

Abstract
CLH-3b is a CLC-1/2/Ka/Kb channel homolog activated by meiotic cell cycle progression and cell swelling. Channel inhibition occurs by GCK-3 kinase-mediated phosphorylation of serine residues on the cytoplasmic C-terminus linker connecting CBS1 and CBS2. Two conserved aromatic amino acid residues located on the intracellular loop connecting membrane helices H and I and α1 of CBS2 are required for transducing phosphorylation changes into changes in channel activity. Helices H and I form part of the interface between the two subunits that comprise functional CLC channels. Using a cysteine-less CLH-3b mutant, we demonstrate that the sulfhydryl reagent reactivity of substituted cysteines at the subunit interface changes dramatically during GCK-3-mediated channel inhibition and that these changes are prevented by mutation of the H-I loop/CBS2 α1 signal transduction domain. We also show that GCK-3 modifies Zn2+ inhibition, which is thought to be mediated by the common gating process. These and other results suggest that phosphorylation of the cytoplasmic C-terminus inhibits CLH-3b by inducing subunit interface conformation changes that activate the common gate. Our findings have important implications for understanding CLC regulation by diverse signaling mechanisms and for understanding the structure/function relationships that mediate intraprotein communication in this important family of Cl− transport proteins.
Introduction
CLC anion channels and Cl−/H+ exchangers perform diverse and essential physiological functions including regulation of cytoplasmic and organelle Cl− and H+ levels, regulation of cell membrane potential, and transepithelial Cl− transport in organisms ranging from archaebacteria to humans. CLC channels and transporter are homodimers. Each monomer of a CLC channel forms an independently gated pore that is opened and closed by a fast gating process, whereas a common gating mechanism functions to close both pores simultaneously.
A CLC monomer consists of 18 α-helical domains (designated A–R). Helices B through R span or are embedded in the lipid bilayer. Membrane helices D, F, N, and R form the pore and a glutamate residue on the F-helix likely functions as the pore fast gate (1–3). Eukaryotic CLC monomers also have large cytoplasmic C-termini containing a pair of cystathionine-β-synthase (CBS) motifs (4,5). The structural basis of common gating is not well understood, but may involve conformational changes at the subunit interface, which comprises helices H, I, P, and Q (1–3), and/or the cytoplasmic C-terminus (6–9).
Numerous CLCs are regulated by phosphorylation (10–14), by binding with intracellular adenosine ligands (15–21), by interaction with accessory proteins (22–25), and by extracellular Ca2+ (26,27). However, the cell signaling and biophysical mechanisms by which regulation occurs are not well understood. We have addressed this important problem using the genetically tractable model organism Caenorhabditis elegans. CLH-3b is a splice variant of the C. elegans CLC gene clh-3 and is a member of the CLC-1/2/Ka/Kb anion channel subfamily. The channel is expressed in the worm oocyte where it is activated during meiotic cell cycle progression or in response to cell swelling (28) by type 1 serine/threonine phosphatase-mediated dephosphorylation (29). Channel inactivation requires concomitant phosphorylation of S742 and S747 mediated by the Ste20 kinase GCK-3 (30–32). GCK-3 is a homolog of the SPAK and OSR1 kinases, both of which play key roles in cellular and systemic ion and water homeostasis (33).
S742 and S747 are part of a ∼14 amino acid activation domain that is located on a ∼176 amino acid linker connecting the two cytoplasmic CBS motifs. Deletion of the activation domain inhibits CLH-3b to the same extent as GCK-3-mediated phosphorylation. Alanine mutation of two highly conserved aromatic amino acid residues located on the first α-helix (α1) of the second CBS domain (CBS2) and a short intracellular loop connecting membrane helices H and I (H-I loop) completely prevents channel inactivation by GCK-3 (34). The crystal structure of a CLC Cl−/H+ exchanger from the thermophilic red alga Cyanidioschyzon merolae (CmCLC) suggest that CBS2 and the H-I loop interact (3).
CBS domains play important regulatory roles in diverse proteins (35,36) and undergo regulatory interactions with adenosyl compounds (18,37,38), ions (39), and charged membrane domains (40). We have postulated that the dephosphorylated activation domain interacts with one or both CLH-3b CBS motifs, and that this interaction is disrupted by S742 and S747 phosphorylation or activation domain deletion. Disruption of this interaction induces a conformational change in the cytoplasmic C-terminus that inactivates CLH-3b. We have further proposed that the interface between CBS2 α1 and the H-I loop functions as a conserved signal transduction module that mediates long-range intraprotein communication from cytoplasmic to membrane domains in CLC proteins (34). The involvement of the H-I loop in signal transduction suggests that the subunit interface may play an important role in regulating channel activity.
What are the structure/function mechanisms by which changes in C-terminus conformation are transduced into changes in channel activity? In the current studies, we tested the hypothesis that C-terminus phosphorylation induces extracellular conformation changes in the subunit interface and channel pore that mediate inactivation of CLH-3b. Using the substituted cysteine accessibility method in a cysteine-less CLH-3b mutant, we demonstrate that the sulfhydryl reagent reactivity of amino acid residues comprising the subunit interface changes dramatically during GCK-3-mediated channel inhibition and that these changes are prevented by mutation of the H-I loop/CBS2 α1 signal transduction interface. We also show that GCK-3 modifies Zn2+ inhibition, which is thought to act through the common gating process (41–43). These and other results suggest that phosphorylation of the CLH-3b cytoplasmic C-terminus inhibits the channel by inducing subunit interface conformation changes that activate the common gate. Our findings have important implications for understanding CLC regulation by intracellular nucleotides, extracellular Ca2+ and accessory proteins, and for understanding the structure/function relationships that mediate intraprotein communication in this important family of channels and transporters.
