

Abstract
Leukotriene B4 (LTB4) is a neutrophil chemotactic molecule with important involvement in the inflammatory responses of chronic obstructive pulmonary disease (COPD). Airway epithelium is emerging as a regulator of innate immune responses to a variety of insults including cigarette smoke, the major risk factor for COPD. In this study we have explored whether cigarette smoke extracts (CSE) or soluble mediators present in distal lung fluid samples (mini-bronchoalveolar lavages) from smokers alter the expression of the LTB4 receptor 2 (BLT2) and peroxisome proliferator-activated receptor-α (PPAR-α) in bronchial epithelial cells. We also evaluated the effects of CSE on the expression of intercellular adhesion molecule 1 (ICAM-1) and on the binding of signal transducer and activator of transcription 1 (STAT-1) to ICAM-1 promoter as well as the adhesiveness of neutrophils to bronchial epithelial cells. CSE and mini-bronchoalveolar lavages from smokers increased BLT2 and ICAM-1 expression as well as the adhesiveness of neutrophils to bronchial epithelial cells and decreased PPAR-α expression. CSE induced the activation of STAT-1 and its binding to ICAM-1 promoter. These findings suggest that, in bronchial epithelial cells, CSE promote a prevalent induction of pro-inflammatory BLT2 receptors and activate mechanisms leading to increased neutrophil adhesion, a mechanism that contributes to airway neutrophilia and to tissue damage.
Keywords: bronchial epithelial cells, cigarette smoke, chronic obstructive pulmonary disease, inflammation, leukotriene B4
Introduction
The airway epithelium plays a role in airway defence mechanisms by releasing cytoprotective mucus and defensins and exerts an important role in co-ordinating local inflammation and immune responses through the release of cytokines and chemokines.1 Leukotriene B4 (LTB4) is a mediator with important involvement in the inflammatory responses in chronic obstructive pulmonary disease (COPD).2 Chronic neutrophilic inflammation is observed in COPD and mediates extensive tissue damage contributing to organ dysfunction.3 Activated neutrophils release elastase, a protease that correlates with LTB4,4 that actively contributes to generating the lung damage in COPD patients. Purulent exacerbations in these patients are related to bacterial infections, and are associated with further increased neutrophilic inflammation and increased LTB4 concentrations.5
Leukotriene B4 is able to interact with two cell-surface receptors LTB4 receptor 1 (BLT1) and LTB4 receptor 2 (BLT2) and with a nuclear receptor named peroxisome proliferator-activated receptor-α (PPAR-α). The interaction of LTB4 with the cell-surface receptors induces LTB4-mediated inflammatory activities while the interaction with PPAR-α induces the catabolism of LTB4 and in turn limits LTB4-related inflammation.6 We have previously demonstrated, in a model of pleural inflammation, that the prevalent activation of pro-inflammatory LTB4 receptors in the presence of an inflammatory milieu may lead to up-regulation of intercellular adhesion molecule 1 (ICAM-1) and to increased adhesion between neutrophils and the mesothelium, resulting in neutrophil retention on the surface of mesothelial cells.7
Although increased expression of BLT1 and of PPAR-α has been observed in distal airways of patients with COPD,8 the molecular mechanisms involved in this phenomenon are largely unknown. The aims of the present study were to test whether cigarette smoke extracts (CSE) alter the expression of both BLT-2 and PPAR-α and to assess whether this alteration is the result of a direct effect of CSE or of the inflammatory responses promoted by cigarette smoke. The experiments were performed in bronchial epithelial cells stimulated with CSE or with mini-bronchoalveolar lavage (BAL) from smokers and the expression of BLT2 and PPAR-α as well as the neutrophil–epithelial cell adhesion were assessed.
Materials and methods
Patient population
Smoking subjects (> 15 packs/year) (n = 8) and non-smoking subjects (n = 4) with acute respiratory failure upon surgery for abdominal or thoracic aneurysm were recruited. Patients with X-ray or clinical evidence of sepsis or pneumonia at the time of mini-BAL collection were not included. All recruited subjects required mechanical ventilation and underwent therapy with antibiotics and systemic corticosteroids (no significantly different doses among the patients included in the study). Tissue specimens from central bronchi from smoking (> 15 packs/year) and from non-smoking subjects were also collected. The study fulfilled the criteria of the Ethics Committee of Policlinico-Giaccone Hospital, Palermo and was in agreement with the Helsinki Declaration. Informed written consent from either the patients or their closest relatives was obtained.
Mini-BAL collection and processing
Distal lung fluid samples (mini-BALs) were obtained using bronchoscopic aspirate sampling catheters (Kimberly-Clark Health Care, West Malling, Kent, UK) within 1 hr from the intubation.9 The protected catheter was blindly advanced through the endotracheal tube until it was wedged into a distal airway and two aliquots of 10 ml sterile 0·9% NaCl were instilled and gently suctioned (recovered volume about 70% of the instilled volume). Mini-BAL samples were filtered through a sterile gauze and then centrifuged at 300 g for 10 min to separate cells from supernatants. The supernatants were used for stimulating bronchial epithelial cells.
Immunohistochemistry
Tissue specimens from central bronchi were selected, fixed with 10% neutral buffered formalin and embedded in paraffin wax. Three-micrometer tissue sections were attached to poly-l-lysine-coated microscope slides and, after dewaxing and rehydration, were stained with haematoxylin and eosin or analysed with immunohistochemistry.
Immunohistochemistry and image analysis were used to determine BLT2 and PPAR-α expression using rabbit polyclonal antibodies (Cayman Chemical, Ann Arbor, MI) in central (internal perimeter > 6 mm) airways. LSAB2 Dako kit (Code No. K0674) (Dako, Glostrup, Denmark) and Fuchsin Substrate-Chromogen System Dako were used for the staining. Non-immune rabbit (Dako) was used as negative control. The immunoreactivity was evaluated blindly by two independent investigators using a Leica (Wetzlar, Germany) microscope × 400 magnification.
Preparation of cigarette smoke extracts
Commercial cigarettes (Marlboro) were used in this study. Cigarette smoke solution was prepared as described previously.10 Each cigarette was smoked for 5 min and one cigarette was used per 25 ml PBS to generate a CSE-PBS solution. The CSE solution was filtered through a 0·22-μm pore filter to remove bacteria and large particles. The smoke solution was then adjusted to pH 7·4 and used within 30 min of preparation. This solution was considered to be 100% CSE and was diluted to obtain the desired concentration in each experiment. The concentration of CSE was calculated spectrophotometrically measuring the optical density (OD) as previously described11 at a wavelength of 320 nm. The pattern of absorbance showed very little difference among different batches and the mean OD of the different batches was 1·37 ± 0·16. The presence of contaminating lipopolysaccharide on undiluted CSE was assessed by a commercially available kit (Cambrex Corporation, East Rutherford, NJ) and was below the detection limit of 0·1 EU/ml.
