

Abstract
Allergen-specific IgE has long been regarded as a major molecular component of allergic asthma. Additionally, there is increasing evidence of the important roles of interleukin-33 (IL-33) in the disease. Here, we show that IL-33 and alveolar macrophages play essential roles in the exacerbation of IgE-mediated airway inflammation and remodelling. BALB/c mice passively sensitized with ovalbumin (OVA)-specific IgE monoclonal antibody (mAb) were challenged with OVA seven times intratracheally. The seventh challenge exacerbated airway inflammation and remodelling compared with the fourth challenge; furthermore, markedly increased expression of IL-33 in the lungs was observed at the fourth and seventh challenges. When anti-IL-33 or anti-ST2 antibody was administered during the fourth to seventh challenge, airway inflammation and remodelling were significantly inhibited at the seventh challenge. Because increases of IL-33+ and ST2+ alveolar macrophages and ST2+ CD4+ T cells in the lungs were observed at the fourth challenge, the roles of macrophages and CD4+ cells were investigated. Depletion of macrophages by 2-chloroadenosine during the fourth to seventh challenge suppressed airway inflammation and remodelling, and IL-33 production in the lung at the seventh challenge; additionally, anti-CD4 mAb inhibited airway inflammation, but not airway remodelling and IL-33 production. Meanwhile, treatment with 2-chloroadenosine or anti-CD4 mAb decreased IL-33-induced airway inflammation in normal mice; airway remodelling was repressed only by 2-chloroadenosine. These results illustrate that macrophage-derived IL-33 contributes to the exacerbation of IgE-mediated airway inflammation by mechanisms associated with macrophages and CD4+ cells, and airway remodelling through the activation of macrophages.
Keywords: airway remodelling, asthma, IgE, interleukin-33, macrophages
Introduction
Patients with allergic asthma have elevated levels of allergen-specific IgE, and the disease is a chronic inflammatory disorder of the airways characterized by reversible airway obstruction, airway hyper-responsiveness (AHR) and eosinophilic airway inflammation.1–4 IgE has been shown to mediate early airway obstruction via the activation of mast cells and basophils, which release various chemical mediators.5 Furthermore, IgE has the capacity to induce infiltration by eosinophils and AHR through the production of T helper type 2 (Th2) cytokines, such as interleukin-4 (IL-4), IL-5 and IL-13,6,7 indicating that allergen-specific IgE can be indispensable for the chronic pathogenesis of asthma. Meanwhile, many studies have suggested that chronic airway inflammation observed in the asthmatic airways causes airway remodelling, such as airway wall thickening due to goblet cell hyperplasia, sub-epithelial fibrosis and airway smooth muscle thickening.8,9 However, the precise mechanisms leading to airway remodelling are still unknown, although it is thought to result from an injury-repair response driven by many inflammatory cells and cellular elements in the airway.
There is increasing evidence of the important roles of IL-33 in allergic asthma. Interleukin-33, which is a member of the IL-1 family of cytokines that includes IL-1 and IL-18, has been identified as a ligand for ST2.10,11 Increased levels of IL-33 have been found in serum and the lungs of patients with allergic asthma;12–14 furthermore, in the lungs of mice with antigen-induced airway inflammation, IL-33 is expressed at substantial levels.13 The pathogenic role of IL-33 in airway inflammation emerged from experiments with intranasal administration of IL-33 into naive mice that then exhibited the production of Th2 cytokines, eosinophilic pulmonary inflammation and AHR.13,15,16 Additionally, in allergic models of asthma, administration of neutralizing antibodies against IL-33 or ST2 at the time of airway challenge with antigen attenuated the production of Th2 cytokines, eosinophilic inflammation and AHR.17–19 Hence, IL-33 can contribute to the development of allergic asthma; however, it has not been fully defined whether IL-33 contributes to IgE-mediated chronic allergic asthma, especially in airway remodelling.
On the other hand, alveolar macrophages are the most abundant cells in the alveolar spaces and conducting airways and are known to be involved in immune homeostasis in the respiratory tract.20 Several reports have suggested that alveolar macrophages suppress the allergen-specific immune response and airway inflammation,21–24 whereas the definition and function of alveolar macrophages have been expanded, particularly with regard to their role in regulating Th2-type inflammatory responses, as alveolar macrophages are involved in the development of Th2-dependent immunity;25,26 however, the role of alveolar macrophages in allergic asthma has not been fully defined.
We have previously reported that BALB/c mice passively sensitized with an intraperitoneal injection of ovalbumin (OVA) -specific IgE monoclonal antibody (mAb) showed not only airway inflammation but also airway remodelling by seven repeated intratracheal OVA challenges.27,28 In the current study, we aimed to define the cells and molecules essential to the development of these IgE-mediated responses, focusing on the roles of IL-33, macrophages and CD4+ cells. In this IgE-sensitized model, a key feature was that the seventh antigen challenge exacerbated airway inflammation and remodelling in comparison with the fourth challenge; furthermore, markedly increased expression of IL-33 in the lungs was observed at the fourth and seventh challenges. First, we examined the roles of IL-33 and ST2 in airway inflammation and remodelling during the fourth to seventh challenges by conducting pathological analyses using anti-IL-33 and anti-ST2 antibodies. Second, we assessed the roles of macrophages and CD4+ cells in IL-33 production in the lungs and these responses by conducting pathological analyses using 2-chloroadenosine (2-CA) and anti-CD4 mAb. Third, we assessed whether macrophages and CD4+ cells contribute to repeated intratracheal administration of IL-33-induced airway inflammation and remodelling in normal mice by conducting analyses using 2-CA and anti-CD4 mAb.
Materials and methods
Animals
Male 7-week-old BALB/c mice were obtained from Japan SLC (Hamamatsu, Japan). These mice were maintained in a temperature-controlled environment with free access to standard rodent chow and water. All of the experimental procedures were approved by the Experimental Animal Research Committee at Kobe Pharmaceutical University.
OVA-specific IgE mAb
The OVA-specific IgE mAb (OE-1) was derived from a B-cell hybridoma producing murine IgE, as described previously.27,28 The hybridoma was grown in CELLine CL1000 with BD-Cell-MAb medium (BD Biosciences, San Diego, CA) supplemented with 20% heat-inactivated fetal bovine serum, 1% l-glutamine and 1% penicillin–streptomycin. Levels of OE-1 in culture supernatants of hybridoma were assayed by ELISA; OE-1 was detected using plates coated with anti-mouse IgE antibody and by adding biotin-labelled anti-mouse IgE antibody. Alkaline phosphate anti-biotin was added, and then the plate was developed with p-nitrophenyl phosphate, and measurements were made at 405 nm using a microplate reader. OE-1 levels were calculated by comparison with mouse IgE standards (Southern Biotech, Birmingham, AL).
Passive sensitization with specific IgE mAb
Passive sensitization with OE-1 was performed with modification of the protocol described previously.27 As shown in Fig. 1(a), BALB/c mice were passively sensitized with repeated intraperitoneal injections of a hybridoma supernatant containing OE-1 (100 μg/mouse) on days 0, 1 and 2. Non-sensitized mice were injected with a culture supernatant of the parental myeloma cell line. Both the sensitized and non-sensitized mice were challenged on days 1, 2, 3, 8, 9, 10 and 15 under anaesthesia with escain (Mylan, Osaka, Japan) with 1% OVA (grade V; Sigma-Aldrich, St Louis, MO) in a volume of 20 μl by intratracheal administration as reported previously.27,28 Additionally, we have reported that purified OE-1 also induced allergic airway inflammation by repeated antigen challenges.28.
Figure 1.

