

Abstract
DNA methylation and histone modifications are central to epigenetic gene regulation, which has been shown to play a crucial role in development. Epigenetics has often been discussed in the context of the maintenance of cell identity because of the heritable nature of gene expression status. Indeed, crucial roles of the epigenetic machinery in establishment and maintenance of particular lineages during early development have been well documented. However, unexpected observation of a developmental plasticity retained in mature T lymphocytes, in particular in CD4+ T-cell subsets, by recent studies is accelerating studies that focus on roles of each epigenetic pathway in cell fate decisions of T lymphocytes. Here, we focus on the repressive epigenetic machinery, i.e. DNA methylation, histone deacetylation, H3K9 methylation and Polycomb repressive complexes, and briefly review the studies examining the role of these mechanisms during T-lymphocyte differentiation. We also discuss the current challenges faced when analysing the function of the epigenetic machinery and potential directions to overcome the problems.
Keywords: epigenetics, gene repression, lineage decision, T lymphocytes
Introduction
Cell fate determination, or lineage commitment, which is often initiated by exposure to developmental cues, accompanies both up-regulation of genes specific for one lineage and down-regulation of genes associated with other lineages. Hence, nuclear machinery that is involved in activation or repression of genes constitutes an integral part of lineage commitment. In the last decade, it has become clearer that epigenetic mechanisms play major roles in both activation and repression processes.
Epigenetic mechanisms act through two major substrates, DNA and histones. In higher eukaryotes, DNA can be methylated at cytosine mainly in the context of CpG dinucleotide sequences, producing 5-methylcytosine (5mC), the accumulation of which at promoter regions is often associated with the repressive state of gene loci as well as its stable maintenance, known as gene silencing.1 De novo DNA methylation is mediated through the Dnmt3-class of methyltransferases,2,3 whereas maintenance of a pre-existing methylation pattern during DNA replication is catalysed by Dnmt1.4 5mC is recognized by methyl DNA binding proteins through specific peptide modules such as MBD (methyl-CpG binding domain) or triple-zinc-finger motif.5 It is now becoming apparent that 5mC is not an unchanging modification as it was once thought to be; it can be de-methylated through active processes. Recently, TET-family proteins were identified as enzymes that convert 5mC to 5-hydroxymethylcytosine (5hmC),6,7 and further to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC)8,9 through consecutive oxidation reactions. It is also hypothesized that these oxidized products can be reverted to cytosine through the base excision repair pathway.10 The biological function of 5hmC, 5fC and 5caC still remains to be fully uncovered, but genome-wide studies suggest a role of 5hmC in both transcriptional repression and activation,10 potentially adding further layers to epigenetic regulation mediated by modifications of the cytosine residues on DNA. Recognition of methylated DNA is in some cases followed by recruitment of histone modification enzymes, such as histone deacetylases (HDACs)11–14 and histone methyltransferase.15
Covalent histone modifications are another means of epigenetic regulation. Core histones can be modified at various sites with diverse molecules, which include acetylation, methylation, ubiquitination and phosphorylation. Combinations of these modifications are hypothesized to function as a ‘histone code’ that is recognized by specific binding proteins to execute defined down-stream biological processes.16,17 However, recent data suggest that the role of the code may not be limited to recruitment of factors but may include allosteric regulation of the bound factors.18 Furthermore, it was noted that DNA and histone modifications act from one to the other in both directions.19
Many components of epigenetic machinery were initially identified through genetic screening in Drosophila, in which developmental abnormalities were used as an indicator. Hence, there is no question that the epigenetic machinery plays crucial roles in determining the cell fate in multi-cellular organisms, and T-lymphocyte development would not be the exception. T lymphocytes develop in the thymus, and thymocyte progenitors give rise to several distinct T-cell subsets under various differentiation conditions, and the key transcription factors required for the differentiation of each subset have been identified (Fig. 1). Not surprisingly, these key transcription factors are shown to interact with various molecules involved in epigenetic regulation (Table 1). In the table, we only listed the regulators that were shown to interact directly with the transcription factors. Hence, the list will further expand if it includes indirect association through co-factors. These multi-molecular interactions between transcription factors and epigenetic modifiers further strengthen the idea that epigenetic gene regulation is an integral part of establishing lineage-specific transcriptional programmes during T-lymphocyte differentiation. Below, we present an overview of two different types of approach that were aimed at exploring this idea.