Bronchial epithelial cell cultures
The human bronchial epithelial SV40 immortalized cell line 16HBE was used in this study.12 16HBE cells were maintained in minimium essential medium (MEM; Gibco, Life Technologies, Carlsbad, CA), supplemented with 10% fetal calf serum (Gibco) and 0·5% gentamicin (Gibco). Cell cultures were maintained in a humidified atmosphere of 5% CO2 in air at 37°. The cells were cultured in the presence and absence of CSE and in the presence and absence of mini-BAL from smokers or non-smokers for 18 hr. The concentration of CSE used, 10%, was selected on the basis of cell apoptosis/necrosis experiments. The time-point was selected on the basis of preliminary experiments (data not shown). At the end of stimulation, cell pellets, cell extracts and cell culture supernatants were collected for further evaluations. In some experiments a BLT2 antagonist (LY255283) (10 μm) or a PPAR-α antagonist (MK886) (10 μm) (Cayman Chemical) was added 1 hr before cell stimulation, as previously described.7,13,14 The concentrations of both LY255283 and MK886 were selected by performing a dose–response experiment (see Supplementary material, Fig. S1).The 20-μm concentration for both inhibitors was discarded because it induced cell morphology alterations.
Cell necrosis and cell apoptosis
Cell necrosis and cell apoptosis were evaluated, as previously described,9 by staining with annexin V-FITC and propidium iodide using a commercial kit (Bender MedSystem, Vienna, Austria) following the manufacturer's directions. Cells were analysed using a FACS Calibur (Becton Dickinson, Mountain View, CA) analyser equipped with an Argon ion Laser (Innova 70 Coherent, Santa Clara, CA).
BLT2 and PPAR-α expression
The expression of BLT2 and of PPAR-α in 16HBE cells was evaluated by flow cytometry using rabbit polyclonal antibodies anti-BLT2 and anti-PPAR-α (both from Cayman Chemical) (1 : 250 for 1 hr) followed by an FITC-conjugated anti-rabbit IgG (Dako). To evaluate the expression of BLT2 and of PPAR-α before incubation with rabbit polyclonal antibodies, cells were permeabilized using a commercial fix-perm cell permeabilization kit (Caltag Laboratories, Burlingame, CA). Negative controls were performed using non-immune rabbit (Dako). Data are expressed as GeoMean fluorescence intensity and as percentage of positive cells.
Cell-adhesion assay
Cell-adhesion assay was performed by a method previously described with minor modifications.15 Briefly, 16HBE cells were seeded in 96-well plates at half-confluency and kept for 18 hr alone and with mini-BAL supernatants from smokers. Peripheral blood neutrophils were isolated from peripheral blood of normal donors as previously described,16 labelled for 30 min with 20 μm of fluorocromic dye SFDA (Molecular Probes, Eugene, OR) and washed and resuspended in PBS (1 × 105/ml). Immediately before the addition of neutrophils, the 16HBE cultures were washed with warm PBS. Labelled neutrophils were added in a final volume of 0·1 ml (1 × 104/well). The plates were incubated at 37° for 20 min to allow neutrophils to contact and to adhere to the confluent 16HBE, and total fluorescence was measured using an excitation wavelength of 485 nm and monitoring emission at 530 nm in a Wallac 1420 Victor multilabel counter (PerkinElmer Life and Analytical Sciences-Wallac OY, Turku, Finland). Non-adherent cells were then removed by washing and fluorescence was measured to evaluate bound cells. Adhesion was expressed as percentage of the fluorescence ratio of bound cells over total cells. All test points were performed in triplicate. The baseline values represent the adhesion of neutrophils to unstimulated cells.
Evaluation of ICAM-1 expression
The expression of ICAM-1 on the surface of bronchial epithelial cells was determined by flow cytometry. An FITC-conjugated mouse anti-human ICAM-1 antibody (anti-CD54; clone 6.5B5; Dakopatts, Glostrup, Denmark) was used. Negative controls were performed using a non-immune FITC-conjugated mouse IgG1 (from Dakopatts). Data are expressed as GeoMean fluorescence intensity.
RNA interference and transfection
Cells were plated in 24-well tissue culture plates and grown in medium containing 10% FBS without the use of antibiotic until 60–80% confluency. BLT2 or PPAR-α small interfering (si) RNA (10 μm; Santa Cruz Biotechnology, Inc., Dallas, TX) were added to 40 μl siRNA transfection medium, and the reaction was performed according to the manufacturer's instructions until complete transfection of cells (30 hr at 37°). For optimal siRNA transfection efficiency, control siRNA (10 μm; Santa Cruz Biotechnology Inc.) was used containing a scrambled sequence that did not lead to the specific degradation of any known cellular mRNA. Finally, cells were stimulated with CSE 10% for 18 hr, and ICAM-1 expression was evaluated. The silencing efficacy of the RNA interference was checked by flow cytometry.
Western blot analysis for STAT-1
Western blot analysis for signal transducer and activator of transcription 1 (STAT-1) was performed as previously described7 with some modifications. Briefly, 16HBE cells were lysed (10 mm Tris–HCl, pH 7·4; 50 mm NaCl; 5 mm EDTA; 1% Nonidet P-40; 10 μg/ml PMSF) and protein were extracted. A 50-μg sample total proteins was subjected to SDS–PAGE on 4–12% gradient gels (Novex, San Diego, CA) and blotted onto nitrocellulose membranes. All blots were first probed using a polyclonal antibody anti-phospho-STAT-1 (Tyr701) (Cell Signaling, Boston, MA) (1 : 100, overnight) and a polyclonal antibody anti-STAT-1 (1 : 100; 1 hr) (Cell Signaling Technology Inc). Revelation was performed with an enhanced chemoluminescent system (GE Healthcare, Chalfont St Giles, UK) followed by autoradiography. β-actin (Sigma) was used as housekeeping protein to normalize differences in protein loading. Data underwent densitometric analysis and were expressed as densitometric arbitrary units by normalization with the density of the band obtained for β-actin. STAT-1 activation was assessed by calculating ratio of pSTAT-1/STAT-1 expression.
Chromatin immunoprecipitation analysis
Chromatin immunoprecipitation (ChIP) analysis was performed using the EZ-ChIP kit (Upstate-Millipore Corporate, Billerica, MA) following the manufacturer's directions. The 16HBE cells stimulated with CSE 10% were treated with formaldehyde and the cross-linked chromatin was sonicated to lengths spanning 200–1000 bp. The samples were pre-cleared with 60 μl Protein A–Agarose and then incubated with a polyclonal antibody anti-human STAT-1. Immunocomplexes were precipitated using Protein A–Agarose. After washing, DNA fragments were isolated and purified with columns. The PCR was performed using primers spanning the promoter region 3537 of ICAM-1 gene using the primers: 5′-CAC AGA GTG AGA CTC CAT C-3′ (the forward) and 5′-TGT TGT CCA GGC TGG AGT A-3′ (the reverse).