Changes in interleukin-33 (IL-33) production, airway inflammation and airway remodelling after repeated antigen challenges in IgE-sensitized mice. (a) Experimental protocol for sensitization with OE-1 and challenge with antigen in BALB/c mice. (b) Levels of IL-33 in lung homogenate supernatants 24 hr after the seventh challenge in non-sensitized-challenged [NS-C (7th)] and before the fourth [OE-1 (4th before)] or 24 hr after the fourth [OE-1 (4th)] and seventh [OE-1 (7th)] challenges in mice sensitized with OE-1. (c) Changes in inflammatory cell numbers in bronchoalveolar lavage fluid in NS-C (7th), OE-1 (4th) and OE-1 (7th) groups. (d–g) Changes in airway inflammation (d), goblet cell hyperplasia (e), sub-epithelial fibrosis (f) and smooth muscle cell hypertrophy (g) in NS-C (7th) (i) and OE-1 (7th) (ii) groups. Bar = 100 µm. (h) Changes in airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) in NS-C (7th), OE-1 (4th) and OE-1 (7th) groups. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of six or seven animals. *P < 0·05 and **P < 0·01 compared with the NS-C (7th) group. P < 0·05 and P < 0·01 compared with the S-C (4th) group. &&P < 0·01 compared with the OE-1 (4th before) group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Treatment with neutralizing antibodies against IL-33, ST2 and CD4, and 2-CA
As shown in Fig. 2(a) or 3(a), on days 8, 9, 10 and 15, a dose (5 μg/mouse) of anti-IL-33 mAb (MAB3626; R&D Systems, Minneapolis, MN) or anti-ST2 polyclonal antibody (pAb) (AF1004; R&D Systems) was administered intratracheally 30 min before each of the challenges to mice sensitized with OE-1, and control mice were given the same amount of rat IgG2a (R&D Systems) or goat IgG (R&D Systems), respectively. A dose of anti-IL-33 mAb or anti-ST2 pAb has been reported to significantly reduce the late-phase inflammatory response in passive cutaneous anaphylaxis in mice.29
Figure 2.
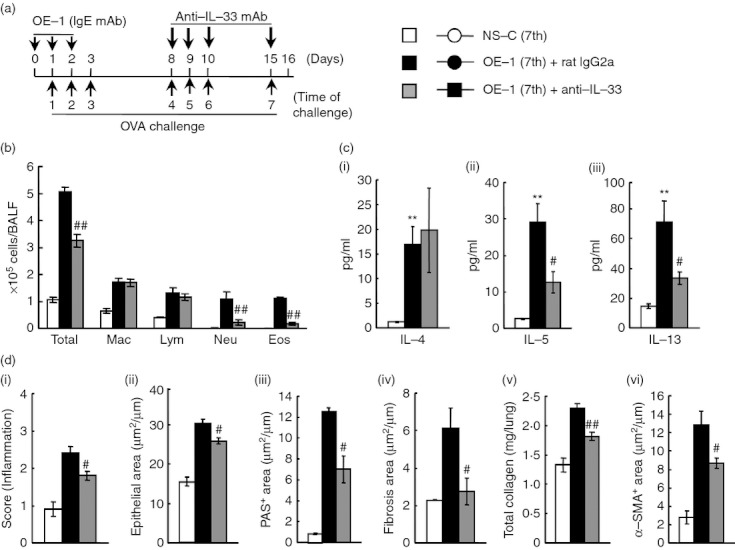
Effect of anti-interleukin-33 monoclonal antibody (IL-33 mAb) on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice. (a) Experimental protocol for treatment with anti-IL-33 mAb. Anti-IL-33 mAb was intratracheally administered on days 8, 9, 10 and 15 [OE-1 (7th) + anti-IL-33]. Negative and positive controls were non-sensitized-challenged [NS-C (7th)] and OE-1-sensitized-challenged, control rat IgG2a mAb-treated [OE-1 (7th) + rat IgG2a] mice, respectively. (b) Effect of treatment with anti-IL-33 mAb on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the seventh challenge. (c) Effect of treatment with anti-IL-33 mAb on increased levels of IL-4 (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the seventh challenge. (d) Effect of treatment with anti-IL-33 mAb on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the seventh challenge. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of six to eight animals. **P < 0·01 compared with the NS-C (7th) group. #P < 0·05 and ##P < 0·01 compared with the OE-1 (7th) + rat IgG2a group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Figure 3.
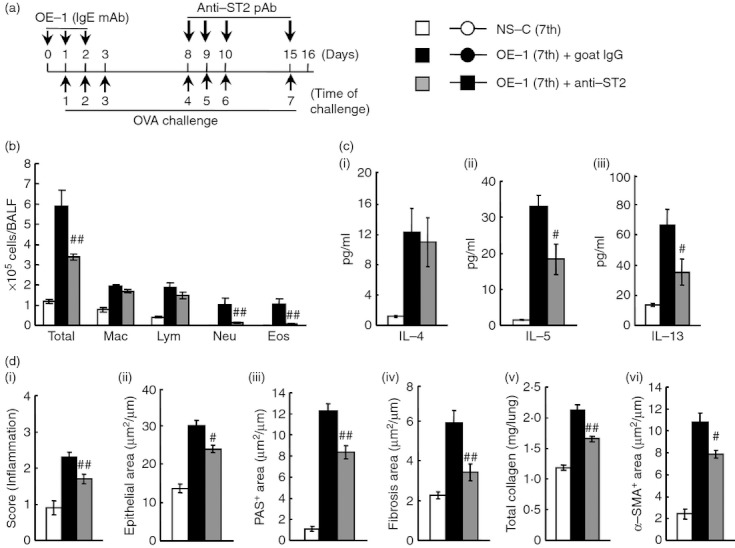
Effect of anti-ST2 polyclnoal antibody (pAb) on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice. (a) Experimental protocol for treatment with anti-ST2 pAb. Anti-ST2 pAb was intratracheally administered on days 8, 9, 10 and 15 [OE-1 (7th) + anti-ST2]. Negative and positive controls were non-sensitized-challenged [NS-C (7th)] and OE-1-sensitized-challenged, control goat IgG-treated [OE-1 (7th) + goat IgG] mice, respectively. (b) Effect of treatment with anti-ST2 pAb on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the seventh challenge. (c) Effect of treatment with anti-ST2 pAb on increased levels of interleukin-4 (IL-4) (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the seventh challenge. (d) Effect of treatment with anti-ST2 pAb on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the seventh challenge. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of five to eight animals. #P < 0·05 and ##P < 0·01 compared with the OE-1 (7th) + goat IgG group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
As shown Fig. 4(e), to deplete macrophages, 4 mm 2-CA (Sigma-Aldrich) in a volume of 20 μl by intratracheal administration was administered 18 h before the fourth to seventh antigen challenge to mice sensitized with OE-1. Intratracheal or aerosolized administration of 2-CA (1–5 mm) has been reported to cause a specific reduction in alveolar macrophages, but not other cells, in asthmatic mice.30,31 Furthermore, we confirmed that treatment with 2-CA (4 mm, 20 μl/mouse) 6 hr after the fourth administration of OVA to mice actively sensitized with OVA + alum significantly reduced the number of macrophages in bronchoalveolar lavage fluid (BALF) 24 hr after the antigen challenge, but not other cells (lymphocytes, neutrophils and eosinophils).
Figure 4.