Figure 1.
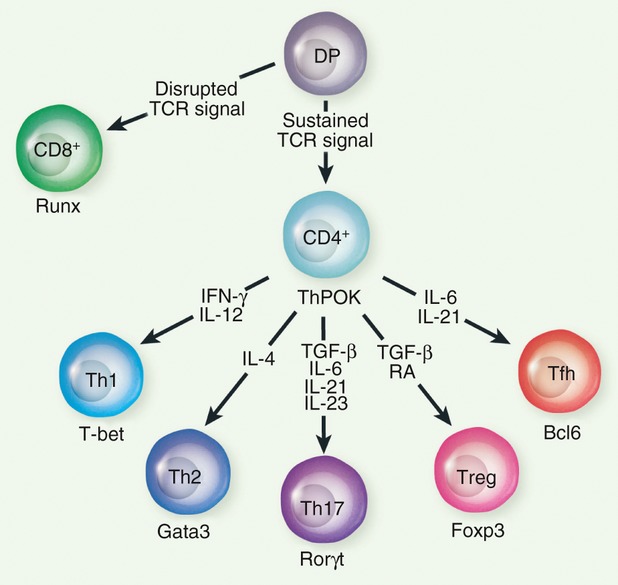
Differentiation of T-cell subsets and the key transcription factors. Signals that induce differentiation are shown on the arrows, whereas the key transcription factor for each lineage is shown below the each subset. DP, double-positive; IFN-γ, interferon-γ; IL-12, interleukin-12; RA, retinoic acid; TCR, T-cell receptor; Tfh, folicular helper T; TGF-β, transforming growth factor-β; Th, T helper; Treg, regulatory T.
Table 1.
The key transcription factors for T-cell subsets and their interacting epigenetic regulators
| Cell subset | Key transcription factor | Interacting epigenetic regulators |
|---|---|---|
| CD4+ | ThPOK | p300,71 HDAC3,4,572 |
| CD8+ | Runx | HDAC1, 2, 3,53,73,74 Bmi1,53 Suv39h1,73,74,75 p300,76 MLL,53 MBD353 |
| Th1 | T-bet | CBP/p300,77 Set7/9,78 Jmjd379 |
| Th2 | Gata3 | HDAC3, 5,77 HDAC4,80 Bmi160 |
| Th17 | RORγt | p30081 |
| Treg | Foxp3 | p300,35 SIRT135 |
| Tfh | Bcl6 | p300,82 HDAC2,82 HDAC4,5,783 |
Approaches from cis-regulatory elements in the genes
In lineage decision, the expression or repression of key transcription factors exclusively determines a cell fate in many cases. Hence, studying the epigenetic regulation of genes encoding such factors can provide deeper insights into the role of the epigenetic machinery in lineage choices. In this regard, the Thpok gene (also known as Zbtb7b gene) encoding the master regulator of CD4+ helper T-cell development20,21 has served as an ideal model for such a study, and the regulatory mechanisms of its expression are being extensively studied. The helper-lineage-specific expression of the Thpok gene is regulated by a silencer element located upstream of the distal promoter.22,23 Removal of this silencer element can direct all post-selected thymocytes into the CD8+ cytotoxic lineage regardless of their MHC specificity through uncontrolled de-repression of ThPOK. Epigenetically, the Thpok locus is in a ‘bivalent’ state, i.e. co-localization of activation-associated tri-methylated histone H3 Lys4 (H3K4me3) mark and repression-associated tri-methylated histone H3 Lys27 (H3K27me3) mark, in the CD4+ CD8+ double-positive thymocytes24 (H. Tanaka, T.N. and I.T., manuscript submitted). As a consequence of helper versus cytotoxic lineage choice, this bivalent state resolves to H3K4me3-only or H3K27me3-only status in CD4+ and CD8+ cells, respectively. Importantly, an increase or attenuation of the silencer activity alters the H3K4me3/H3K27me3 ratio at the Thpok locus (H. Tanaka, T.N. and I.T., manuscript submitted). So it is conceivable that the expression of Thpok gene is under epigenetic control.