Statistics
Data are expressed as mean counts ± standard deviation. Comparison between different experimental conditions was evaluated by paired t-test. P < 0·05 was accepted as statistically significant.
Results
Mini-BAL from smokers increase BLT2 but decrease PPAR-α expression in bronchial epithelial cells
Bronchial epithelial cells play a crucial role in the inflammatory responses to external agents including cigarette smoke. The effects of mini-BAL supernatants from smokers and non-smokers on the expression of pro-(BLT2) and anti-inflammatory (PPAR-α) receptors for LTB4 in bronchial epithelial cells were initially explored. Mini-BALs from smokers and, at lower extent, mini-BALs from non-smokers increased the expression of BLT2 as both percentage of positive cells (13·3 ± 3% and 6 ± 2·5% variation for smokers and non-smokers, respectively) and GeoMean fluorescence intensity (18·7 ± 1·2% and 3·3 ± 3·5% variation for smokers and non-smokers, respectively) (Fig. 1a,b). Mini-BAL from smokers, but not mini-BALs from non-smokers, decreased PPAR-α expression as percentage of positive cells (11 ± 3% of variation) (Fig. 2a,b). The percentage of positive cells indicates the number of positive cells. The GeoMean gives a measure of the average content of the antigen per cell. The findings that mini-BALs reduced the percentage but not the GeoMean fluorescence intensity suggest that mini-BALs are able to reduce the number of PPAR-α-positive cells without modifying the PPAR-α content per cell.
Figure 1.
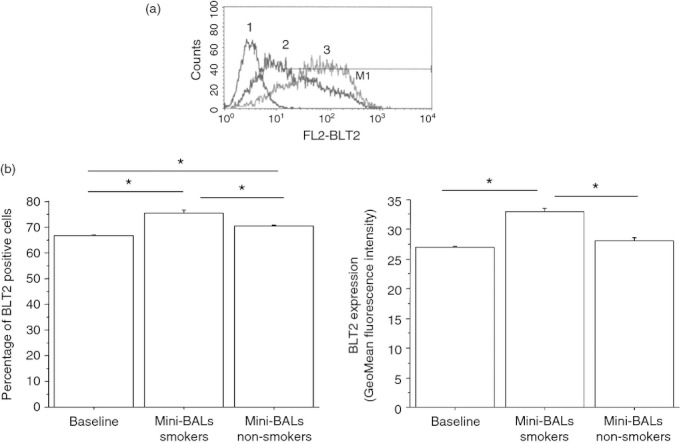
Mini-bronchoalveolar lavages (mini-BALs) from smokers increase BLT2 expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured with or without mini-BALs from smokers (n = 5) and from non-smokers (n = 3) for 18 hr and were used to assess BLT2 expression by flow cytometry (see Materials and methods for details). (a) Representative examples of flow cytometric analysis. 1 = negative control; 2 = BLT2 baseline expression; 3 = BLT2 expression after a mini-BAL from smoker exposure. (b) The results are expressed as percentage of positive cells and as GeoMean fluorescence intensity ± SD. Two replicates were performed from each mini-BAL sample.*P < 0·05
Figure 2.
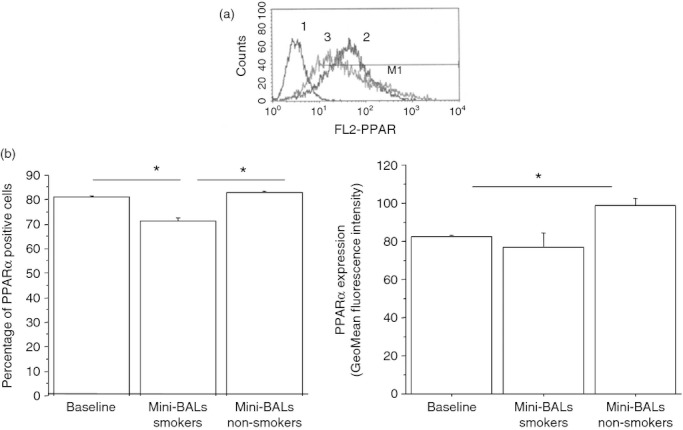
Mini-bronchoalveolar lavages (mini-BALs) from smokers decrease peroxisome proliferator-activated receptor-α (PPAR-α) expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured with/without mini-BALs from smokers (n = 5) and from non-smokers (n = 3) for 18 hr and were used for assessing PPAR-α expression by flow cytometry (see Materials and methods for details). (a) Representative histogram plots. 1 = negative control; 2 = baseline expression of PPAR-α; 3 = a mini-BAL from smoker. (b) The results are expressed as percentage of positive cells and as GeoMean fluorescence intensity ± SD.Two replicates were performed from each mini-BAL sample. *P < 0·05
In tissue samples, bronchial epithelial cells from smokers showed an intense expression of BLT2 but a weak PPAR-α expression. In contrast, bronchial epithelial cells from non-smokers showed an intense expression of PPAR-α but a weak BLT2 expression (Fig. 3a,b).
Figure 3.
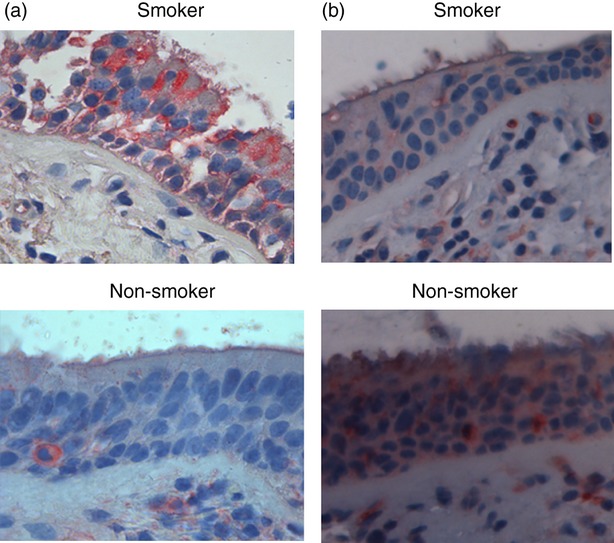
Expression of BLT2 and peroxisome proliferator-activated receptor-α (PPAR-α) molecules in bronchial epithelial cells from surgical samples of smokers. Immunohistochemistry for BLT2 (a) and PPAR-α (b) in central airways from surgical samples of smokers (n = 5) and of non-smokers (n = 3) was performed. Representative BLT2 and PPAR-α immunostaining at 400 × magnification was shown.