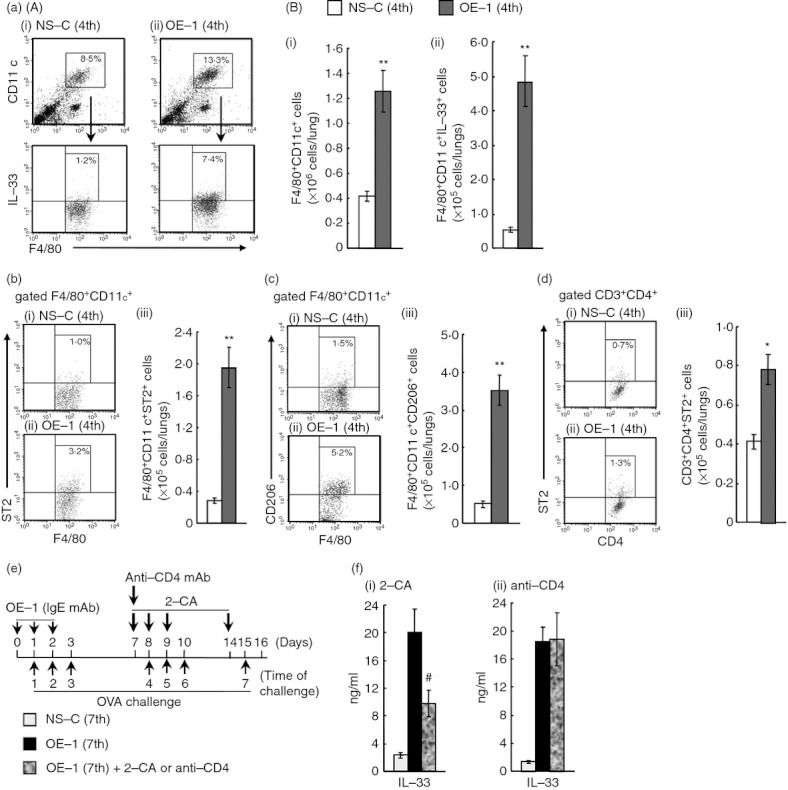
Effect of 2-chloroadenosine (2-CA) on interleukin-33 (IL-33) production in the lungs after the seventh antigen challenge in IgE-sensitized mice. (a) (A) Representative dot plots depicting the percentages of alveolar macrophages (F4/80+ CD11c+) and IL-33+ alveolar macrophages (F4/80+ CD11c+ IL-33+) in the lungs 24 hr after the fourth challenge in non-sensitized [NS-C (4th)] (i) and IgE-sensitized mice [OE-1 (4th)] (ii). (B) Changes in the number of alveolar macrophages (F4/80+ CD11c+ cells) (i) and IL-33+ alveolar macrophages (ii) in the lungs. (b) Representative dot plots depicting the percentage of F4/80+ CD11c+ ST2+ in the lungs 24 hr after the fourth challenge in NS-C (4th) (i) and OE-1 (4th) (ii) groups. Changes in the number of F4/80+ CD11c+ ST2+ in the lungs (iii). (c) Representative dot plots depicting the percentage of F4/80+ CD11c+ CD206+ in the lungs 24 hr after the fourth challenge in NS-C (4th) (i) and OE-1 (4th) (ii) groups. Changes in the number of F4/80+ CD11c+ CD206+ in the lungs (iii). (d) Representative dot plots depicting the percentage of CD3+ CD4+ ST2+ in the lungs 24 hr after the fourth challenge in NS-C (4th) (i) and OE-1 (4th) (ii) groups. Changes in the number of CD3+ CD4+ ST2+ in the lungs (iii). (e) Experimental protocol for treatment with 2-CA or anti-CD4 mAb. 2-CA was intratracheally administered on days 7, 8, 9 and 14 [OE-1 (7th) + 2-CA]. Anti-CD4 monoclonal antibody (mAb) was intraperitoneally administered on day 7 [OE-1 (7th) + anti-CD4]. Negative and positive controls were non-sensitized-challenged [NS-C (7th)] and OE-1-sensitized-challenged, vehicle or control rat IgG2b-treated [OE-1 (7th)] mice, respectively. (f) Effects of 2-CA (i) and anti-CD4 mAb (ii) on IL-33 production in lung homogenate supernatants 24 hr after the seventh challenge. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of four to seven animals. *P < 0·05 and **P < 0·01 compared with the NS-C (4th) group. #P < 0·05 and ##P < 0·01 compared with the OE-1 (7th) group.
A cell line producing rat IgG2b mAbs, which recognizes the murine CD4 molecule (YTS191.1.2), was kindly provided by Prof. David D. Chaplin (University of Alabama at Birmingham, Birmingham, AL). Anti-CD4 mAb was produced and purified as described previously.32,33 On day 7, purified anti-CD4 mAb or rat IgG2b (eBioscience, San Diego, CA) was intraperitoneally administered 18 hr before the fourth antigen challenge at 0·6 mg/mouse (Fig. 4e). It has been reported that treatment with anti-CD4 mAb 18 hr before the first administration of antigen to mice actively sensitized with OVA + alum significantly reduced the number of CD4+ cells in BALF, peripheral blood and spleen at the fourth challenge.32,33
Analysis of cells recovered by BAL
To evaluate airway cellular inflammation, we examined the accumulation of inflammatory cells in BALF as detailed previously.34 Animals were killed with diethyl ether. The trachea was cannulated and the left bronchi were tied for histological examination. The right air lumen was washed twice with 0·5 ml Hanks’ balanced salt solution containing 2% heat-inactivated fetal bovine serum. The cell pellet was suspended with a defined volume (200 μl/sample) of Hanks’ balanced salt solution. The total leucocyte count in the lavage fluid was determined by staining with Turk's solution. For differential cell counts, BAL cells were stained with Diff-Quik solution (Sysmex International Reagents, Kobe, Japan).
Histological study
Left lungs were fixed in 10% neutral-buffered formalin, then dissected, embedded in paraffin, and cut 4-μm thick. Sections were stained with haematoxylin & eosin (H&E), periodic acid-Schiff (PAS), and Masson's trichrome.
Two to four specimens of the stained histological preparations of the left lobe (the total length of the epithelial basement membrane of the bronchioles was 1·0–2·5 mm) were photographed, and then the epithelial area (H&E), PAS+ area (PAS), and sub-epithelial fibrotic area beneath the basement membrane at 20 μm depth (Masson's trichrome) were measured using image j (version 1·42; NIH, Bethesda, MD). The mean scores (μm2/μm) of the areas divided by basement membrane length in two to four preparations of one mouse were calculated, and then the mean scores were calculated in each group.
The degree of airway inflammation was analysed as follows: Each section was evaluated for inflammation (H&E) on a scale of 0–4 with increments of 0·5 by a blinded observer.
Immunohistochemistry
Sections of left lungs fixed in 10% neutral-buffered formalin were used for immunohistochemistry. Endogenous peroxidase was blocked with 3% H2O2 in water for 30 min. After blocking non-specific binding with diluted normal rabbit serum in PBS for 20 min, the sections were incubated for 1 hr at room temperature with biotinylated anti-α-SMA mAb (clone 1A4; LifeSpan BioSciences, Seattle, WA). The slides were stained with streptavidin-horseradish peroxidase, and the colour was developed using the diaminobenzidine substrate kit for peroxidase (Vector Laboratories, Burlingame, CA). Counterstaining was performed using Mayer's haematoxylin. As a negative control, biotinylated mouse IgG2a (Cedarlane Laboratories, Burlington, NC) was used. As described above, the mean scores (μm2/μm) were calculated in each group.
Measurement of cytokines and total collagen in lung tissue supernatants
The frozen right lobe (after BAL) was homogenized in 1 ml T-PER (Thermo Scientific, Rockford, IL) containing a Complete Mini Protease Inhibitor Cocktail tablet (Roche, Mannheim, Germany; 1 tablet/10 ml T-PER stock reagent). Lung homogenates were centrifuged at 9000 g for 10 min at 4°. The levels of IL-4, IL-5, IL-13 (R&D Systems) and IL-33 (BioLegend, San Diego, CA) in supernatants of lung homogenates were measured using quantitative colorimetric sandwich ELISA kits.
The total collagen content in supernatants of lung homogenates was determined using the Sircol Collagen Assay kit (Biocolor, Carrickfergus, UK) according to the manufacturer's protocols.