There is also evidence for the involvement of repressive epigenetic machinery in the regulation of other lineage-determining genes. For instance, the Foxp3 locus has a conserved non-coding sequence that is differentially methylated between natural regulatory T (Treg) cells and induced Treg or naive CD4+ T cells.25 The demethylation at this region correlates well with stable Foxp3 expression, and thereby stable Treg cell identity.26 In the Gata3 locus, a distal promoter region is covered by H3K27me3 and polycomb complexes in naive CD4+ T cells, which express the Gata3 gene at a modest level. Upon differentiation toward the T helper type 2 (Th2) subset, H3K4me3 and Menin–trithorax complex take over H3K27me3 and Polycomb complexes, and induce a concomitant increase in Gata3 expression level.27 These observations strongly support the idea that epigenetic changes from a repressive state into an active one play a crucial role in regulating cell fate decisions.
Approaches from trans-acting factors that regulate epigenetic modifications
Another approach complementary to the one described above is to knock out constituent(s) of the epigenetic machinery and examine the resulting effect on T-cell differentiation and function. As covering all the molecules listed in Table 1 is beyond the scope and space of this article, we focus on the epigenetic machinery involved in gene repression and discuss how perturbation of parts of it affects T-lymphocyte differentiation and function.
DNA methylation
Dnmt1 is a DNA cytosine methyltransferase that recognizes a hemi-methylated DNA and introduces methyl-groups on the unmethylated strand.4 Given this conservative mode of DNA methylation, Dnmt1 is often called the ‘maintenance methylase’. Germline knockout of Dnmt1 causes embryonic lethality.28 To investigate its role in T-cell differentiation and function, an experimental system that enables T-cell-specific Dnmt1 inactivation was created using the Cre/LoxP system. Dnmt1 deletion in the early stage of T-cell differentiation using the Lck-Cre transgene resulted in reduced cell survival.29 Although the Dnmt1-deficiency did not significantly influence thymocyte differentiation including CD4 versus CD8 lineage choice, the number of γδ T cells was increased, the majority of which were atypical CD8+ γδ T cells. In the mutant using a Cd4-Cre transgene, which acts slightly later than Lck-Cre at the transition stage from late DN4 to double-positive thymocytes, a decrease in the number of CD44hi memory T cells in both CD4+ and CD8+ subsets as well as increased cytokine mRNA expression from activated T cells were observed.29 Repression of Th2 cytokine (i.e. Il4) genes was reversed in both CD4+ and CD8+ T cells. Similarly, expression of the Th1 cytokine interferon-γ (IFN-γ) was increased in the absence of Dnmt1. However, analyses of the cytokine level after exposure to Th1- or Th2-polarizing conditions indicated that DNA methylation would determine the basal level of expression but not the responsiveness to a specific cue.30 Additionally, loss of Dnmt1 did not significantly affect the expression of T-bet and Gata3,30,31 the key transcription factors required for Th1 and Th2 cell differentiation, respectively, nor the cytotoxic lineage-affiliated genes,30 suggesting that the deregulated production of high amounts of cytokines is a result of the loss of direct repressive regulation of these cytokine genes by Dnmt1 rather than an indirect secondary effect via impairment in the lineage choice.
The knockout of a methyl-DNA binding protein, MBD2, shows a similar phenotype to that of Dnmt1. MBD2 knockout mice did not show any significant developmental arrest during T-lymphocyte development.32 However Mbd2−/− CD4+ T cells produced higher levels of IFN-γ and interleukin-4 (IL-4) in both Th1- and Th2-skewing conditions compared with wild-type cells. MBD2 regulates Il4 by directly competing with Gata3 protein at the key regulatory regions in the Il4 gene, such as CNS-1 and an intronic enhancer. Similar to Dnmt1 knockout, increased IL-4 production was not accompanied by an increase of Gata3 mRNA, again suggesting that the observed effect might not be the result of a lineage diversion.