Mini-BALs from smokers increase neutrophil adhesion and ICAM-1 expression in bronchial epithelial cells
The increased expression of BLT2 may lead to increased neutrophil adhesion and to increased ICAM-1 expression.7 Mini-BALs from smokers increased both neutrophil adhesion to bronchial epithelial cells (Fig. 4) (43 ± 19% of variation) and ICAM-1 expression (71 ± 31% of variation) in bronchial epithelial cells (Fig. 5). The effect on ICAM-1 expression was related to BLT2/PPAR-α deregulated expression, since BLT2 antagonist reduced while PPAR-α antagonist further increased the ICAM-1 expression in bronchial epithelial cells stimulated with mini-BALs from smokers (Fig. 5).
Figure 4.
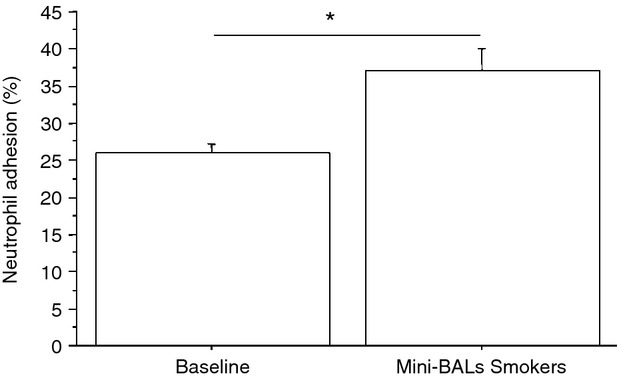
Mini-bronchoalveolar lavages (mini-BALs) from smokers increase the adhesiveness between bronchial epithelial cells and neutrophils. Bronchial epithelial cells (16HBE) were cultured with/without mini-BALs from smokers (n = 5) for 18 hr and were used for assessing neutrophil adhesion to bronchial epithelial cells (see Materials and methods for details). Two replicates were performed from each mini-BAL sample. The results are expressed as % of the fluorescence ratio of bound cells on total cells ± SD. *P < 0·05
Figure 5.
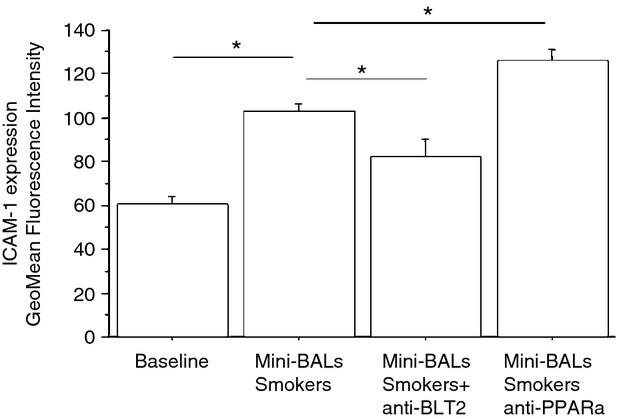
Mini-bronchoalveolar lavages (mini-BALs) from smokers increase intercellular adhesion molecule 1 (ICAM-1) expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured with/without mini-BALs from smokers (n = 5) and in the presence of an inhibitor for BLT2 (LY255283; 1 hr pre-treatment) or an inhibitor for peroxisome proliferator-activated receptor-α (PPAR-α) (MK886; 1 hr pre-treatment) for 18 hr and were used for assessing ICAM-1 expression by flow-cytometry (see Materials and methods for details). The results are expressed as GeoMean fluorescence intensity ± SD. Two replicates were performed from each mini-BAL sample. *P < 0·05
CSE increase BLT2 and neutrophil adhesion but decrease PPAR-α expression in bronchial epithelial cells
To assess whether the exposure to cigarette smoke was responsible for the effects induced by mini-BALs from smokers, the direct effect of CSE was tested. The effects of two different CSE concentrations (10 or 20%) on cell necrosis/apoptosis were initially assessed. Since CSE 20% increased cell apoptosis more than CSE 10% (Fig. 6), this latter concentration was used for assessing BLT2 or PPAR-α expression. The CSE increased BLT2 (29 ± 13% of variation) (Fig. 7) but decreased PPAR-α (13 ± 8% of variation) expression (Fig. 8) in bronchial epithelial cells. Moreover, when bronchial epithelial cells were stimulated with CSE, an increased neutrophil adhesion occurred (29 ± 19% of variation) (Fig. 9).
Figure 6.
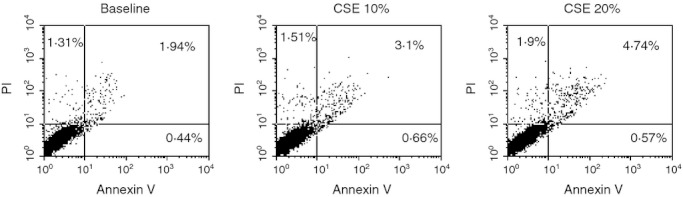
Effect of cigarette smoke extract (CSE) in the apoptosis of bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE (10%, 20%) and for 18 hr and then were used for assessing cell necrosis and cell apoptosis by annexin V/propidium iodide (PI) method by flow cytometry (see Materials and methods for details). Representative dot plots showing the percentage of annexin V, PI and annexin V/PI positive 16HBE at baseline and following the exposure of CSE (10%, 20%) are shown.
Figure 7.
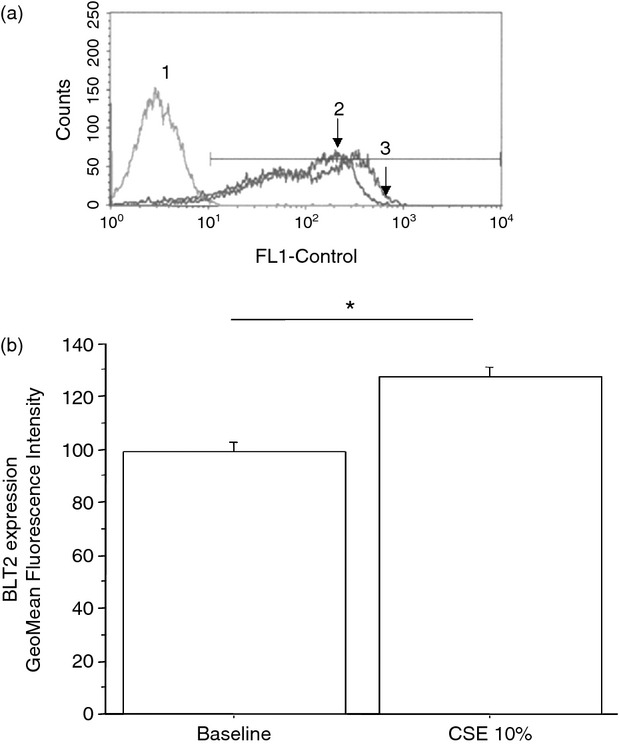
Cigarette smoke extracts (CSE) increase BLT2 expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE 10% for 18 hr and were used for assessing BLT2 expression by flow cytometry (see Materials and methods for details). (a) Representative histogram plots. 1 = negative control; 2 = baseline expression of BLT2; 3 = CSE 10%. (b) The results are expressed as GeoMean fluorescence intensity ± SD. *P < 0·05
Figure 8.