Flow cytometry analysis
The whole lung was isolated, cut into 1-mm3 pieces in digestion buffer [RPMI-1640 containing 150 U/ml collagenase (WAKO, Osaka, Japan), 30 μg/ml DNase I (Sigma-Aldrich) and 10 mm HEPES] and incubated at 37° for 1 hr. The resulting single-cell suspension was washed by centrifugation with PBS supplemented with 2% fetal bovine serum, and cell numbers were determined using staining with trypan blue after treatment with ACK lysis buffer to remove erythrocytes.
Leucocytes recovered from collagenase/DNase I-digested lung tissue were incubated with anti-mouse FcγRII/III mAb (clone 2.4G2; BD Biosciences) to block the binding of subsequent antibodies to FcγRII/III followed by phycoerythrin (PE)- or fluorescein isothiocyanate (FITC)- labelled anti-CD11c mAb, PE-Cy5-labelled anti-F4/80 mAb, FITC-labelled anti-CD3 mAb, FITC-labelled anti-CD206 mAb (all from BioLegend), PE-Cy5-labelled anti-mouse CD4 mAb (BD Biosciences), or PE-labelled anti-ST2 mAb (R&D Systems) in different combinations. After washing, the stained cells were fixed with 4% paraformaldehyde and then analysed using FACSCalibur (BD Biosciences) and cell quest software (version 3·3; BD Biosciences). In addition, to detect IL-33+ alveolar macrophages 24 hr after the fourth challenge, after staining with PE-Cy5-labelled anti-F4/80 mAb and FITC-labelled anti-CD11c mAb, the cells were fixed with 4% paraformaldehyde, made permeable with saponin, and stained with PE-labelled anti-IL-33 mAb (R&D Systems). Alveolar macrophages were sorted based on their expression of F4/80 and CD11c as shown in a previous report.35 Furthermore, M2 (alternative) alveolar macrophages were characterized by the expressions of ST2.36
Changes in airway inflammation and remodelling in response to IL-33
As shown in Figs 7(a) and 8(a), IL-33 (R&D Systems) in solution (200 ng/mouse) was intratracheally administered to normal mice on days 0, 1, 2 and 7. We assessed the effects of the depletion of macrophages and CD4+ cells on the accumulation of inflammatory cells in BALF and airway remodelling 24 hr after the last intratracheal instillation of IL-33 (on day 8). As shown in Fig. 7(a), to deplete macrophages, 4 mm of 2-CA in a volume of 20 μl by intratracheal administration was administered 18 hr before the first to fourth instillations of IL-33 (on day −1, 0, 1, and 6). Furthermore, anti-CD4 mAb (0·6 mg/mouse) was intraperitoneally administered 18 hr before the first instillation of IL-33 (on day −1) (Fig. 8a).
Figure 7.
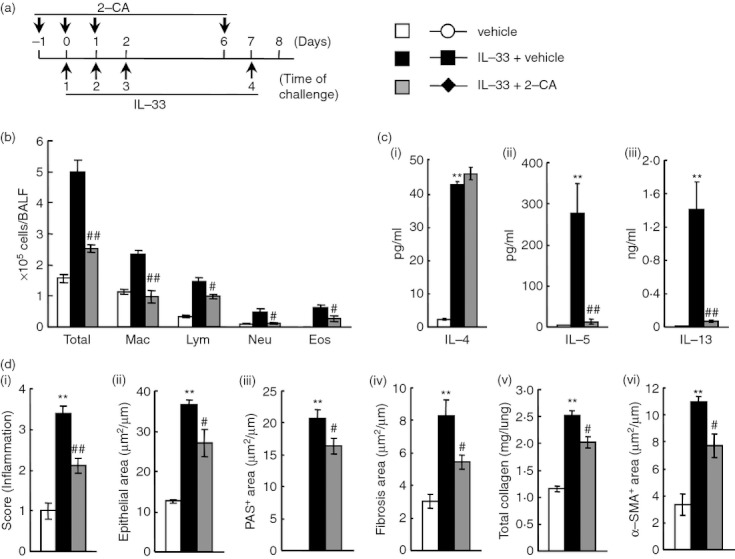
Effect of 2-chloroadenosine (2-CA) on interleukin-33 (IL-33) -induced airway inflammation and remodelling in mice. (a) Experimental protocol for treatment with 2-chloroadenosine (2CA). The 2-CA was intratracheally administered on days –1, 0, 1 and 6 (IL-33 + 2-CA). Negative and positive controls were vehicle-administered (vehicle) and IL-33-administered, vehicle-treated (IL-33 + vehicle) mice, respectively. (b) Effect of treatment with 2-CA on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the last administration of IL-33. (c) Effect of treatment with 2-CA on increased levels of IL-4 (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the last administration. (d) Effect of treatment with 2-CA on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the last administration of IL-33. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of six animals. **P < 0·01 compared with the vehicle group. #P < 0·05 and ##P < 0·01 compared with the IL-33 + vehicle group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Figure 8.
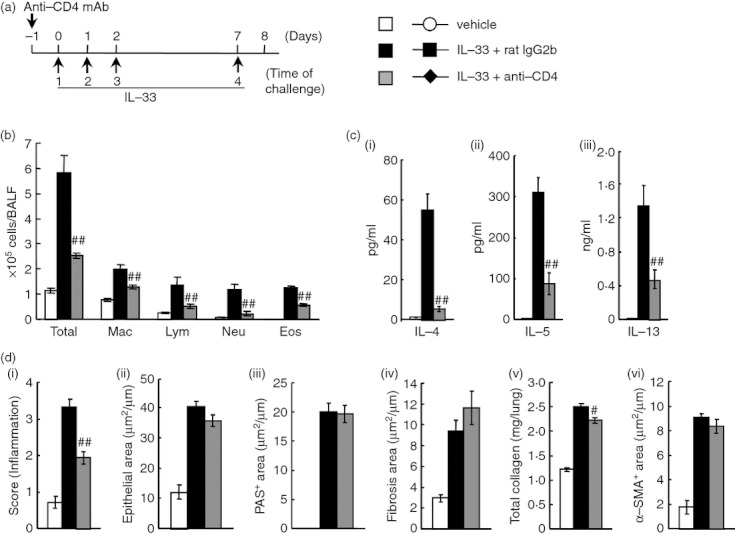
Effect of anti-CD4 monoclonal antibody (mAb) on interleukin-33 (IL-33) -induced airway inflammation and remodelling in mice. (a) Experimental protocol for treatment with anti-CD4 mAb. Anti-CD4 mAb was intraperitoneally administered on day -1 (IL-33 + anti-CD4). Negative and positive controls were vehicle-administered (vehicle) and IL-33-administered, control rat IgG2b-treated (IL-33 + rat IgG2b) mice, respectively. (b) Effect of treatment with anti-CD4 mAb on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the last administration of IL-33. (c) Effect of treatment with anti-CD4 mAb on increased levels of IL-4 (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the last administration. (d) Effect of treatment with anti-CD4 mAb on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the last administration of IL-33. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of six animals. #P < 0·05 and ##P < 0·01 compared with the IL-33 + rat IgG2b group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Statistical analyses
Data are shown as the mean ± SEM. Statistical analyses between the two groups were performed using Student's t-test (two-tailed). To compare more than two groups, Dunnett's test was used after conducting one-way analysis of variance. A probability value of P < 0·05 was considered significant.
Results
Changes in IL-33 production, airway inflammation, and airway remodelling after seven repeated antigen challenges in IgE-sensitized mice
First, to investigate the role of IL-33 in the development of IgE-mediated airway inflammation and remodelling, we assessed whether the production of IL-33 was increased in IgE-sensitized mice. The production of IL-33 in the lung tissue supernatants before the fourth challenge and 24 hr after the fourth and seventh challenges in IgE-sensitized mice was significantly increased in comparison with that in non-sensitized mice; additionally, IL-33 production 24 hr after the fourth and seventh challenges was significantly increased in comparison with that before the fourth challenge (Fig. 1b).