Histone acetylation
Histone acetylation is associated with active transcription, whereas deacetylation is associated with transcriptional repression, and deacetylases, e.g. members of the HDAC family proteins, are found in many co-repressor complexes. SIRT1, which belongs to the Sirtuin family proteins, is an NAD-dependent deacetylase of histones as well as other proteins, and is considered to be a sensor of metabolic state.33 T-cell-specific Sirt1 deletion, using either Cd4-Cre or Foxp3-Cre, did not affect the differentiation of conventional T cells and Foxp3+ Treg cells.34 However, SIRT1 deficiency caused an increase in Foxp3 expression level at both mRNA and protein levels in Treg cells, thereby resulting in enhanced immune suppression in mice. This is consistent with the previous report that SIRT1 destabilizes Foxp3 by directly deacetylating FoxP3 itself.35 Hence SIRT1 is likely to tune Treg cell functions by regulating the amount of FoxP3.
H3K9 methylation
Suv39h1 is the first histone methyltransferase identified,36 and is responsible for tri-methylation of histone H3 Lys9 residue (H3K9me3),37,38 which in turn recruits heterochromatin protein 1 (HP1)39,40 to mediate gene repression. The major role of Suv39h1 has been linked to heterochromatin maintenance,41 but there is also evidence that it regulates euchromatic gene expression as well. Suv39h1 knockout mice did not have an apparent defect in thymocyte differentiation and Th1/Th2 differentiation in primary culture. However, when Th2 cells were re-stimulated under Th1 conditions, substantial expression of Th1-associated molecules, IFN-γ and T-bet, was observed.42 In those Suv39h1-deficient cells, there was a decrease in H3K9me3 depositions at various Ifng regulatory regions, and an increase in the acetylation of histone H3 Lys9 (H3K9ac) at those regions as well as the promoter of the Tbx21 gene, which encodes T-bet. Given a reported interaction between Suv39h1 and HDAC1/2,43 this increase in H3K9ac levels may be explained in part by the loss of HDAC1/2 recruitment to these regions. As there was no major alteration in the chromatin status of other important Th1 or Th2 signature genes, Suv39h1 seems to be required specifically for the repression of Ifng and Tbx21 genes in Th2 cells. In the same report, the knockout of the Hp1α gene is also described to show a similar phenotype to that observed in Suv39h1 knockout mice,42 consistent with the fact that HP1α binds not only to H3K9me3, which is created by Suv39h1, but also to Suv39h1 itself.44
G9a is another H3K9 methyltransferase that preferentially catalyses H3K9 mono-methylation (H3K9me1) and di-methylation (H3K9me2) in the euchromatic region.37,38,45,46 Although overall T-lymphocyte differentiation was not altered in the absence of G9a,47,48 G9a turned out to play a role in regulating cytokine gene expression during differentiation of effector CD4+ T-cell subsets.48 The H3K9me2 level was decreased at both Th1 and Th2 cytokine genes in the absence of G9a. Despite this H3K9me2 reduction, expression levels of Th2 cytokines were reduced in cells that were differentiated under both neutral and Th2-skewing conditions, whereas IFN-γ production was significantly increased in neutral conditions. Interestingly, a chemical compound that inhibits methyltransferase activity of G9a did not induce deregulated expression of the above-mentioned cytokines, suggesting a possibility that the catalytic activity of G9a might be dispensable for regulation of these cytokine loci.
Polycomb complexes
Polycomb group genes were initially identified as genes that caused homeotic transformation in Drosophila. Further genetic and biochemical characterization showed that Polycomb group proteins are repressors of gene expression and exert their function by forming two distinct protein complexes, namely Polycomb repressive complex (PRC) 1 and PRC2. The core components of Drosophila PRC1 are Polycomb, Posterior sex comb, polyhomeotic and RING1.49 In mammals there are two or more homologues for each of these subunits. Through its chromodomain-containing subunit, PRC1 can be recruited to tri-methylated histone H3 Lys27 (H3K27me3), which is catalysed by PRC2.50 However, there is evidence indicating that PRC1 can function independently of PRC2/H3K27me3.51–53 Another important biochemical function of PRC1 is to mono-ubiquitinate histone H2A Lys119.54,55 This reaction is catalysed by the Ring1B subunit, a mammalian homologue of dRING1,55,56 and is considered to be important for gene repression. T-cell-specific deletion of Ring1b did not apparently affect thymocyte differentiation.57 However, a decrease in Th2 cytokine production was observed in Ring1B-deficient Th2 cells, in which Gata3 protein, but not Gata3 mRNA, was decreased. This suggests that direct regulation of Gata3 by Ring1B might not be responsible for the observed lower production of Th2 cytokines. Also, Ring1B-deficient Th2 cells were more susceptible to apoptotic cell death, in part because of elevated expression of pro-apoptotic factor Bim.57 The lower Th2 cytokine production and higher susceptibility for apoptosis result in attenuated Th2-type responses, and so antigen-induced allergic airway inflammation was reduced in the Ring1b−/− mouse.
