (Fig. 1, A–C), after the addition of a final concentration of 10 μM ionomycin there is a short delay, ranging from a few seconds to a minute, before the AnxA4-mTq starts to localize to the plasma membrane. Concomitant with this relocalization, the fluorescence lifetime was reduced (see Movie S2). The total reduction in lifetime averaged 0.92 ± 0.09 ns for the phase lifetime, τφ, and 0.59 ± 0.08 ns for the modulation lifetime, τM (n = 11), indicating significant FRET. Intriguingly, measurements in some cells suggested that there might be a slight lag between the start and finish of AnxA4-mTq localization to the plasma membrane and the lifetime drop associated with FRET.
Figure 1.
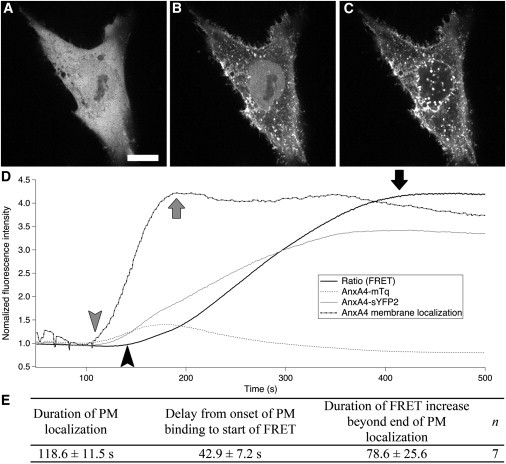
Timing of translocation and self-association of AnxA4 at the plasma membrane. Ionomycin-induced membrane binding of AnxA4-sGFP2 in HeLa cells (A–C). AnxA4-sGFP2 shows a largely uniform distribution throughout the nucleus and cytoplasm (A). After the addition of ionomycin induces a rise in intracellular Ca2+ levels, AnxA4 relocalizes from the cytoplasm to the plasma membrane as well as to some internal membranes (B). This is followed by a relocalization of the nuclear pool of AnxA4 to the nuclear membrane (C). The relative timing of AnxA4-sGFP2 binding to the plasma membrane and self-association was followed with ratio FRET imaging in TIRF (D). The ratio of the normalized intensity traces (solid line) from the mTq channel (grey dotted line) over the sYFP2 channel (grey solid line) shows the steady increase in FRET during AnxA4 self-association. There is a short delay after membrane binding begins (shaded arrowhead) before the start of self-association (solid arrowhead) and the assembly of oligomers continues for up to two minutes (solid arrow) following the complete relocation of the cytoplasmic pool (shaded arrow). The table (E) shows the mean values (mean ± SE) for these parameters. Scale bar = 25 μm.
Table 1.
Measurement of self-association with TIR-FLIM-FRET
| τφ ns | τM ns | |
|---|---|---|
| AnxA4-mTq (cytoplasm) | 3.75 ± 0.10 | 3.84 ± 0.09 |
| AnxA4-mTq (plasma membrane) | 2.83 ± 0.09 | 3.25 ± 0.07 |
| Δτ(n = 11) | 0.92 ± 0.09 | 0.59 ± 0.08 |
Phase and modulation lifetime values for mTq fused to AnxA4 in the cytoplasm and after ionomycin-induced membrane localization. In both cases sYFP2 fused to AnxA4 is the acceptor construct. Values are mean ± SE for 11 cells from four separate experiments.
To get a higher temporal resolution on the dynamics of the self-assembly, we utilized the more qualitative, but faster method of ratio-FRET in a TIRF modality. Using the same pair of constructs, we imaged mTq and sYFP2 simultaneously at one frame/s. After the addition of ionomycin to the cell media, the start of AnxA4 membrane binding was evident by an increase in fluorescent signal in both the donor (mTq) and acceptor (sYFP2) channels (Fig. 1 D, grey lines). To account for the effects of donor quenching and sensitized emission in these respective signals, we generated a third curve that was reflective of the localization kinetics by summing the intensity of the two detection channels and renormalizing the trace (Fig. 1 D, dotted line). For easier comparison to the FRET signal (Fig. 1 D, solid line), the localization trace was scaled so its peak value matched the peak value of the ratio trace. The onset of AnxA4 localization to the plasma membrane (Fig. 1 D, shaded arrowhead) does precede the first detectable FRET signal (Fig. 1 D, solid arrowhead) with an average value of 43 ± 7 s, mean ± SE (Fig. 1 E). Additionally, the FRET signal continues to increase after AnxA4 membrane localization was complete (Fig. 1 D, shaded arrow). The duration of this gap was highly variable, ranging between 20 and 220 s, with an average of 79 ± 26 s (Fig. 1 E). The initial delay between plasma membrane localization and FRET suggests that self-association does not begin immediately upon membrane binding, but instead requires a certain concentration threshold before beginning in earnest. The second delay is even more notable, as in some cases it is quite prolonged (Fig. 1 D). This could reflect the continued formation of trimers from a membrane bound pool of monomers, or perhaps the continued clustering of higher-order AnxA4 aggregates from the basic trimeric form.