We investigated the changes in infiltration by inflammatory cells in BALF and airway remodelling in the lungs of IgE-sensitized mice. Infiltration by inflammatory cells such as macrophages, lymphocytes and neutrophils in BALF, but not eosinophils, was observed 24 hr after the fourth challenge; furthermore, infiltration by eosinophils was recognized 24 hr after the seventh challenge, although infiltration by macrophages, lymphocytes and neutrophils did not further increase (Fig. 1c). As defined by histological analysis using H&E staining, airway inflammation at the seventh challenge was higher than that at the fourth challenge (Fig. 1d,h). In addition to airway inflammation, we examined changes in airway remodelling. Epithelial cell hyperplasia (epithelial area), goblet cell hyperplasia (PAS+ area), the increase of total collagen, and smooth muscle cell hypertrophy (α-SMA+ area) in lungs were significantly observed at the fourth challenge; these responses further increased at the seventh challenge (Fig. 1d–f,h). Additionally, sub-epithelial fibrosis (fibrosis area) was recognized at the seventh challenge, although it was not significantly observed at the fourth challenge (Fig. 1g,h).
Effects of anti-IL-33 mAb or anti-ST2 pAb on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice
In this IgE-sensitized model, IL-33 production in the lungs was strongly increased during the fourth to seventh challenges. Therefore, we assessed by pathological analysis using anti-IL-33 mAb whether IL-33/ST2 responses during the fourth to seventh challenges contribute to the development of IgE-mediated airway inflammation and remodelling. Treatment with anti-IL-33 mAb during the fourth to seventh challenges inhibited eosinophil and neutrophil accumulations in BALF at the seventh challenge (Fig. 2b). Additionally, IL-5 and IL-13 production in the lung tissue supernatants, but not IL-4, were suppressed (Fig. 2c). Meanwhile, airway inflammation (H&E) and airway remodelling, such as epithelial cell hyperplasia (epithelial area), goblet cell hyperplasia (PAS+ area), sub-epithelial fibrosis (fibrosis area), the increase of total collagen, and smooth muscle cell hypertrophy (α-SMA+ area) in the lung, were significantly suppressed by the treatment (Fig. 2d).
Subsequently, we examined the effects of anti-ST2 pAb on airway inflammation and remodelling in IgE-sensitized mice. Treatment with anti-ST2 pAb inhibited eosinophil and neutrophil accumulations in BALF (Fig. 3b). Furthermore, the amounts of IL-5 and IL-13 in the lung tissue supernatants were decreased, but not IL-4 production (Fig. 3c). Additionally, airway remodelling was inhibited by the treatment (Fig. 3d); hence, anti-ST2 pAb had similar effects to anti-IL-33 mAb.
Effect of 2-CA on the increased production of IL-33 in lungs of IgE-sensitized mice
In IgE-sensitized mice, IL-33 contributed to the development of IgE-mediated airway inflammation and remodelling; furthermore, IL-33 production and infiltration by alveolar macrophages were significantly observed in the lungs at the fourth challenge. Based on these data, we hypothesized that alveolar macrophages may produce IL-33 in the lungs; therefore, we examined whether an increased number of IL-33+ alveolar macrophages was observed. There were increases in the percentages and the absolute numbers of alveolar macrophages (F4/80+ CD11c+) and IL-33+ alveolar macrophages 24 hr after the fourth challenge in comparison with those in non-sensitized mice (Fig. 4a). Furthermore, the depletion of macrophages by treatment with 2-CA inhibited the increased production of IL-33 in the lung, but not anti-CD4 mAb (Fig. 4f).
Furthermore, in IgE-sensitized mice, the percentages and the absolute numbers of M2 macrophages (F4/80+ CD11c+ ST2+) and ST2+ CD4+ T cells (CD3+ CD4+ ST2+) cells were increased in comparison with non-sensitized challenged mice (Fig. 4b,d). Additionally, when lung cells were stained with CD206/mannose receptor instead of ST2 as a surface marker of M2 macrophages, an increased percentage and number of F4/80+ CD11c+ CD206+ cells in the lungs of IgE-sensitized mice were also observed (Fig. 4c).
Effects of 2-CA on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice
The results shown in Fig. 4 indicate that alveolar macrophages were major sources of IL-33 production; therefore, we hypothesized that macrophages may lead to the development of airway inflammation and remodelling at the seventh challenge. As expected, the depletion of macrophages inhibited eosinophil and neutrophil accumulation in BALF at the seventh challenge (Fig. 5a). Additionally, IL-5 and IL-13 production in the lung tissue supernatants, but not IL-4 production, was significantly suppressed (Fig. 5b). Meanwhile, airway inflammation (H&E) and airway remodelling, such as epithelial cell hyperplasia (epithelial area), goblet cell hyperplasia (PAS+ area), sub-epithelial fibrosis (fibrosis area), the increase of total collagen, and smooth muscle cell hypertrophy (α-SMA+ area) in the lungs, were significantly suppressed by the treatment (Fig. 5c).
Figure 5.
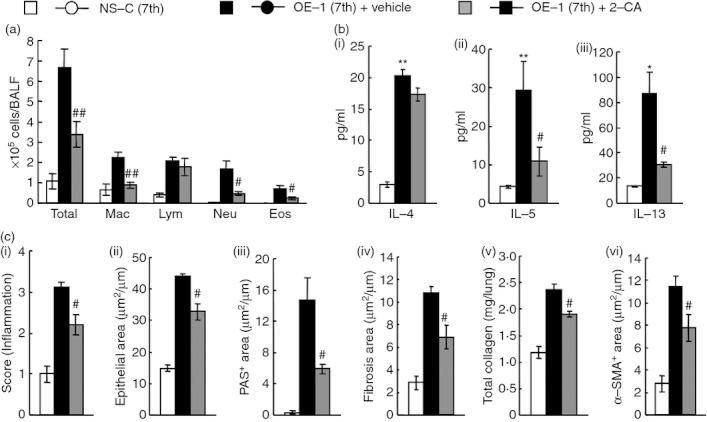
Effect of 2-chloroadenosine (2-CA) on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice. (a) Effect of treatment with 2-CA on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the seventh challenge. (b) Effect of treatment with 2-CA on increased levels of interleukin-4 (IL-4) (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the seventh challenge. (c) Effect of treatment with 2-CA on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the seventh challenge. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of six or seven animals. #P < 0·05 and ##P < 0·01 compared with the OE-1 (7th) + vehicle group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Effects of anti-CD4 mAb on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice
We have reported that CD4+ cells are important for the induction of allergic responses, such as eosinophilic inflammation in a murine model of asthma32,33 and, in this study, an increase of CD4+ cells expressing ST2 was observed at the fourth challenge; therefore, we examined whether CD4+ cells contributed to the development of IgE-mediated airway inflammation and remodelling during the fourth to seventh challenges. Similar to the treatment with anti-IL-33 mAb, treatment with anti-CD4 mAb inhibited airway inflammation (H&E) in the lungs, and eosinophil and neutrophil accumulations in BALF (Fig. 6a,c). Furthermore, the production of IL-5 and IL-13 in the lung tissue supernatants, but not IL-4 production, was decreased (Fig. 6b).
Figure 6.