Mel-18 is also a subunit of PRC1 and possesses a Ring-finger domain. Mel-18 inactivation resulted in impaired Th2 cytokine production and Gata3 expression under Th2 conditions. The defect was rescued by introducing Gata3, demonstrating that impaired Th2 cytokine production is mainly a result of decreased Gata3 expression.58 Bmi-1 is another subunit of PRC1, and is structurally similar to Mel-18. It is proposed that Bmi-1 and Mel-18 are present in highly similar but distinct PRC1 sub-complexes.59 Bmi-1 over-expression led to an enhancement of Th2 cytokine production in a RING-finger domain-dependent manner. Conversely, Bmi1−/− CD4+ T cells were deficient in Th2 cytokine production with a concomitant increase in Th1 cells. Given a putative role of Bmi-1 in stabilizing Gata3 protein by direct interaction,60 impaired Th2 cell differentiation in Bmi-1−/− mice might be attributed, to some extent, to the instability of Gata3 protein. It is intriguing that Ring1B/Mel-18 and Bmi-1 play different roles in Th2 cytokine production, one through transcriptional and the other through post-translational regulation of Gata3, despite their structural similarities. Also, it is worth noting that the Ring1B and Mel-18 are likely to regulate the Gata3 gene in different ways despite the fact that these factors could be in the same complex.
Polycomb repressive complex 2 is another Polycomb complex, which catalyses H3K27 di-methylation (H3K27 me2) and tri-methylation (H3K27me3). Ezh2, a SET-domain containing protein, is a known catalytic subunit of PRC2.50,61–63 T-cell-specific inactivation of Ezh2 at double-positive thymocyte stage did not cause appreciable lineage skewing to either helper or cytotoxic lineage.64 However, the H3K27me3 level was not significantly changed, possibly because of the stability of the H3K27me3 mark or compensation by Ezh1, an Ezh2 homologue. Ezh2 deletion at the thymocyte progenitor stage caused a severe developmental arrest at CD4− CD8− double-negative stage, which was probably the result of defective pre-TCR signalling caused by impaired actin polymerization. This study suggests a difficulty in examining the function of PRC2 in epigenetic regulation during T-lymphocyte differentiation because of its putative role in regulating TCR signalling, which is critical for both T-lymphocyte differentiation and function, besides its involvement in H3K27 methylation.
Current challenges and future directions
As described above, gene knockout approaches have so far provided insights into the roles of the epigenetic machinery in regulating lineage choice and later in lineage maintenance during T-lymphocyte development. Despite an assumption of dramatic effects because of their global roles in gene regulation, a lack of one factor does not result in an apparent developmental arrest or severe lineage skewing in most cases. It is likely that this reflects redundant functions among related factors and pathways, which in turn secure robustness in regulating lineage-specific gene programmes. The majority of the factors described above have at least one homologue or functionally similar molecule, which may compensate and mask the true impact of a single ablated repressive pathway. In addition, there is an alternative possibility that co-activators and co-repressors recruited directly by transcription factors are sufficient to guide cells to their appropriate lineages. If that is the case, what is the role of the epigenetic machinery in lineage determination? One possibility is that it fine-tunes a threshold by which to select one lineage, i.e. modifying a length of time window for lineage commitment. Such a function has been reported in neurogenesis65 and in vernalization of Arabidopsis.66 Alternatively, epigenetic modification might act mainly as a stabilizer of gene programmes that are established by transcription factor circuits. These scenarios are not necessarily mutually exclusive.