Oligomerization state of AnxA4 at the plasma membrane as measured by number-and-brightness analysis
Although we were able to follow AnxA4 self-association at the plasma membrane in real time, using FRET, we were also interested in determining the oligomerization state of these AnxA4 assemblies. Although in some cases, homo-FRET has been used to distinguish dimers from larger assemblages (42–44), the technique is largely fettered in situations where more precise details on complexes larger than dimers are sought. To achieve this, we made use of the fluorescence fluctuation-based technique known as number-and-brightness analysis (26). The neuroblastoma cell line, N1E-115 (45), was utilized for these experiments because, unlike HeLa cells, the N1E-115 line shows no native expression of AnxA4 (14) and the presence of substantial levels of endogenous proteins can significantly confound brightness-based methods such as number-and-brightness analysis. These cells were initially transfected with either the monomeric sGFP2 or a tandem construct consisting of two sGFP2 coding sequences joined by a short flexible linker to validate our acquisition and analysis protocols in cells. Results from one representative experiment are shown in Table 2 (additional results are shown in Table S1). The brightness value (ε) for the sGFP2 control in this experiment was 0.035 ± 0.003 kHz/molecule∗bin (n = 6). The tandem sGFP2 construct exhibited an ε-value of 0.062 ± 0.005 (n = 6), or ∼1.8-fold increase in brightness. In the same experiment, N1E-115 cells were also transfected with an AnxA4-sGFP2 construct. As with the HeLa cells, the AnxA4-sGFP2 construct showed uniform distribution throughout the cytoplasm and nucleus (Fig. 2 A). The measured ε-value for cytoplasmic/nucleoplasmic AnxA4-sGFP2 was 0.032 ± 0.004 (n = 6, Table 2, Fig. 2 B). This is comparable to the values for monomeric sGFP2 alone, indicating that AnxA4 exists as a monomer when it is in the cytoplasmic, nonCa2+-bound state. After ionomycin-induced Ca2+ binding of AnxA4-sGFP2 and relocation of the protein to the plasma membrane, the ε-value for AnxA4-sGFP2 at the plasma membrane averaged 0.108 ± 0.005—3.09-fold brighter than monomeric sGFP2 (Table 1, Fig. 2, B and C).
Figure 2.
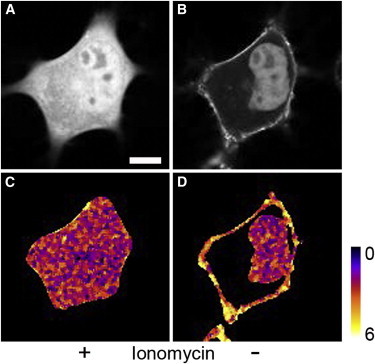
Increase in molecular brightness of AnxA4-sgfp2 after translocation to the plasma membrane. The average intensity (A and B) and pseudo-colored normalized brightness values (C and D) for a representative N1E-115 cell are shown before (A and C) and after (B and D) the addition of ionomycin. There is a marked increase in molecular brightness for AnxA4-sGFP2 upon binding the plasma membrane, while the soluble AnxA4-sGFP2 in the nucleus remains monomeric (D). Scale bar = 10 μm.
The approximately threefold increase in the ε-values measured for AnxA4-sGFP2 at the plasma membrane over the monomeric values suggests the formation of AnxA4-sGFP2 trimers upon membrane binding. It is important to note that the number-and-brightness analysis technique only reports a single brightness for each individual pixel. A mixed population will yield a value that is the weighted average of brightness of all the individual molecules in that detection volume, so it is possible that some monomeric species and larger multimers are also present. Yet, given the consistency of our molecular brightness measurements (Table 3, and see Table S1), our experiments suggest that the predominant mobile species of membrane-bound AnxA4-sGFP2 is the trimeric form.