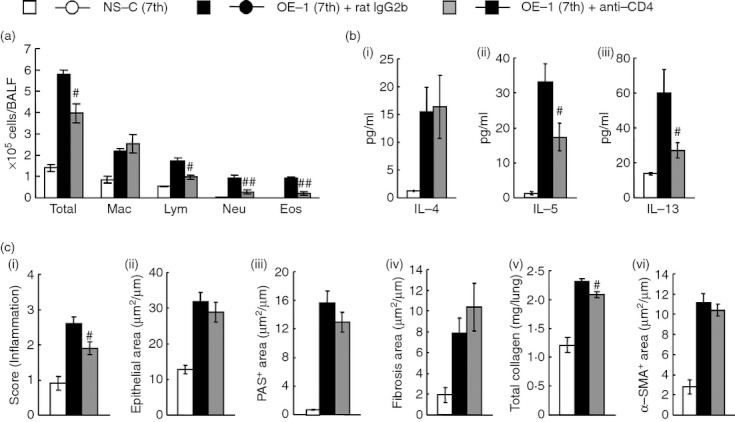
Effect of anti-CD4 monoclonal antibody (mAb) on airway inflammation and remodelling after the seventh antigen challenge in IgE-sensitized mice. (a) Effect of treatment with anti-CD4 mAb on inflammatory cell number in bronchoalveolar lavage fluid 24 hr after the seventh challenge. (b) Effect of treatment with anti-CD4 mAb on increased levels of interleukin-4 (IL-4) (i), IL-5 (ii) and IL-13 (iii) in the lung tissue supernatant 24 hr after the seventh challenge. (c) Effect of treatment with anti-CD4 mAb on airway inflammation (i), epithelial area (ii), PAS+ area (iii), fibrosis area (iv), total collagen (v) and α-SMA+ area (vi) 24 hr after the seventh challenge. Results shown are from one experiment representative of two independent trials. Each value is the mean ± SEM of five to seven animals. #P < 0·05 and ##P < 0·01 compared with the OE-1 (7th) + rat IgG2b group. Total, all cells; Mac, macrophages; Lym, lymphocytes; Neu, neutrophils; Eos, eosinophils.
Meanwhile, suppression of airway remodelling, such as epithelial cell hyperplasia (epithelial area), goblet cell hyperplasia (PAS+ area), sub-epithelial fibrosis (fibrosis area), and smooth muscle cell hyperplasia (α-SMA+ area), by the treatment was not significantly observed, although the increase of total collagen in the lungs was inhibited (Fig. 6c).
Effects of 2-CA and anti-CD4 mAb on IL-33-induced airway inflammation and remodelling in mice
Next, we investigated whether IL-33-induced airway inflammation and remodelling in normal mice were mediated by the activation of macrophages and CD4+ cells. When macrophages or CD4+ cells were depleted by treatment with 2-CA or anti-CD4 mAb, respectively, both treatments significantly reduced airway inflammation (H&E) in the lungs and infiltration by macrophages, lymphocytes, neutrophils and eosinophils in BALF (Figs 7b,d and 8b,d). Furthermore, treatment with 2-CA inhibited airway remodelling, such as epithelial cell hyperplasia (epithelial area), goblet cell hyperplasia (PAS+ area), sub-epithelial fibrosis (fibrosis area), the increase in total collagen in the lungs, and smooth muscle cell hyperplasia (α-SMA+ area) (Fig. 7d); however, anti-CD4 mAb failed to inhibit airway remodelling, although the increase in total collagen in the lungs was suppressed (Fig. 8d). Additionally, the increased levels of IL-5 and IL-13 in the lung tissue supernatants caused by IL-33 were inhibited by both treatments; however, anti-CD4 mAb, but not 2-CA, inhibited the production of IL-4 (Figs 7c and 8c).
Discussion
Allergen-specific IgE plays a critical role in the pathogenesis of allergic asthma; however, the precise mechanisms underlying the development of IgE-mediated chronic allergic asthma remain unclear. In this study, we focused on the role of IL-33 and alveolar macrophages in the development of airway inflammation and remodelling by repeated antigen challenges in mice sensitized with allergen-specific IgE mAb. The seventh antigen challenge induced the exacerbation of airway inflammation and remodelling in comparison with the fourth challenge; furthermore, increased expression of IL-33 in the lungs was strongly observed at the fourth and seventh challenges. These findings prompted us to examine the effect of anti-IL-33 mAb or anti-ST2 pAb during the fourth to seventh challenges. The treatment reduced infiltrations of neutrophils and eosinophils in BALF and airway remodelling at the seventh challenge. Additionally, increased levels of IL-33+ and ST2+ alveolar macrophages in the lungs at the fourth challenge were observed; furthermore, the depletion of macrophages by treatment with 2-CA during the fourth to seventh challenges significantly repressed the production of IL-33 in the lungs as well as these responses at the seventh challenge. Meanwhile, intratracheal administration of IL-33 induced airway inflammation and remodelling via activation of macrophages in normal mice. These results clearly show that IL-33 and macrophages play critical roles in the exacerbation of IgE-mediated airway inflammation and remodelling.
The production of IL-33 was strongly increased in the lungs 24 hr after the fourth and seventh administrations of antigen to IgE-sensitized mice compared with these before the fourth challenge (Fig. 1), suggesting that the first three challenges elicited changes that conditioned the high production of IL-33 after the fourth challenge in IgE-sensitized mice. Indeed, infiltration by macrophages in the lungs was significantly observed at the fourth challenge (Fig. 1); furthermore, the number of IL-33+ alveolar macrophages in the lungs was increased (Fig. 4). Based on these data, we hypothesized that further increased production of IL-33 in the lungs after the fourth challenge could be derived from alveolar macrophages. As expected, the depletion of macrophages by treatment with 2-CA reduced the increased production of IL-33 in the lungs 24 hr after the seventh challenge in the lungs of IgE-sensitized mice (Fig. 4), indicating that alveolar macrophages were regarded as major sources of IL-33 production during the fourth to seventh challenges. Treatments with not only anti-IL-33 mAb but also 2-CA suppressed the development of airway inflammation and remodelling at the seventh challenge (Figs 2 and 5). Hence, alveolar macrophage-derived IL-33 was critical for the development of these IgE-mediated responses.
It has been reported that ST2 receptors are expressed on inflammatory cells such as Th2 cells,37–39 and in IgE-sensitized mice, the number of CD4+ T cells expressing ST2 was increased in the lungs at the fourth challenge (Fig. 4); therefore, we investigated the roles of CD4+ T cells in IgE-mediated airway inflammation. The infiltration of eosinophils and production of Th2 cytokines such as IL-5 and IL-13 were suppressed by treatment with anti-CD4 mAb in IgE-sensitized mice, and the inhibitory effects were similar to those of anti-IL-33 mAb (Figs 2 and 6). Additionally, the treatment showed the inhibition of IL-33-induced infiltration by eosinophils associated with the reduction of Th2 cytokine production (Fig. 8), indicating that IL-33 induced eosinophil accumulation via Th2 cytokines produced by activation of CD4+ T cells in IgE-sensitized mice. On the other hand, it has been reported that ST2 was expressed on eosinophils,40 indicating that IL-33 directly induces the migration of eosinophils, owing to their expression of ST2.
Alternatively activated (M2) macrophages are polarized by cytokines IL-4 and/or IL-13 and thereby mirror the Th2 polarization of T cells.41–43 Furthermore, repeated intranasal administration of IL-33 in normal mice induces differentiation of alveolar macrophages toward M2 macrophages, which show increased expressions of ST2 and CD206.13 In IgE-sensitized mice, we found that the number of M2 macrophages (F4/80+ CD11c+ ST2+ and F4/80+ CD11c+ CD206+) in the lungs was increased at the fourth challenge (Fig. 4). In our previous27,28 and present studies, the production of IL-4, IL-13 and IL-33 in the lungs was observed until at least 24 hr after the fourth challenge, suggesting that M2 macrophages may be polarized by these cytokines produced during the first to fourth challenges in IgE-sensitized mice. Meanwhile, it has been reported that M2 macrophages enhance the secretion of Th2 cytokines from CD4+ T cells.44 In IgE-sensitized and IL-33-treated mice, the depletion of macrophages suppressed infiltration by eosinophils and Th2 cytokines such as IL-5 and IL-13 (Figs 5 and 7), suggesting that IL-33 may stimulate CD4+ T lymphocytes to secrete these Th2 cytokines, which contribute to eosinophil accumulation, through the activation of M2 macrophages in IgE-sensitized mice; furthermore, M2 macrophages themselves secrete IL-13,45 indicating that M2 macrophage-derived IL-13 may induce infiltration by eosinophils.