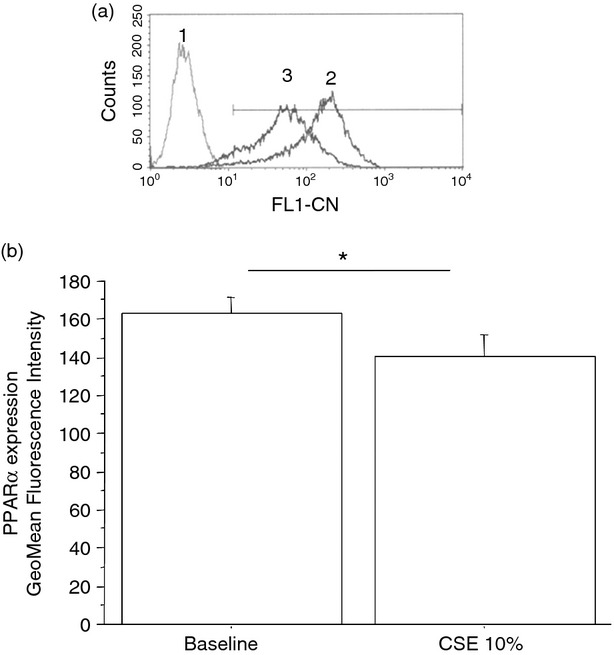
Cigarette smoke extracts (CSE) decrease peroxisome proliferator-activated receptor-α (PPAR-α) expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE 10% for 18 hr (n = 6) and were used for assessing PPAR-α expression by flow cytometry (see Materials and methods for details). (a) Representative histogram plots. 1 = negative control; 2 = baseline expression of PPAR-α; 3 = CSE 10%. (b) The results are expressed as GeoMean fluorescence intensity ± SD. *P < 0·05
Figure 9.
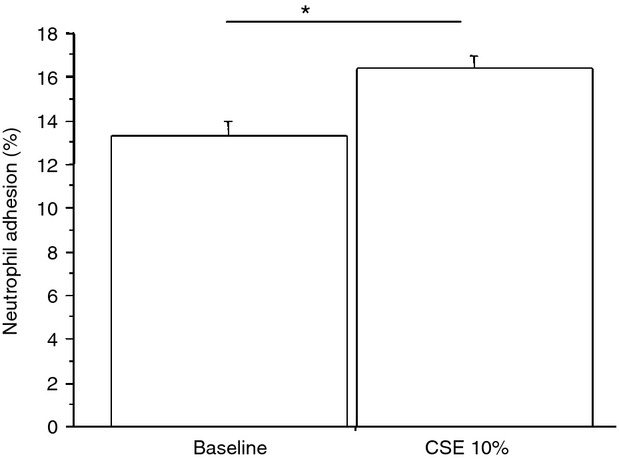
Cigarette smoke extracts (CSE) increase neutrophil adhesion to bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE 10% for 18 hr (n = 6) and were used for assessing neutrophil adhesion to bronchial epithelial cells. The results are expressed as % of the fluorescence ratio of bound cells on total cells. *P < 0·05
CSE increase ICAM-1, STAT-1 activation and the binding of STAT-1 to ICAM-1 promoter in bronchial epithelial cells
Exposure to CSE increased ICAM-1 expression (35 ± 12% of variation) in bronchial epithelial cells and this phenomenon was reduced by a BLT2 antagonist (20 ± 7% of variation) which was further increased by a PPAR-α antagonist (22 ± 2% of variation) (Fig. 10). Silencing experiments for BLT2 and for PPAR-α confirmed the results obtained with BLT2 or PPAR antagonists. Moreover, the mechanisms promoting the increased ICAM-1 expression in CSE-treated cells were explored by assessing the activation of STAT-1 and by assessing the binding of STAT-1 to ICAM-1 promoter. When bronchial epithelial cells were exposed to CSE, an increased expression of phosphorylated STAT-1 occurred (Fig. 11a) and an increased pSTAT-1/STAT-1 ratio was observed (Fig. 11b). ChIP assays with anti-STAT-1-specific antibodies showed that STAT-1 was detected on the promoter region of ICAM-1 in bronchial epithelial cells to a greater extent after CSE exposure (Fig. 11c). These data provide further evidence that ICAM-1 expression is regulated by STAT-1 activation.
Figure 10.
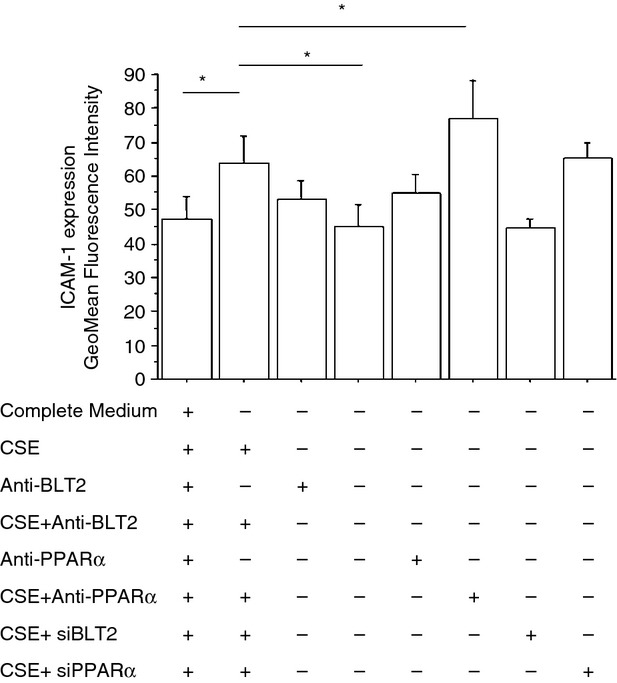
Cigarette smoke extracts (CSE) increase intercellular adhesion molecule 1 (ICAM-1) expression in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE 10%, of an inhibitor for BLT2 (LY255283; 1 hr pre-treatment) or of an inhibitor for peroxisome proliferator-activated receptor-α (PPAR-α) (MK886; 1 hr pre-treatment) (n = 6) and then were used for assessing ICAM-1 expression by flow cytometry. In some experiments (n = 2), RNA interference for BLT2 or PPAR-α was also performed. The results are expressed as GeoMean fluorescence intensity ± SD.*P < 0·05
Figure 11.