FRAP analysis reveals multicomponent recovery and is correlated with cell morphology
Because number-and-brightness analysis is a fluctuation-based technique, it cannot provide data on molecules that are immobile. Therefore it is possible that AnxA4-sGFP2 also forms higher-order aggregates in cells as it does on lipid layers in vitro, but that these higher-order aggregates are largely immobile. In vitro FRAP studies measuring the diffusion of fluorescently-labeled lipid analogs in a planar bilayer have shown that the binding of AnxA4 to the layer reduced lateral diffusion coefficients by up to 35-fold (15). A qualitative assessment of FRAP data in HeLa cells indicated that AnxA4 showed very little recovery compared to a YFP-labeled PH domain from PLCδ1 (14). Additionally, the same study showed that the overexpression and translocation of AnxA4 profoundly reduced the fluorescence recovery of other membrane proteins. The authors conclude that AnxA4, once at the membrane, is largely immobile due to the formation of surface arrays, and that these arrays function to impede general lateral mobility of other membrane components. However, data from the fluctuation-based number-and-brightness analysis techniques indicates that there must be a substantial proportion of AnxA4 undergoing membrane-bound diffusion. To reconcile this apparent contradiction and to characterize the mobile population of AnxA4 at the plasma membrane, we performed a set of quantitative FRAP experiments. HeLa cells expressing AnxA4-sGFP2 were treated with 10 μM ionomycin to induce Ca2+-mediated membrane binding. Optical sections containing the top membrane were selected and a uniform circular region with a radius of 4.14 μm was bleached using the tornado scanning mode, bleaching from the center outwards (Fig. 3 A). In a method similar to that employed by Mueller et al. (28,46), intensity averages were obtained from a series of radial rings centered on the middle of the bleached region. In this way, the spatial characteristics of the recovery could be factored into the fitting models (see Movie S3). Strikingly, there was rather marked variation in recovery characteristics between different cells (Fig. 3, A and B). The characteristics of the recovery curves seemed correlated with the morphology of the plasma membrane region subjected to FRAP. In areas where the membrane was flat, broadly spread out, and AnxA4-sGFP2 was evenly distributed, the recovery was quite robust (Fig. 3, A and C). Conversely, in regions where the membrane did not have these characteristics, especially when visible puncta or aggregates of AnxA4 were present, the recovery was more limited (Fig. 3, B–D, and Fig. 4). This did not seem to be a concentration-based effect, as cells with equitable AnxA4-sGFP2 expression levels were selected for analysis.
Figure 3.
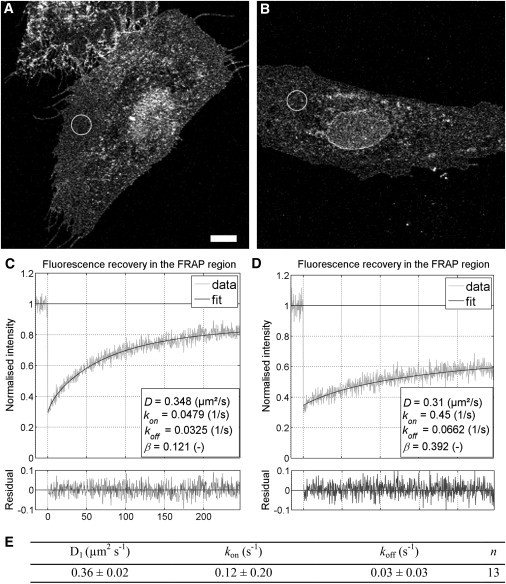
Fluorescence recovery after photobleaching correlates with membrane morphology. Two representative HeLa cells (A and B) exhibiting different membrane morphology and the corresponding fluorescence recovery curves (C and D) are shown. (Open circles) Approximate FRAP regions. The extent of the recovery varied greatly. Note the difference in β (immobile fraction) values, 0.12 in panel C and 0.39 in panel D; all curves could be fit with a model describing one diffusion component, a binding component, and an immobile fraction. The table (E) shows the mean values (± SD) of these estimated parameters from 13 cells from two experiments. The immobile fraction (β) ranged from 0.08 to 0.48 whereas the other parameters were less variable. Scale bar = 10 μm.
Figure 4.
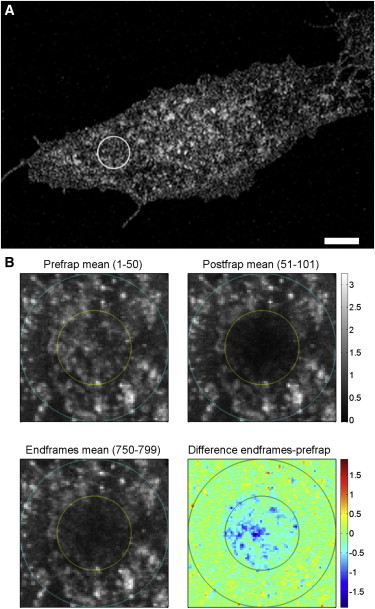
Recovery in regions with high levels of AnxA4 aggregates. (A) Image of an HeLa cell expressing AnxA4-sGFP2 with tornado FRAP region highlighted. (B) Average intensity (50 frames each) of prebleach, immediately postbleach, and ∼4 min postbleach. The difference image illustrates heterogeneous recovery. (Dark-blue areas, −1 to −1.5) Highest difference (little to no recovery). (Light areas, >−0.5) Regions where recovery has occurred. Scale bar = 10 μm.