Meanwhile, treatment with anti-CD4 mAb did not inhibit IL-4 production at the seventh challenge in IgE-sensitized mice (Fig. 6). This may be related to the timing of administration of the mAb, which was administered at the fourth challenge in IgE-sensitized mice, because we have reported that treatment with anti-CD4 mAb at the first challenge inhibited the production of IL-4 at the fourth challenge in IgE-sensitized mice.41 It has been reported that mast cells and/or basophils as well as Th2 cells are potential cellular sources of IL-4 after an antigen challenge in sensitized mice,46 suggesting that the increase in IL-4 level might be closely linked to the increase in mast cells and/or basophils during the fourth to seventh challenges in IgE-sensitized mice.
Airway remodelling, including goblet cell hyperplasia, sub-epithelial fibrosis, and smooth muscle cell hypertrophy, are characteristic features of severe and chronic asthma.8,9 As demonstrated in our previous27 and present studies, IgE mediated the development of airway remodelling. Treatment with anti-CD4 mAb at the fourth challenge did not inhibit airway remodelling at the seventh challenge in IgE-sensitized mice, although the treatment significantly inhibited airway inflammation (Fig. 6). Previous findings47,48 from studies with protocols that have depleted CD4+ cells at the time of the first challenge showed the reduction in both airway remodelling and inflammation. Hence, the timing of CD4 depletion is critical to the development of remodelling, suggesting that CD4+ cells are required for initiation, but not progression or maintenance of airway remodelling. On the other hand, treatments with anti-IL-33 mAb and 2-CA (depletion of macrophages) during the fourth to seventh challenges suppressed the exacerbation of airway remodelling at the seventh challenge in IgE-sensitized mice (Figs 2 and 5); furthermore, IL-33-induced airway remodelling was inhibited by the depletion of macrophages (Fig. 7). Hence, after initiation of the remodelling process, which requires CD4+ cells, mechanisms associated with IL-33 and macrophages are necessary for the exacerbation of airway remodelling in IgE-sensitized mice. Additionally, M2 macrophages promote wound healing and fibrosis through the production of matrix metalloproteinase 12, tissue inhibitor of metalloproteinase 1, growth factors including platelet-derived growth factor, and cytokines such as transforming growth factor-β1,49 indicating that IL-33 may contribute to the exacerbation of airway remodelling via these M2 macrophage-derived molecules in IgE-sensitized mice.
It has been reported that IL-13 contributes to the development of airway remodelling50; however, although treatment with anti-CD4 mAb inhibited IL-13 production in IgE-sensitized and IL-33-treated mice, inhibitory effects on airway remodelling were not observed (Figs 6 and 8), suggesting that IL-13 may not be related to the exacerbation of airway remodelling. Meanwhile, experimental studies using IL-5 and eotaxin over-expressing transgenic mice have shown that eosinophils can elicit many of the hallmark pathologies of allergic diseases, including airway remodelling51; however, additional investigations of mouse models examining the requirement of eosinophils in the development of airway remodelling indirectly by interfering with IL-5 function have yielded disparate results.32,52,53 In the present study, although treatment with anti-CD4 mAb suppressed the infiltration of eosinophils, the inhibition of airway remodelling was not recognized in IgE-sensitized and IL-33-treated mice (Figs 6 and 8). Hence, it is suggested that airway remodelling occurs independently of infiltration by eosinophils in IgE-sensitized mice; however, treatment with anti-CD4 mAb inhibited the increase of total collagen in the lungs of IgE-sensitized and IL-33-treated mice (Figs 6 and 8); additionally, treatment inhibited lung parenchymal fibrosis as well as airway inflammation, indicating that the inhibition of total collagen in the lungs may be related to the suppression of lung parenchymal fibrosis; therefore, IL-13 and eosinophils may contribute to the development of lung parenchymal fibrosis in IgE-sensitized mice.
In conclusion, we demonstrated that (i) IgE mediates the increased levels of IL-33+ and ST2+ alveolar macrophages and ST2+ CD4+ T cells in the lungs, (ii) macrophage-derived IL-33 plays an essential role in the development of IgE-mediated airway inflammation and remodelling, and (iii) IL-33 contributes to the development of airway inflammation by mechanisms associated with macrophages, and CD4+ T cells, and airway remodelling via the activation of macrophages. These findings suggest that IL-33 and alveolar macrophages contribute to the mechanisms underlying the exacerbation of IgE-mediated airway inflammation and remodelling and so provide new insights into the underlying pathogenesis of chronic allergic asthma and a potential therapeutic strategy for the disease.
Acknowledgments
Part of this work was supported by a Grant-in-aid for Young Scientists (B) (23790164, to N. Mizutani), and a Grant-in-aid for Scientific Research (C) (23590093, to T. Nabe) from the Japan Society for the Promotion of Science. We thank Hirofumi Goshima, Yuki Fujimoto, Kaori Kubota, and Mio Sasaki (Kobe Pharmaceutical University) for their assistance with sample measurements.
Glossary
- BALF
bronchoalveolar lavage fluid
- 2-CA
2-chloroadenosine
- PAS
periodic acid-Schiff
- PE
phycoerythrin
- FITC
fluorescein isothiocyanate
- α-SMA
α-smooth muscle actin
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 2.Robinson DS, Hamid Q, Ying S, et al. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 3.Cockcroft DW. Allergen-induced increase in nonallergic airway responsiveness: a citation classic revisited. Can Respir J. 2000;7:182–7. doi: 10.1155/2000/846798. [DOI] [PubMed] [Google Scholar]
- 4.Durham SR. The significance of late responses in asthma. Clin Exp Allergy. 1991;21:3–7. doi: 10.1111/j.1365-2222.1991.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 5.Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992;101:916–21. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- 6.Coyle AJ, Wagner K, Bertrand C, Tsuyuki S, Bews J, Heusser C. Central role of immunoglobulin (Ig) E in the induction of lung eosinophil infiltration and T helper 2 cell cytokine production: inhibition by a non-anaphylactogenic anti-IgE antibody. J Exp Med. 1996;183:1303–10. doi: 10.1084/jem.183.4.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshiba A, Hamelmann E, Takeda K, Brandley KL, Loader JE, Larsen GL, Gelfand EW. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig) E and IgG1 in mice. J Clin Invest. 1996;97:1398–408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–6. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 9.Jeffery PK, Godfrey RW, Adelroth E, Nelson F, Rogers A, Johansson SA. Effects of treatment on airway inflammation and thickening of basement membrane reticular collagen in asthma. A quantitative light and electron microscopic study. Am Rev Respir Dis. 1992;145:890–9. doi: 10.1164/ajrccm/145.4_Pt_1.890. [DOI] [PubMed] [Google Scholar]
- 10.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179:2551–5. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 12.Sakashita M, Yoshimoto T, Hirota T, et al. Association of serum interleukin-33 level and the interleukin-33 genetic variant with Japanese cedar pollinosis. Clin Exp Allergy. 2008;38:1875–81. doi: 10.1111/j.1365-2222.2008.03114.x. [DOI] [PubMed] [Google Scholar]