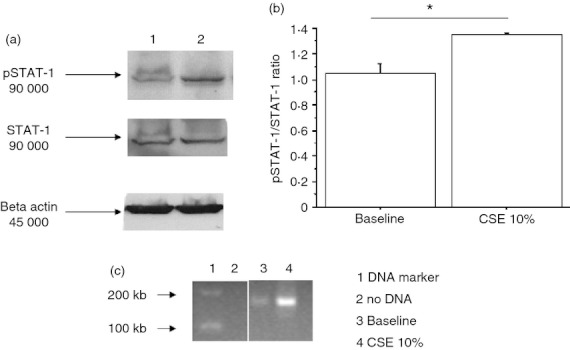
Cigarette smoke extracts (CSE) increase signal transducer and activator of transcription 1 (STAT-1) activation and its interaction with the promoter of intercellular adhesion molecule 1 (ICAM-1) in bronchial epithelial cells. Bronchial epithelial cells (16HBE) were cultured in the presence and in the absence of CSE 10% for 18 hr (n = 3) and were used for assessing STAT-1 or pSTAT-1 by Western blot analysis and for chromatin immunoprecipitation assay (ChIP). (a) Representative Western blots of STAT-1, pSTAT-1 and β-actin expression. Lane 1 = baseline; lane 2 = CSE 10%. (b) Signals corresponding to pSTAT-1 or to STAT-1 were semi-quantified by densitometric scanning, normalized for β-actin and results were expressed as pSTAT-1/STAT-1 ratio.*P < 0·05. (c) ChIP assay using anti-STAT1 antibody and PCR using primers spanning the promoter region 3537 of intercellular adhesion molecule 1 (ICAM-1) gene were performed (see Materials and methods for details) (n = 3). Lane 1 = DNA marker; lane 2 = negative control of PCR; lane 3 = baseline; lane 4 = CSE 10%.
Discussion
Long-term cigarette smoking is the major aetiological factor for the development of COPD. Airway inflammation and oxidative stress are implicated in the pathogenesis of COPD. Leukotriene B4, a potent lipid mediator of inflammation generated from arachidonic acid via the 5-lipoxygenase pathway, actively contributes to the inflammatory processes in smokers and in COPD patients.17
The present study demonstrates for the first time that the response of the bronchial epithelium to LTB4 is the result of a balance between the activation of receptors for LTB4 with a pro-inflammatory outcome (BLT2) and with an anti-inflammatory outcome (PPAR-α). Cigarette smoke generates an inflammatory milieu that promotes the preferential activation of pro-inflammatory LTB4 receptors, activation of STAT-1, an up-regulation of ICAM-1, and an increased adhesiveness between neutrophils and bronchial epithelial cells.
Neutrophils are a critical component of the host defence against micro-organisms18; however, cessation of neutrophil recruitment and clearance by apoptosis is mandatory to restore homeostasis and limit host tissue damage.19
Chronic neutrophilic inflammation is a frequent feature in lung diseases including COPD; it mediates extensive tissue damage and contributes to organ dysfunction.3 Leukotriene B4 induces neutrophil recruitment and activation in patients with COPD,17 it is higher in those with COPD than in controls, it correlates with the number of neutrophils20 and contributes to disease progression.21 Leukotriene B4 uses a dual-receptor system because it mediates its activity through the activation of pro-inflammatory transmembrane receptors (BLT1 and BLT2) and of an anti-inflammatory nuclear receptor (PPAR-α). Our group has previously demonstrated that pleural mesothelial cells, expressing both pro-inflammatory transmembrane BLT2 receptor and anti-inflammatory nuclear PPAR-α receptor, may provide an integrated response to pleural LTB4 present in parapneumonic effusions.7 The transmembrane BLT2 receptor is expressed, other than by leucocytes, by structural cells in a variety of body compartments and has a low binding affinity for LTB4.7,22 A previous study reports that BLT2 activation led to elevation of reactive oxygen species and subsequent activation of nuclear factor-κB), so inducing the expression of vascular cell adhesion molecule-1, which is known to be involved in eosinophil infiltration into the airways.23 The down-regulation of BLT2 with anti-sense BLT2 oligonucleotides markedly attenuated airway inflammation and airway hyper-responsiveness.23 The PPARs are transcription factors that belong to the superfamily of nuclear receptors. PPAR-α reduces pain and inflammation, inhibits the release of several pro-inflammatory and pro-angiogenic enzymes24,25 and has also been reported to bind and catabolise LTB4 and to limit inflammation. PPAR-α-deficient mice exhibit a prolonged inflammatory response when challenged with LTB426,27 and PPAR-α activation inhibits nuclear factor-κB activation28 and down-regulates chemoattractant production and neutrophil infiltration.29 In a rat model of CSE-induced emphysema, the treatment with PPAR-α+γ agonist for 4 weeks prevented the progression of emphysematous lung destruction.30
It has been previously demonstrated that BLT1 and PPAR-α immunoreactivity is increased in the distal airways of COPD patients compared with control subjects but it does not significantly differ in smokers without COPD.8 No information is available on BLT2 expression and on the mechanisms promoted by the unbalanced expression of BLT2 and PPAR-α on bronchial epithelial cells.