The recovery curves could all be fit reasonably well with a model describing one diffusion component, a binding component, and an immobile component. The estimated values for the effective lateral diffusion coefficient, , and , are shown in Fig. 3 E. Notably, the values for the diffusion coefficient and were quite consistent between the different regions analyzed, whereas the value showed more variability. The calculated immobile fraction (β) varied greatly, ranging from 0.07 to 0.48 (n = 13). In cells where large aggregates of AnxA4 were visible, limited recovery was observed, with high immobile fractions β > 0.40 (Fig. 4, A and B). Notably, when regions containing these AnxA4 clusters are bleached (Fig. 4), there is almost no recovery in the aggregates themselves, although the areas between and around these structures do show fluorescence recovery, as seen in the difference image (Fig. 4 B, lower right panel) that highlights the intensity difference between prebleach and 4-min postbleach. AnxA4 structures that can be seen in Fig. 4 B, upper left panel show the least amount of recovery (dark blue pixels, -1.0 to -1.5 in Fig. 4 B, lower right panel). This seems to indicate that these larger aggregates are quite stable on the minutes timescale, with very limited exchange of protein. Because recovery is still observed in these regions, it suggests that there remains a mobile proportion of AnxA4 and there may be exchange of proteins from the peripheries of these structures. We attribute the binding kinetics ( and ) to the association and disassociation of trimer subunits from the edges of these aggregates.
Discussion
Although there have been many studies linking AnxA4 to a range of cellular functions, these connections have largely been phenomenological and despite a wealth of detailed biophysical data (especially for the closely related family member, AnxA5) there is limited information on how AnxA4 mechanistically functions at the plasma membrane. For example, AnxA4 seems to play a role in negatively regulating Ca2+-activated Cl− conductance (12,13). It has been suggested that the AnxA4 two-dimensional arrays, which have been observed to form in vitro, might act to physically segregate the ion channels and block the activating phosphorylation by calmodulin-dependent protein kinase II. Piljić and Schultz (14) found that AnxA4 can reduce the mobility of proteins at the plasma membrane and similar effects were seen on the diffusion of lipids in a supported bilayer (15). Although this is intriguing and consistent with the notion that AnxA4 follows the in vitro-based model in cells, it does not quantify many important parameters that would more completely describe the behavior of the protein—namely specifics on its mobility and oligomerization state at the plasma membrane. In this study, we have been able to provide details on these characteristics.
Timing and nature of self-assembly of AnxA4 at the plasma membrane
In vitro studies on the hydra annexin B12 showed that trimer assembly occurs within milliseconds of membrane association (37) and in the case of both AnxA5 and AnxB12, almost all the membrane-bound protein is in a trimeric state (36). Contrary to this study, single-particle tracking of dye-labeled AnxA5 on liquid-phase, supported lipid bilayers indicated that monomers can persist even at high, but nonsaturating, concentrations (39) and a FRET-based in vitro assay also suggested that AnxA5 does not form multimers at low membrane occupancy (47). In our time-series FRET experiments on AnxA4 in cells, we observed a delay between the start of membrane binding and the first measureable signs of self-assembly (Fig. 1 D, Table 1 and Table 2; and see Movie S2). This suggests that at the low concentrations of protein that initially exist at the plasma membrane, AnxA4 may be largely monomeric. It has been suggested that the formation of annexin trimers may increase the membrane-binding affinity of the protein (36), although there have also been indications in the literature that self-assembly, at least in the case for AnxA5, does not enhance affinity for membranes (47,48). Number-and-brightness analysis suggests that trimers predominately constitute the mobile fraction of AnxA4 at the membrane (Table 2). However, like all fluctuations techniques, the method is disproportionately influenced by molecules with higher molecular brightness (49,50), so it remains possible that a subpopulation of membrane-bound monomers exists. Our ratio-FRET data also indicates that AnxA4 continues to assemble and/or aggregate for some time after the cytoplasmic pool has completely relocated to the plasma membrane. The duration of this period varies greatly between cells; we measured periods as short as 20 s and as long as 220 s (Fig. 1 D, Table 2). This suggests that the assembly and aggregation of AnxA4 continues over a long, but highly variable period, possibly as smaller subunits continue to assemble into larger arrays or are incorporated into cellular membrane structures, which are evolving more slowly.