There are several directions that would further address the role of epigenetics in lineage choice. First, a conventional and time-consuming, but potentially the most fruitful, approach is to analyse the function of each individual regulatory element for its role in regulating epigenetic modifications of key transcription factor loci. This approach can benefit from the astonishing progress in genomics and epigenomics, assisted by the mass-sequencing technique. This technique has enabled us to identify potential regulatory elements and their putative regulators, so deducing their complex regulatory networks as exemplified by the ENCODE project (see ref. 67 and references therein). Such genome-wide approaches will provide useful information and generate hypotheses to be further explored by conventional reverse-genetics approaches. Currently, several new methods that enable us to identify proteins associated with regions of interest are reported.68,69 Although these methods are in their infancy, they will become powerful tools with which to explore the epigenomic landscape in coming years as they mature. Another important challenge is to manipulate specific epigenetic modifications at a single-locus level and examine the outcome. This approach has a precedent,70 and is becoming more feasible thanks to the advance in zinc-finger engineering technology. Combining these techniques will enable us to dissect the molecular events occurring on the key regulatory elements and to examine the role of each event in closer detail. Second, extensive and comprehensive knockout approaches are also to be considered. For instance, to overcome the redundancy problem, generating double-, triple-, or more if necessary, mutant mice for the related epigenetic factors will bring new insights. The potential problem of this approach is that these factors have not only shared functions but also distinct functions in many cases. Hence, interpreting the results from multiple-mutant mice will need extra caution. So far, most of the knockout approaches have focused on enzymes that add epigenetic modifications to their substrates. However, modification-recognizing or de-modifying factors remain largely untouched. Hence, targeting and analysing those factors is another important path to unveil the function of each modification. Third, it will be important to examine the stability of a given lineage by challenging the cell identity, as was performed recently in Suv39h1 mutant mice in the context of Th1/2 lineages.42 It is possible to further extend this point by assessing the reprogramming potential of mutant cells in an assay that examines whether lineage conversion by ectopic transduction of lineage-inducing transcription factors is facilitated by the mutation.
Even though the forest of epigenetics is far wider and deeper than we previously thought, it will become possible to map every single tree in the forest through the use of all the tools and resources that we have now and will have in the future.
Acknowledgments
We are grateful to W. Seo and S. Nieke for critical reading and discussion of the manuscript. The work was supported by a Grant-in-Aid for Scientific Research (S) and for Scientific Research on Priority Areas (I.T.).
Disclosures
The authors declare no competing financial interests.
References
- 1.Lewis J, Bird A. DNA methylation and chromatin structure. FEBS Lett. 1991;285:155–9. doi: 10.1016/0014-5793(91)80795-5. [DOI] [PubMed] [Google Scholar]
- 2.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–20. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 3.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 4.Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971–83. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–65. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 12.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–35. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng HH, Zhang Y, Hendrich B, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- 15.Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem. 2003;278:24132–8. doi: 10.1074/jbc.M302283200. [DOI] [PubMed] [Google Scholar]
- 16.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 17.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr Opin Genet Dev. 2012;22:148–55. doi: 10.1016/j.gde.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 20.He X, Dave VP, Zhang Y, Hua X, Nicolas E, Xu W, Roe BA, Kappes DJ. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433:826–33. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 21.Sun G, Liu X, Mercado P, Jenkinson SR, Kypriotou M, Feigenbaum L, Galera P, Bosselut R. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat Immunol. 2005;6:373–81. doi: 10.1038/ni1183. [DOI] [PubMed] [Google Scholar]