- 13.Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 14.Prefontaine D, Lajoie-Kadoch S, Foley S, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 15.Kurowska-Stolarska M, Kewin P, Murphy G, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 16.Kondo Y, Yoshimoto T, Yasuda K, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 17.Lohning M, Stroehmann A, Coyle AJ, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998;95:6930–5. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coyle AJ, Lloyd C, Tian J, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. 1999;190:895–902. doi: 10.1084/jem.190.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun. 2009;386:181–5. doi: 10.1016/j.bbrc.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Wissinger E, Goulding J, Hussell T. Immune homeostasis in the respiratory tract and its impact on heterologous infection. Semin Immunol. 2009;21:147–55. doi: 10.1016/j.smim.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Thepen T, McMenamin C, Oliver J, Kraal G, Holt PG. Regulation of immune response to inhaled antigen by alveolar macrophages: differential effects of in vivo alveolar macrophage elimination on the induction of tolerance vs. immunity. Eur J Immunol. 1991;21:2845–50. doi: 10.1002/eji.1830211128. [DOI] [PubMed] [Google Scholar]
- 22.Thepen T, McMenamin C, Girn B, Kraal G, Holt PG. Regulation of IgE production in pre-sensitized animals: in vivo elimination of alveolar macrophages preferentially increases IgE responses to inhaled allergen. Clin Exp Allergy. 1992;22:1107–14. doi: 10.1111/j.1365-2222.1992.tb00137.x. [DOI] [PubMed] [Google Scholar]
- 23.Tang C, Inman MD, van Rooijen N, Yang P, Shen H, Matsumoto K, O'Byrne PM. Th type 1-stimulating activity of lung macrophages inhibits Th2-mediated allergic airway inflammation by an IFN-γ-dependent mechanism. J Immunol. 2001;166:1471–81. doi: 10.4049/jimmunol.166.3.1471. [DOI] [PubMed] [Google Scholar]
- 24.Careau E, Turmel V, Lauzon-Joset JF, Bissonnette EY. Alveolar macrophages reduce airway hyperresponsiveness and modulate cytokine levels. Exp Lung Res. 2010;36:255–61. doi: 10.3109/01902140903410757. [DOI] [PubMed] [Google Scholar]
- 25.Moon KA, Kim SY, Kim TB, Yun ES, Park CS, Cho YS, Moon HB, Lee KY. Allergen-induced CD11b+ CD11cint CCR3+ macrophages in the lung promote eosinophilic airway inflammation in a mouse asthma model. Int Immunol. 2007;19:1371–81. doi: 10.1093/intimm/dxm108. [DOI] [PubMed] [Google Scholar]
- 26.Herbert C, Scott MM, Scruton KH, et al. Alveolar macrophages stimulate enhanced cytokine production by pulmonary CD4+ T-lymphocytes in an exacerbation of murine chronic asthma. Am J Pathol. 2010;177:1657–64. doi: 10.2353/ajpath.2010.100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizutani N, Goshima H, Nabe T, Yoshino S. Establishment and characterization of a murine model for allergic asthma using allergen-specific IgE monoclonal antibody to study pathological roles of IgE. Immunol Lett. 2012;141:235–45. doi: 10.1016/j.imlet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Mizutani N, Goshima H, Nabe T, Yoshino S. Complement C3a-induced IL-17 plays a critical role in an IgE-mediated late-phase asthmatic response and airway hyperresponsiveness via neutrophilic inflammation in mice. J Immunol. 2012;188:5694–705. doi: 10.4049/jimmunol.1103176. [DOI] [PubMed] [Google Scholar]
- 29.Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. 2010;5:e11944. doi: 10.1371/journal.pone.0011944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song C, Luo L, Lei Z, et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J Immunol. 2008;181:6117–24. doi: 10.4049/jimmunol.181.9.6117. [DOI] [PubMed] [Google Scholar]
- 31.Yang M, Kumar RK, Foster PS. Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-γ and TLR4/MyD88 pathways. J Immunol. 2009;182:5107–15. doi: 10.4049/jimmunol.0803468. [DOI] [PubMed] [Google Scholar]
- 32.Nabe T, Morishita T, Matsuya K, Ikedo A, Fujii M, Mizutani N, Yoshino S. Complete dependence on CD4+ cells in late asthmatic response, but limited contribution of the cells to airway remodeling in sensitized mice. J Pharmacol Sci. 2011;116:373–83. doi: 10.1254/jphs.11083fp. [DOI] [PubMed] [Google Scholar]
- 33.Nabe T, Hosokawa F, Matsuya K, et al. Important role of neutrophils in the late asthmatic response in mice. Life Sci. 2011;88:1127–35. doi: 10.1016/j.lfs.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizutani N, Nabe T, Yoshino S. Complement C3a regulates late asthmatic response and airway hyperresponsiveness in mice. J Immunol. 2009;183:4039–46. doi: 10.4049/jimmunol.0901468. [DOI] [PubMed] [Google Scholar]
- 35.Bedoret D, Wallemacq H, Marichal T, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergic in mice. J Clin Invest. 2009;119:3723–38. doi: 10.1172/JCI39717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trajkovic V, Sweet MJ, Xu D. T1/ST2–an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 2004;15:87–95. doi: 10.1016/j.cytogfr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, Robinson JH, Liew FY. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cell. J Exp Med. 1998;187:787–94. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CC, Grimbaldeston MA, Tsai M, Weissmann IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci USA. 2005;102:11408–13. doi: 10.1073/pnas.0504197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. ST2 protein induced by inflammatory stimuli can modulate acute lung inflammation. Biochem Biophys Res Commun. 2002;299:18–24. doi: 10.1016/s0006-291x(02)02578-0. [DOI] [PubMed] [Google Scholar]
- 40.Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:1526–34. doi: 10.1182/blood-2008-05-157818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anthony RM, Urban JF, Jr, Alem F, Hamed HA, Rozo CT, Boucher JL, Van Rooijen N, Gause WC. Memory TH2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. 2006;12:955–60. doi: 10.1038/nm1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–82. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 43.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 44.Donnelly S, Stack CM, O'Neill SM, Sayed AA, Williams DL, Dalton JP. Helminth 2-Cys peroxiredoxin drives Th2 responses through a mechanism involving alternatively activated macrophages. FASEB J. 2008;22:4022–32. doi: 10.1096/fj.08-106278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim EY, Battaile JT, Patel AC, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–40. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luccioli S, Brody DT, Hasan S, Keane-Myers A, Prussin C, Metcalfe DD. IgE+, Kit–, I-A/I-E– myeloid cells are the initial source of IL-4 after antigen challenge in a mouse model of allergic pulmonary inflammation. J Allergy Clin Immunol. 2002;110:117–24. doi: 10.1067/mai.2002.125828. [DOI] [PubMed] [Google Scholar]
- 47.Foster PS, Yang M, Herbert C, Kumar RK. CD4+ T-lymphocytes regulate airway remodeling and hyper-reactivity in a mouse model of chronic asthma. Lab Invest. 2002;82:455–62. doi: 10.1038/labinvest.3780438. [DOI] [PubMed] [Google Scholar]
- 48.Komai M, Tanaka H, Masuda T, Nagao K, Ishizaki M, Sawada M, Nagai H. Role of Th2 responses in the development of allergen-induced airway remodelling in a murine model of allergic asthma. Br J Pharmacol. 2003;138:912–20. doi: 10.1038/sj.bjp.0705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanone S, Zheng T, Zhu Z, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–74. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochkur SI, Jacobsen EA, Protheroe CA, et al. Coexpression of IL-5 and eotaxin-2 in mice creates an eosinophil dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol. 2007;178:7879–89. doi: 10.4049/jimmunol.178.12.7879. [DOI] [PubMed] [Google Scholar]
- 52.Blyth DI, Wharton TF, Pedrick MS, Savage TJ, Sanjar S. Airway subepithelial fibrosis in a murine model of atopic asthma: suppression by dexamethasone or anti-interleukin-5 antibody. Am J Respir Cell Mol Biol. 2000;23:241–6. doi: 10.1165/ajrcmb.23.2.3999. [DOI] [PubMed] [Google Scholar]
- 53.Cho JY, Miller M, Baek KJ, et al. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest. 2004;113:551–60. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]