With experiments that rely on exogenously transfected protein constructs, care should be taken to avoid excessive levels of protein production that may lead to expression-related artifacts. In FRET studies along membranes, such overproduction might lead to energy transfer that is not due to a specific protein-protein interaction. In this study, we were careful to choose cells for the FRET experiments where the expression level was moderate. In addition, we consistently observed a continuous increase of FRET after the point of maximum membrane occupancy (Fig. 1), indicating that the energy transfer we measure is due to bona fide AnxA4-AnxA4 interactions and not to aspecific overproduction-related bystander FRET. Our number-and-brightness analysis, like fluorescence correlation spectroscopy (51), requires that the proteins under study are kept at a low concentration (in the ten-hundreds nM range) and the molecules under study must be codiffusing as a single physical entity. Utilizing this low concentration regime, overproduction-related artifacts are avoided.
Mobility and aggregation of AnxA4
These higher-order aggregates are thought to be highly immobile and may be the mechanism that restricts mobility and access of other membrane proteins (12,14). In partial agreement, our FRAP studies did show membrane regions where AnxA4 exhibited very low recovery and a high-immobile fraction. These regions were often characterized by visible AnxA4 aggregates, ranging in size from ∼0.5–2 μm (Fig. 4 A). In contrast, we also found plasma membrane regions in which AnxA4 exhibits a high degree of recovery. This was most pronounced in flattened, extended regions of the membrane, where the lack of visible aggregation of AnxA4 was observed (Fig. 3, A and C). We propose that this fast component reflects the freely mobile trimeric state of AnxA4. The diffusion coefficient of ∼0.36 μm2·s−1 for AnxA4 that we measured is consistent with earlier in vitro FRAP measurements of AnxA5 trimers on supported lipid bilayers (52) and what was measured (0.4 μm2·s−1) by single-particle tracking for AnxA5 monomers (39). Despite this consistency, a recent in vitro FCS-based study (53), on AnxA5 mobility on supported lipid layers, reported two diffusing components—one with a very fast diffusion coefficient of 3.1 μm2·s−1 and a second, much slower component with a diffusion coefficient of 0.02 μm2·s−1. The reason for the full magnitude discrepancy is not clear to us. Whereas our study represents (to our knowledge) the first direct in vivo, quantitative measurements of AnxA4 diffusion on membranes, a study of the mobility of fluorescently labeled lipid analogs in a supported planar bilayer showed that the binding of AnxA4 to this membrane resulted in a, greatly reduced, two-component lateral diffusion of these lipids (15). The diffusion coefficients in the latter study were in the range of 1.6–3.0 μm2·s−1 before AnxA4 binding and slowed to ∼0.4 μm2·s−1 for a fast component and 0.05 μm2·s−1 for the slower component (depending upon the type of lipid and the composition of the bilayer). The close agreement of the mobility of the fast lipid component and the diffusion coefficient for the AnxA4 measured here raises the intriguing possibility that, once the electrostatic interaction occurs between the negatively-charged phospholipid and the Ca2+ bound protein, this association is quite stable, with very little exchange of lipid from underneath the protein—resulting in effective comigration. Gilmanshin et al. (15) suggest that the slower lipid component in their study could be the result of lipid exchange between annexin-rich and annexin-poor domains, which would be consistent with a model where AnxA4 can trap certain lipid species. Although purely speculative, if this is borne out by future studies, it might imply a role for AnxA4 in sequestering phospholipids. It has also been suggested that AnxA4 may segregate and enmesh other membrane components through the formation of immobile two-dimensional arrays. Indeed, it appears that when aggregates of AnxA4 are prominent on the plasma membrane, these structures are immobile over the timescales studied (Fig. 4). The interior regions of these aggregates are also quite stable, although there appears to be some protein exchange from the peripheries. This on/off exchange would account for the binding component in our FRAP analysis. The high degree of deviation in our fit parameter for k∗on indicates that this pseudo on-rate (a function of on-rate and the available bindings sites (54)) varies as there are more aggregates or larger aggregates with greater edge area to bind.
Taken together, our data allow us to propose a hypothetical model for the biophysical behavior of AnxA4 at the plasma membrane, which is summarized in Fig. 5. As confirmed by both our FRET and number-and-brightness analysis measurements, in resting cells, AnxA4 exists as a monomer in the cytoplasm.
-
Step 1
Upon binding Ca2+, these monomers bind anionic phospholipids in the membrane.
-
Step 2.
The monomers diffuse laterally along the membrane until chance encounters lead to trimer formation, hence an effect dependent upon (local) membrane concentration. Once a certain concentration has been reached, these trimers are the predominate mobile fraction at the membrane, exhibiting an lateral diffusion coefficient of ∼0.36 μm2/s.
-
Step 3.
Larger aggregates of AnxA4 may eventually form from these basic trimeric building blocks (55), a process that we believe contributes to the longer timescale increase in FRET shown in Fig. 1 D.
Figure 5.