- 22.Setoguchi R, Tachibana M, Naoe Y, Muroi S, Akiyama K, Tezuka C, Okuda T, Taniuchi I. Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science. 2008;319:822–5. doi: 10.1126/science.1151844. [DOI] [PubMed] [Google Scholar]
- 23.He X, Park K, Wang H, Zhang Y, Hua X, Li Y, Kappes DJ. CD4-CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity. 2008;28:346–58. doi: 10.1016/j.immuni.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Jackson AF, Naito T, et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat Immunol. 2012;13:86–94. doi: 10.1038/ni.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol. 2007;37:2378–89. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 26.Polansky JK, Kretschmer K, Freyer J, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–63. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 27.Onodera A, Yamashita M, Endo Y, et al. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J Exp Med. 2010;207:2493–506. doi: 10.1084/jem.20100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 29.Lee PP, Fitzpatrick DR, Beard C, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–74. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 30.Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol. 2004;173:4402–6. doi: 10.4049/jimmunol.173.7.4402. [DOI] [PubMed] [Google Scholar]
- 31.Makar KW, Perez-Melgosa M, Shnyreva M, Weaver WM, Fitzpatrick DR, Wilson CB. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T cells. Nat Immunol. 2003;4:1183–90. doi: 10.1038/ni1004. [DOI] [PubMed] [Google Scholar]
- 32.Hutchins AS, Mullen AC, Lee HW, Sykes KJ, High FA, Hendrich BD, Bird AP, Reiner SL. Gene silencing quantitatively controls the function of a developmental trans-activator. Mol Cell. 2002;10:81–91. doi: 10.1016/s1097-2765(02)00564-6. [DOI] [PubMed] [Google Scholar]
- 33.Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31:212–20. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, Hancock WW. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol. 2011;31:1022–9. doi: 10.1128/MCB.01206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Loosdregt J, Vercoulen Y, Guichelaar T, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115:965–74. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 36.Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 37.Peters AH, Kubicek S, Mechtler K, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–89. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 38.Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–8. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 39.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 40.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 41.Peters AH, O'Carroll D, Scherthan H, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–37. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 42.Allan RS, Zueva E, Cammas F, et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature. 2012;487:249–53. doi: 10.1038/nature11173. [DOI] [PubMed] [Google Scholar]
- 43.Vaute O, Nicolas E, Vandel L, Trouche D. Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res. 2002;30:475–81. doi: 10.1093/nar/30.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aagaard L, Laible G, Selenko P, et al. Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3–9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J. 1999;18:1923–38. doi: 10.1093/emboj/18.7.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309–17. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- 46.Tachibana M, Sugimoto K, Nozaki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–91. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas LR, Miyashita H, Cobb RM, et al. Functional analysis of histone methyltransferase g9a in B and T lymphocytes. J Immunol. 2008;181:485–93. doi: 10.4049/jimmunol.181.1.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, Rossi FM, Zaph C. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J Exp Med. 2010;207:915–22. doi: 10.1084/jem.20100363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 50.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 51.Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 2006;25:3110–22. doi: 10.1038/sj.emboj.7601187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tavares L, Dimitrova E, Oxley D, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664–78. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu M, Mazor T, Huang H, et al. Direct recruitment of polycomb repressive complex 1 to chromatin by core binding transcription factors. Mol Cell. 2012;45:330–43. doi: 10.1016/j.molcel.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Napoles M, Mermoud JE, Wakao R, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. 2004;7:663–76. doi: 10.1016/j.devcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 56.Elderkin S, Maertens GN, Endoh M, et al. A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol Cell. 2007;28:107–20. doi: 10.1016/j.molcel.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki A, Iwamura C, Shinoda K, et al. Polycomb group gene product Ring1B regulates Th2-driven airway inflammation through the inhibition of Bim-mediated apoptosis of effector Th2 cells in the lung. J Immunol. 2010;184:4510–20. doi: 10.4049/jimmunol.0903426. [DOI] [PubMed] [Google Scholar]