Our results suggest that cigarette smoke and mini-BALs from smokers, in vitro, in bronchial epithelial cells, shift the emphasis of LTB4 towards a pro-inflammatory phenotype up-regulating BLT2 expression and down-regulating PPAR-α. In addition, bronchial epithelial cells from smokers highly express BLT2 but not PPAR-α molecules. These findings may be relevant to clarify some pathogenetic events leading to COPD because several studies provide evidence that the in vitro exposure to CSE reproduces the in vivo changes observed within the airways of smoking humans.31 Exposure to CSE up-regulated the surface expression of ICAM-1 in human umbilical vein endothelial cells32 and in bronchial epithelial cells.33 Exposure of bronchial epithelial monolayers to the combination of cigarette smoke followed by C5a results in an additive response for ICAM-1 expression and mononuclear cell adhesion compared with smoke or C5a challenge alone.33
The contribution of LTB4 activity on ICAM-1 up-regulation because of CSE is largely unexplored. Here, we demonstrate that the treatment with CSE and mini-BALs from smokers resulted in an up-regulated expression of BLT2, which in turn increased ICAM-1 expression. Treatment with a BLT2 antagonist reduced the generated increased expression of ICAM-1. Moreover, CSE and mini-BALs from smokers reduce the activation of PPAR-α leading to increased ICAM-1 expression because the presence of a PPAR-α antagonist increases the effects of CSE and of mini-BALs from smokers in ICAM-1 expression. This finding is consistent with previous results showing that mice lacking PPAR-α have increased expression of ICAM-1 in the lungs.34 The increased ICAM-1 expression, increasing the adhesiveness of bronchial epithelial cells, may have several pathological consequences in subjects who smoke. Cigarette smoke exposure may play a critical role in rhinovirus-induced exacerbations,35 compromising the innate responses against rhinovirus infection36 and increasing the adhesion of viruses to airway epithelial cells because ICAM-1 acts as cellular receptor for the major group of rhinoviruses.37 In our model, the increased ICAM-1 expression is associated with increased neutrophil adhesion to bronchial epithelial cells. This phenomenon may promote an increased accumulation of neutrophils in the airways of smokers, so contributing to airway neutrophilia, a common feature in smokers and in patients with COPD. In this regard, it has been previously demonstrated that epithelial ICAM-1 and ICAM-2 regulate the passage of human T cells across the bronchial epithelium.38
Finally, we investigated the molecular mechanisms leading to the increased ICAM-1 expression in CSE-stimulated bronchial epithelial cells. The ICAM-1 is regulated by tyrosine phosphorylation activity through STAT-1-dependent signalling pathways.39 The effect of cigarette smoke on STAT-1 activation is unclear and it is unknown whether CSE per se interfere with STAT-1 activation in bronchial epithelial cells. It has been demonstrated that the pre-incubation with cigarette smoke reduces the interferon-induced STAT-1 phosphorylation in bronchial epithelial cells40 and that alveolar macrophages from smokers have reduced STAT-1 phosphorylation,41 but interferon-γ synergistically enhances lipopolysaccharide signalling in alveolar macrophages from patients with COPD via STAT1 activation.42 Here, when bronchial epithelial cells were exposed to CSE, an increased expression of phosphorylated STAT-1 and an increased expression of STAT-1 on the promoter region of ICAM-1 are detected.
Conclusions
Cigarette smoke and cigarette smoke-induced inflammation promote an imbalance between pro-inflammatory and anti-inflammatory LTB4 receptors by up-regulating BLT2 and down-regulating PPAR-α in bronchial epithelial cells. The prevalence of pro-inflammatory activities of BLT2 upon cigarette smoke exposure in turn leads to increased neutrophil adhesion, a mechanism contributing to airway neutrophilia and to lung tissue damage. These phenomena are frequently observed in smokers and in patients with COPD.
Acknowledgments
This work was supported by the Italian National Research Council. Elisabetta Pace and Maria Ferraro designed the study, performed the statistical analysis of the data and wrote the manuscript and declare that they have had access to and take responsibility for the integrity of the data and the accuracy of the data analysis. Serena Di Vincenzo, Luana Lipari, Valeria Scafidi, Carolina Sciarrino performed all the experiments of the study and participated in the interpretation of the data. Antonino Giarratano, Denis Di Benedetto contributed to the patient selection, and collected and managed biological samples. Mark Gjomarkaj and Andreina Bruno contributed to the writing of the manuscript.
Glossary
- BAL
bronchoalveolar lavages
- COPD
chronic obstructive pulmonary disease
- CSE
cigarette smoke extracts
- ICAM-1
intercellular adhesion molecule 1
- LTB4
leukotriene B4
- NF-κB
nuclear factor-κB
- PPAR-α
peroxisome proliferator-activated receptor-α
Disclosures
The authors have no financial disclosures or competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Cigarette smoke extract (CSE) increase intercellular adhesion molecule 1 (ICAM-1) expression in 16HBE cells.
References
- 1.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242:186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- 2.Crooks SW, Bayley DL, Hill SL, Stockley RA. Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. Eur Respir J. 2000;15:274–80. doi: 10.1034/j.1399-3003.2000.15b09.x. [DOI] [PubMed] [Google Scholar]
- 3.O'Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax. 2006;61:448–54. doi: 10.1136/thx.2004.024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill AT, Bayley D, Stockley RA. The interrelationship of sputum inflammatory markers in patients with chronic bronchitis. Am J Respir Crit Care Med. 1999;160:893–8. doi: 10.1164/ajrccm.160.3.9901091. [DOI] [PubMed] [Google Scholar]
- 5.Gompertz S, O'Brien C, Bayley DL, Hill SL, Stockley RA. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur Respir J. 2001;17:1112–9. doi: 10.1183/09031936.01.99114901. [DOI] [PubMed] [Google Scholar]
- 6.Fiedler J, Simon FR, Iwahashi M, Murphy RC. Effect of peroxisome proliferator-activated receptor α activation on leukotriene B4 metabolism in isolated rat hepatocytes. J Pharmacol Exp Ther. 2001;299:691–7. [PubMed] [Google Scholar]
- 7.Pace E, Ferraro M, Mody CH, et al. Pleural mesothelial cells express both BLT2 and PPARa and mount an integrated response to pleural LTB4. J Immunol. 2008;181:7292–9. doi: 10.4049/jimmunol.181.10.7292. [DOI] [PubMed] [Google Scholar]
- 8.Marian E, Baraldo S, Visentin A, Papi A, Saetta M, Fabbri LM, Maestrelli P. Up-regulated membrane and nuclear leukotriene B4 receptors in COPD. Chest. 2006;129:1523–30. doi: 10.1378/chest.129.6.1523. [DOI] [PubMed] [Google Scholar]