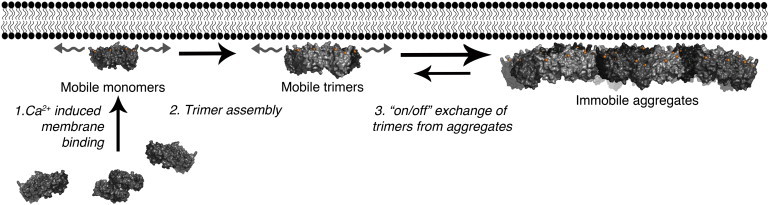
A hypothetical model for AnxA4 biophysical behavior at the plasma membrane. See text for details.
The mechanism through which this occurs in cells is not yet clear, although it may be through some stochastic process or, alternatively, some as-yet-to-be-described seeding event serves to initiate their formation. Because the relative predominance of one form or another (mobile trimers versus immobile aggregates) appears to be correlated with cell membrane morphology, this implies a functional link between AnxA4 aggregation and membrane structure. Although these aggregates form quite readily by the high levels of intracellular Ca2+ induced by ionomycin, these relatively immutable assemblages may form only rarely under physiological conditions, such as in the case of plasma membrane rupture or in the vicinity of open Ca2+ channels. This type of response might mediate a role for the array-forming annexins (AnxA4 and AnxA5) to effect plasma membrane repair (48) and regulate ion transport (12,13,56).
Acknowledgments
The authors thank the members of the Molecular Cytology group, Carsten Schultz for helpful discussions and for sharing the initial annexin constructs, and Florian Mueller/James McNally for sharing their “Circle FRAP” code with us.
K.C.C. is supported by the European Science Foundation (EuroMembrane, TraPPs). This work was also supported by a Middelgroot investment grant to M.A.H. (grant No. 834-09-003) of the Netherlands Organization for Scientific Research (NWO) and a NWO Earth and Life Sciences Council (NWO-ALW) VIDI fellowship to M.P.
Supporting Material
References
- 1.Moss S.E., Morgan R.O. The annexins. Genome Biol. 2004;5:219. doi: 10.1186/gb-2004-5-4-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerke V., Moss S.E. Annexins: from structure to function. Physiol. Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 3.Gerke V., Creutz C.E., Moss S.E. Annexins: linking Ca2+ signaling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 4.Monastyrskaya K., Babiychuk E.B., Draeger A. The annexins: spatial and temporal coordination of signaling events during cellular stress. Cell. Mol. Life Sci. 2009;66:2623–2642. doi: 10.1007/s00018-009-0027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rescher U., Gerke V. Annexins—unique membrane binding proteins with diverse functions. J. Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 6.Dreier R., Schmid K.W., Riehemann K. Differential expression of annexins I, II and IV in human tissues: an immunohistochemical study. Histochem. Cell Biol. 1998;110:137–148. doi: 10.1007/s004180050275. [DOI] [PubMed] [Google Scholar]
- 7.Zaks W.J., Creutz C.E. Ca2+-dependent annexin self-association on membrane surfaces. Biochemistry. 1991;30:9607–9615. doi: 10.1021/bi00104a007. [DOI] [PubMed] [Google Scholar]
- 8.Willshaw A., Grant K., Gawler D. Identification of a novel protein complex containing annexin A4, rabphilin and synaptotagmin. FEBS Lett. 2004;559:13–21. doi: 10.1016/S0014-5793(03)01513-8. [DOI] [PubMed] [Google Scholar]
- 9.Jeon Y.J., Kim D.H., Bae K.H. Annexin A4 interacts with the NF-κB p50 subunit and modulates NF-κB transcriptional activity in a Ca2+-dependent manner. Cell. Mol. Life Sci. 2010;67:2271–2281. doi: 10.1007/s00018-010-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim A., Enomoto T., Naka T. Enhanced expression of Annexin A4 in clear cell carcinoma of the ovary and its association with chemoresistance to carboplatin. Int. J. Cancer. 2009;125:2316–2322. doi: 10.1002/ijc.24587. [DOI] [PubMed] [Google Scholar]
- 11.Duncan R., Carpenter B., Murray G.I. Characterization and protein expression profiling of annexins in colorectal cancer. Br. J. Cancer. 2008;98:426–433. doi: 10.1038/sj.bjc.6604128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan H.C., Kaetzel M.A., Nelson D.J. Annexin IV inhibits calmodulin-dependent protein kinase II-activated chloride conductance. A novel mechanism for ion channel regulation. J. Biol. Chem. 1994;269:32464–32468. [PubMed] [Google Scholar]
- 13.Kaetzel M.A., Chan H.C., Nelson D.J. A role for annexin IV in epithelial cell function. Inhibition of calcium-activated chloride conductance. J. Biol. Chem. 1994;269:5297–5302. [PubMed] [Google Scholar]
- 14.Piljić A., Schultz C. Annexin A4 self-association modulates general membrane protein mobility in living cells. Mol. Biol. Cell. 2006;17:3318–3328. doi: 10.1091/mbc.E06-01-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilmanshin R., Creutz C.E., Tamm L.K. Annexin IV reduces the rate of lateral lipid diffusion and changes the fluid phase structure of the lipid bilayer when it binds to negatively charged membranes in the presence of calcium. Biochemistry. 1994;33:8225–8232. doi: 10.1021/bi00193a008. [DOI] [PubMed] [Google Scholar]