- 58.Kimura M, Koseki Y, Yamashita M, et al. Regulation of Th2 cell differentiation by mel-18, a mammalian polycomb group gene. Immunity. 2001;15:275–87. doi: 10.1016/s1074-7613(01)00182-0. [DOI] [PubMed] [Google Scholar]
- 59.Wiederschain D, Chen L, Johnson B, et al. Contribution of polycomb homologues Bmi-1 and Mel-18 to medulloblastoma pathogenesis. Mol Cell Biol. 2007;27:4968–79. doi: 10.1128/MCB.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosokawa H, Kimura MY, Shinnakasu R, et al. Regulation of Th2 cell development by Polycomb group gene bmi-1 through the stabilization of GATA3. J Immunol. 2006;177:7656–64. doi: 10.4049/jimmunol.177.11.7656. [DOI] [PubMed] [Google Scholar]
- 61.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muller J, Hart CM, Francis NJ, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 63.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 64.Su IH, Dobenecker MW, Dickinson E, et al. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell. 2005;121:425–36. doi: 10.1016/j.cell.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 65.Hirabayashi Y, Suzki N, Tsuboi M, et al. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63:600–13. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 66.Angel A, Song J, Dean C, Howard M. A Polycomb-based switch underlying quantitative epigenetic memory. Nature. 2011;476:105–8. doi: 10.1038/nature10241. [DOI] [PubMed] [Google Scholar]
- 67.Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E. Genomics: ENCODE explained. Nature. 2012;489:52–5. doi: 10.1038/489052a. [DOI] [PubMed] [Google Scholar]
- 68.Dejardin J, Kingston RE. Purification of proteins associated with specific genomic loci. Cell. 2009;136:175–86. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoshino A, Fujii H. Insertional chromatin immunoprecipitation: a method for isolating specific genomic regions. J Biosci Bioeng. 2009;108:446–9. doi: 10.1016/j.jbiosc.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 70.Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol. 2002;12:2159–66. doi: 10.1016/s0960-9822(02)01391-x. [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Zhang J, Rui J, Liu X. p300-mediated acetylation stabilizes the Th-inducing POK factor. J Immunol. 2010;185:3960–9. doi: 10.4049/jimmunol.1001462. [DOI] [PubMed] [Google Scholar]
- 72.Rui J, Liu H, Zhu X, Cui Y, Liu X. Epigenetic silencing of Cd8 genes by ThPOK-mediated deacetylation during CD4 T cell differentiation. J Immunol. 2012;189:1380–90. doi: 10.4049/jimmunol.1201077. [DOI] [PubMed] [Google Scholar]
- 73.Reed-Inderbitzin E, Moreno-Miralles I, Vanden-Eynden SK, et al. RUNX1 associates with histone deacetylases and SUV39H1 to repress transcription. Oncogene. 2006;25:5777–86. doi: 10.1038/sj.onc.1209591. [DOI] [PubMed] [Google Scholar]
- 74.Philipot O, Joliot V, Ait-Mohamed O, Pellentz C, Robin P, Fritsch L, Ait-Si-Ali S. The core binding factor CBF negatively regulates skeletal muscle terminal differentiation. PLoS ONE. 2010;5:e9425. doi: 10.1371/journal.pone.0009425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chakraborty S, Sinha KK, Senyuk V, Nucifora G. SUV39H1 interacts with AML1 and abrogates AML1 transactivity. AML1 is methylated in vivo. Oncogene. 2003;22:5229–37. doi: 10.1038/sj.onc.1206600. [DOI] [PubMed] [Google Scholar]
- 76.Kitabayashi I, Yokoyama A, Shimizu K, Ohki M. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J. 1998;17:2994–3004. doi: 10.1093/emboj/17.11.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen GY, Osada H, Santamaria-Babi LF, Kannagi R. Interaction of GATA-3/T-bet transcription factors regulates expression of sialyl Lewis X homing receptors on Th1/Th2 lymphocytes. Proc Natl Acad Sci U S A. 2006;103:16894–9. doi: 10.1073/pnas.0607926103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008;22:2980–93. doi: 10.1101/gad.1689708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han S, Lu J, Zhang Y, et al. Recruitment of histone deacetylase 4 by transcription factors represses interleukin-5 transcription. Biochem J. 2006;400:439–48. doi: 10.1042/BJ20061085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dang EV, Barbi J, Yang HY, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32:606–13. doi: 10.1038/ng1018. [DOI] [PubMed] [Google Scholar]
- 83.Lemercier C, Brocard MP, Puvion-Dutilleul F, Kao HY, Albagli O, Khochbin S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J Biol Chem. 2002;277:22045–52. doi: 10.1074/jbc.M201736200. [DOI] [PubMed] [Google Scholar]