- 9.Pace E, Giarratano A, Ferraro M, Bruno A, Siena L, Mangione S, Johnson M, Gjomarkaj M. TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum Immunol. 2011;72:54–62. doi: 10.1016/j.humimm.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–25. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 11.Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer WI, Rabe KF, Hiemstra PS. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir Res. 2005;6:140. doi: 10.1186/1465-9921-6-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cozens AL, Yezzi MJ, Yamaya M, et al. A transformed human epithelial cell line that retains tight junctions post crisis. In Vitro Cell Dev. Biol. 1992;28A:735–44. doi: 10.1007/BF02631062. [DOI] [PubMed] [Google Scholar]
- 13.Seo JM, Cho KJ, Kim EY, Choi MH, Chung BC, Kim JH. Up-regulation of BLT2 is critical for the survival of bladder cancer cells. Exp Mol Med. 2011;43:129–37. doi: 10.3858/emm.2011.43.3.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Li L, Prabhakaran K, Zhang L, Leavesley HB, Borowitz JL, Isom GE. Uncoupling protein-2 up-regulation and enhanced cyanide toxicity are mediated by PPARα activation and oxidative stress. Toxicol Appl Pharmacol. 2007;223:10–9. doi: 10.1016/j.taap.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeidler R, Csanady M, Gires O, Lang S, Schmitt B, Wollenberg B. Tumor cell-derived prostaglandin E2 inhibits monocyte function by interfering with CCR5 and Mac-1. FASEB J. 2000;14:661–8. doi: 10.1096/fasebj.14.5.661. [DOI] [PubMed] [Google Scholar]
- 16.Pace E, Profita M, Melis M, et al. LTB4 is present in exudative pleural effusions and contributes actively to neutrophil recruitment in the inflamed pleural space. Clin Exp Immunol. 2004;135:519–27. doi: 10.1111/j.1365-2249.2003.02387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corhay JL, Henket M, Nguyen D, Duysinx B, Sele J, Louis R. Leukotriene B4 contributes to exhaled breath condensate and sputum neutrophil chemotaxis in COPD. Chest. 2009;136:1047–54. doi: 10.1378/chest.08-2782. [DOI] [PubMed] [Google Scholar]
- 18.Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. 2005;22:539–50. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 20.Profita M, Di Giorgi R, Sala A, et al. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy. 2005;60:1361–9. doi: 10.1111/j.1398-9995.2005.00892.x. [DOI] [PubMed] [Google Scholar]
- 21.Parr DG, White AJ, Bayley DL, Guest PJ, Stockley RA. Inflammation in sputum relates to progression of disease in subjects with COPD: a prospective descriptive study. Respir Res. 2006;7:136. doi: 10.1186/1465-9921-7-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B4 receptors. Prostaglandins Leukot Essent Fatty Acids. 2003;69:123–34. doi: 10.1016/s0952-3278(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 23.Cho KJ, Seo JM, Shin Y, et al. Blockade of airway inflammation and hyperresponsiveness by inhibition of BLT2, a low-affinity leukotriene B4 receptor. Am J Respir Cell Mol Biol. 2010;42:294–303. doi: 10.1165/rcmb.2008-0445OC. [DOI] [PubMed] [Google Scholar]
- 24.D'Agostino G, La Rana G, Russo R, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-α agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–43. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- 25.De Filippis D, D'Amico A, Cinelli MP, Esposito G, Di Marzo V, Iuvone T. Adelmidrol, a palmitoylethanolamide analogue, reduces chronic inflammation in a carrageenin-granuloma model in rats. J Cell Mol Med. 2009;13:1086–95. doi: 10.1111/j.1582-4934.2008.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devchand PR, Ijpenberg A, Devesvergne B, Wahli W. PPARs: nuclear receptors for fatty acids, eicosanoids, and xenobiotics. Adv Exp Med Biol. 1999;469:231–6. doi: 10.1007/978-1-4615-4793-8_34. [DOI] [PubMed] [Google Scholar]
- 27.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto H, Iwamoto T, Kotake S, Momohara S, Yamanaka H, Kamatani N. Inhibition of NF-κB signaling by fenofibrate, a peroxisome proliferator-activated receptor-α ligand, presents a therapeutic strategy for rheumatoid arthritis. Clin Exp Rheumatol. 2005;23:323–30. [PubMed] [Google Scholar]
- 29.Delayre-Orthez C, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N, Pons F. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005;6:91. doi: 10.1186/1465-9921-6-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Hanaoka M, Kitaguchi Y, Kraskauskas D, Shapiro L, Voelkel NF, Taraseviciene-Stewart L. Imbalance of apoptosis and cell proliferation contributes to the development and persistence of emphysema. Lung. 2012;190:69–82. doi: 10.1007/s00408-011-9326-z. [DOI] [PubMed] [Google Scholar]
- 31.Winkler AR, Nocka KH, Sulahian TH, Kobzik L, Williams CM. In vitro modeling of human alveolar macrophage smoke exposure: enhanced inflammation and impaired function. Exp Lung Res. 2008;34:599–629. doi: 10.1080/01902140802366261. [DOI] [PubMed] [Google Scholar]
- 32.Chen HW, Chien ML, Chaung YH, Lii CK, Wang TS. Extracts from cigarette smoke induce DNA damage and cell adhesion molecule expression through different pathways. Chem Biol Interact. 2004;150:233–41. doi: 10.1016/j.cbi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Floreani AA, Wyatt TA, Stoner J, Sanderson SD, Thompson EG, Allen-Gipson D, Heires AJ. Smoke and C5a induce airway epithelial intercellular adhesion molecule-1 and cell adhesion. Am J Respir Cell Mol Biol. 2003;29:472–82. doi: 10.1165/rcmb.2002-0143OC. [DOI] [PubMed] [Google Scholar]
- 34.Cuzzocrea S, Bruscoli S, Mazzon E, et al. PPAR-α contributes to the anti-inflammatory activity of glucocorticoids. Mol Pharmacol. 2008;73:323–37. doi: 10.1124/mol.107.041475. [DOI] [PubMed] [Google Scholar]
- 35.Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-κB-mediated transcription. J Biol Chem. 1999;274:9707–20. doi: 10.1074/jbc.274.14.9707. [DOI] [PubMed] [Google Scholar]
- 36.Bauer CM, Dewitte-Orr SJ, Hornby KR, Zavitz CC, Lichty BD, Stampfli MR, Mossman KL. Cigarette smoke suppresses Type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J Interferon Cytokine Res. 2008;28:167–79. doi: 10.1089/jir.2007.0054. [DOI] [PubMed] [Google Scholar]
- 37.Greve JM, Davis G, Meyer AM, Forte CP, Connolly Yost S, Marlor CW, Kamarck ME, McClelland A. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–47. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 38.Porter JC, Hall A. Epithelial ICAM-1 and ICAM-2 regulate the egression of human T cells across the bronchial epithelium. FASEB J. 2009;23:492–502. doi: 10.1096/fj.08-115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Audette M, Larouche L, Lussier I, Fugere N. Stimulation of the ICAM-1 gene transcription by the peroxovanadium compound [bpV(Pic)] involves STAT-1 but not NF-κB activation in 293 cells. Eur J Biochem. 2001;268:1828–36. [PubMed] [Google Scholar]
- 40.Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol. 2011;44:118–26. doi: 10.1165/rcmb.2009-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhillon NK, Murphy WJ, Filla MB, Crespo AJ, Latham HA, O'Brien-Ladner A. Down-modulation of IFN-γ signaling in alveolar macrophages isolated from smokers. Toxicol Appl Pharmacol. 2009;237:22–8. doi: 10.1016/j.taap.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Southworth T, Metryka A, Lea S, Farrow S, Plumb J, Singh D. IFN-γ synergistically enhances LPS signalling in alveolar macrophages from COPD patients and controls by corticosteroid resistant STAT1 activation. Br J Pharmacol. 2012;166:2070–83. doi: 10.1111/j.1476-5381.2012.01907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.