- 16.Zanotti G., Malpeli G., Berni R. Structure of the trigonal crystal form of bovine annexin IV. Biochem. J. 1998;329:101–106. doi: 10.1042/bj3290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaetzel M.A., Mo Y.D., Seaton B.A. Phosphorylation mutants elucidate the mechanism of annexin IV-mediated membrane aggregation. Biochemistry. 2001;40:4192–4199. doi: 10.1021/bi002507s. [DOI] [PubMed] [Google Scholar]
- 18.Newman R., Tucker A., Crumpton M.J. Crystallization of p68 on lipid monolayers and as three-dimensional single crystals. J. Mol. Biol. 1989;206:213–219. doi: 10.1016/0022-2836(89)90534-2. [DOI] [PubMed] [Google Scholar]
- 19.Newman R.H., Leonard K., Crumpton M.J. 2D crystal forms of annexin IV on lipid monolayers. FEBS Lett. 1991;279:21–24. doi: 10.1016/0014-5793(91)80240-4. [DOI] [PubMed] [Google Scholar]
- 20.Shaner N.C., Campbell R.E., Tsien R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 21.Goedhart J., van Weeren L., Gadella T.W., Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods. 2010;7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]
- 22.Axelrod D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981;89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadella T., Jovin T., Clegg R. Fluorescence lifetime imaging microscopy (FLIM): spatial resolution of microstructures on the nanosecond time scale. Biophys. Chem. 1993;48:221–239. [Google Scholar]
- 24.Gadella T.W. Total internal reflection fluorescence lifetime imaging microscopy. In: Gadella T., editor. FRET and FLIM Techniques. Elsevier; New York: 2008. pp. 395–412. [Google Scholar]
- 25.van Munster E.B., Gadella T.W. Suppression of photobleaching-induced artifacts in frequency-domain FLIM by permutation of the recording order. Cytometry A J. Intl. Soc. Anal. Cytol. 2004;58:185–194. doi: 10.1002/cyto.a.20013. [DOI] [PubMed] [Google Scholar]
- 26.Digman M.A., Dalal R., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalal R.B., Digman M.A., Gratton E. Determination of particle number and brightness using a laser scanning confocal microscope operating in the analog mode. Microsc. Res. Tech. 2008;71:69–81. doi: 10.1002/jemt.20526. [DOI] [PubMed] [Google Scholar]
- 28.Mueller F., Wach P., McNally J.G. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J. 2008;94:3323–3339. doi: 10.1529/biophysj.107.123182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phair R.D., Gorski S.A., Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. In: Allis C.D., Carl W., editors. Methods in Enzymology. Academic Press; New York: 2003. pp. 393–414. [DOI] [PubMed] [Google Scholar]
- 30.Sprague B.L., Pego R.L., McNally J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang M., Kenworthy A. Complex applications of simple FRAP on membranes. In: Faller R., Longo M.L., Risbud S.H., Jue T., editors. Biomembrane Frontiers. Humana Press; Louisville, KY: 2009. pp. 187–221. [Google Scholar]
- 32.Savitzky A., Golay M.J.E. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964;36:1627–1639. [Google Scholar]
- 33.Pietraszewska-Bogiel A., Gadella T.W. FRET microscopy: from principle to routine technology in cell biology. J. Microsc. 2011;241:111–118. doi: 10.1111/j.1365-2818.2010.03437.x. [DOI] [PubMed] [Google Scholar]
- 34.Kremers G.J., Goedhart J., Gadella T.W., Jr. Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry. 2007;46:3775–3783. doi: 10.1021/bi0622874. [DOI] [PubMed] [Google Scholar]
- 35.Monastyrskaya K., Babiychuk E.B., Draeger A. Annexins as intracellular calcium sensors. Cell Calcium. 2007;41:207–219. doi: 10.1016/j.ceca.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Patel D.R., Isas J.M., Haigler H.T. The conserved core domains of annexins A1, A2, A5, and B12 can be divided into two groups with different Ca2+-dependent membrane-binding properties. Biochemistry. 2005;44:2833–2844. doi: 10.1021/bi047642+. [DOI] [PubMed] [Google Scholar]
- 37.Langen R., Isas J.M., Hubbell W.L. Membrane-mediated assembly of annexins studied by site-directed spin labeling. J. Biol. Chem. 1998;273:22453–22457. doi: 10.1074/jbc.273.35.22453. [DOI] [PubMed] [Google Scholar]
- 38.Richter R.P., Him J.L.K., Brisson A.R. On the kinetics of adsorption and two-dimensional self-assembly of annexin A5 on supported lipid bilayers. Biophys. J. 2005;89:3372–3385. doi: 10.1529/biophysj.105.064337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han J.J., Boo D.W. Reversible immobilization of diffusive membrane-associated proteins using a liquid-gel bilayer phase transition: a case study of annexin V monomers. Langmuir. 2009;25 doi: 10.1021/la803903j. 3083–2088. [DOI] [PubMed] [Google Scholar]
- 40.Reference deleted in proof.
- 41.Kremers G.J., Goedhart J., Gadella T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry. 2006;45:6570–6580. doi: 10.1021/bi0516273. [DOI] [PubMed] [Google Scholar]
- 42.Bader A., Hoetz S., Gerritsen H. Homo-FRET imaging as a tool to quantify protein and lipid clustering. ChemPhysChem. 2011;12:475–483. doi: 10.1002/cphc.201000801. [DOI] [PubMed] [Google Scholar]
- 43.Bader A.N., Hofman E.G., Gerritsen H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009;97:2613–2622. doi: 10.1016/j.bpj.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofman E.G., Bader A.N., van Bergen en Henegouwen P.M. Ligand-induced EGF receptor oligomerization is kinase-dependent and enhances internalization. J. Biol. Chem. 2010;285:39481–39489. doi: 10.1074/jbc.M110.164731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amano T., Richelson E., Nirenberg M. Neurotransmitter synthesis by neuroblastoma clones (neuroblast differentiation-cell culture-choline acetyltransferase-acetylcholinesterase-tyrosine hydroxylase-axons-dendrites) Proc. Natl. Acad. Sci. USA. 1972;69:258–263. doi: 10.1073/pnas.69.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller F., Karpova T.S., McNally J.G. Monitoring dynamic binding of chromatin proteins in vivo by fluorescence recovery after photobleaching. Methods Mol. Biol. 2012;833:153–176. doi: 10.1007/978-1-61779-477-3_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeppesen B., Smith C., Tait J.F. Entropic and enthalpic contributions to annexin V-membrane binding: a comprehensive quantitative model. J. Biol. Chem. 2008;283:6126–6135. doi: 10.1074/jbc.M707637200. [DOI] [PubMed] [Google Scholar]
- 48.Bouter A., Gounou C., Brisson A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat Commun. 2011;2:270. doi: 10.1038/ncomms1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y., Müller J.D., Gratton E. Probing ligand protein binding equilibria with fluorescence fluctuation spectroscopy. Biophys. J. 2000;79:1074–1084. doi: 10.1016/S0006-3495(00)76361-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brock R., Hink M.A., Jovin T.M. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 1998;75:2547–2557. doi: 10.1016/S0006-3495(98)77699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwille, P., and E. Haustein. 2002. Fluorescence Correlation Spectroscopy. A Tutorial for the Biophysics Textbook Online. BTOL. http://www.biophysics.org/Portals/1/PDFs/Education/schwille.pdf.
- 52.Cézanne L., Lopez A., Tocanne J.F. Organization and dynamics of the proteolipid complexes formed by annexin V and lipids in planar supported lipid bilayers. Biochemistry. 1999;38:2779–2786. doi: 10.1021/bi9818568. [DOI] [PubMed] [Google Scholar]
- 53.Vats K., Knutson K., Sheets E.D. Peripheral protein organization and its influence on lipid diffusion in biomimetic membranes. ACS Chem. Biol. 2010;5:393–403. doi: 10.1021/cb900303s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sprague B.L., McNally J.G. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 2005;15:84–91. doi: 10.1016/j.tcb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 55.Oling F., Bergsma-Schutter W., Brisson A. Trimers, dimers of trimers, and trimers of trimers are common building blocks of annexin a5 two-dimensional crystals. J. Struct. Biol. 2001;133:55–63. doi: 10.1006/jsbi.2000.4337. [DOI] [PubMed] [Google Scholar]
- 56.Hill W.G., Kaetzel M.A., Zeidel M.L. Annexin A4 reduces water and proton permeability of model membranes but does not alter aquaporin 2-mediated water transport in isolated endosomes. J. Gen. Physiol. 2003;121:413–425. doi: 10.1085/jgp.200308803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.