Material and Methods
Transfection and whole-cell patch-clamp recording
Human embryonic kidney (HEK293) cells were cultured and patch clamped as described previously (44). Cells were transfected using FuGENE 6 or X-tremeGENE HP (Roche Diagnostics, Indianapolis, IN) with 0.5 μg GFP, 3 μg CLH-3b, and 1.5 μg of functional or kinase dead (KD) GCK-3 ligated into pcDNA3.1. Channel properties are indistinguishable when CLH-3b is expressed without kinase or coexpressed with KD GCK-3 (30,31). Point mutations were generated using the QuikChange Lightning Multi Site-Directed mutagenesis kits (Agilent Technologies, Santa Clara, CA). All mutations were confirmed by DNA sequencing. Experimental protocols were performed on at least two independently transfected groups of cells.
Transfected cells were identified by GFP fluorescence and patch clamped using a bath solution containing 90 mM NMDG-Cl, 5 mM MgSO4, 1 mM CaCl2, 12 mM Hepes free acid titrated to pH 7.0 with CsOH, 8 mM Tris, 5 mM glucose, 90 mM sucrose, and 2 mM glutamine (pH 7.4, 300 mOsm), and a pipette solution containing 116 mM NMDG-Cl, 2 mM MgSO4, 20 mM Hepes, 6 mM CsOH, 1 mM EGTA, 2 mM ATP, 0.5 mM GTP, and 10 mM sucrose (pH 7.2, 275 mOsm). Patch electrodes were pulled from 1.5 mm outer diameter silanized borosilicate microhematocrit tubes; electrode resistance ranged from 4 to 8 MΩ. Currents were measured with an Axopatch 200A (Axon Instruments, Foster City, CA) patch clamp amplifier. Electrical connections to the patch clamp amplifier were made using Ag/AgCl wires and 3 M KCl/agar bridges. Data acquisition and analysis were performed using pClamp 10 software (Axon Instruments).
Quantification of CLH-3b current properties
Whole cell currents were elicited by stepping membrane potential from a holding voltage of 0 mV to test voltages of −140 to +60 mV in 20 mV increments. Test voltages were maintained for 1 s and cells were then returned to the holding voltage of 0 mV for 1 s. The channel is strongly inwardly rectifying and does not exhibit tail currents due to rapid inactivation at positive potentials. Current-to-voltage plots were therefore used to estimate channel activation voltage (31). A line was first drawn by linear regression analysis of currents measured between 0 and 60 mV where channel activity is low. A second line was then drawn by linear regression analysis of currents measured between the first test voltage at which inward current was detected and a second voltage 20 mV more negative. The point at which these two lines intersect is defined as the activation voltage, which is the voltage at which current activation is first detected.
The kinetics of hyperpolarization-induced activation of CLH-3b are characterized by either mono- or biexponential fits describing slow or fast and slow time constants, respectively. The nature of the fit is dictated by channel phosphorylation (31). To simplify presentation and interpretation of activation kinetics under different experimental conditions, time constants are not used. Instead, the time required for whole cell current to reach 50% activation when membrane voltage is stepped from 0 to −100 mV for 1 s is quantified. This time is defined as the 50% rise time.
Sulfhydryl reagent and Zn2+ experiments
The methanethiosulfonate (MTS) sulfhydryl reactive reagents 2-(trimethylammonium)ethyl methanethiosulfonate bromide (MTSET) and sodium (2-sulfonatoethyl) methanethiosulfonate (MTSES; Toronto Research Chemicals, Toronto, Ontario, Canada) were dissolved in water as a 400 mM stock and stored in 40 μl aliquots at –80°C until use. Single aliquots were added to 16 ml of control bath immediately before each experiment. The final MTSET and MTSES working concentrations were 1 mM.
Whole cell current amplitude in MTSET/MTSES and Zn2+ experiments was recorded by stepping membrane voltage to –100 mV for 500 ms every 1–2 s from a holding potential 0 mV. The bath was perfused continuously with control solution during this voltage clamp protocol. Cells were exposed to MTSET/MTSES- or Zn2+-containing bath solution after whole cell current reached stable levels. Continuous bath perfusion was maintained in MTSET/MTSES experiments until the effects of the reagents were complete.
Time constants describing the inhibitory or stimulatory effects of MTSET and MTSES were determined by fitting a single exponential function to time course data. For Zn2+ inhibition and washout studies, time constants were determined using the Compare Models function of pClamp 10 (Axon Instruments), which determines the number of terms required for the best fit by statistical analysis.
Statistical analyses
Electrophysiological data are presented as means ± SE and n represents the number of patch-clamped cells from which CLH-3b currents were recorded. Statistical significance was determined using Student’s t-test for unpaired means.
Results
Effects of MTSET on wild-type (WT) and cys-less CLH-3b
MTSET inhibits CLH-3b whole cell current (45). To determine whether GCK-3-mediated phosphorylation of S742 and S747 induces conformational changes in channel extracellular domains, we expressed CLH-3b with functional or KD GCK-3 and characterized the effects of 1 mM MTSET on current properties. As described previously (30,31), coexpression of CLH-3b with GCK-3 reduced current amplitude, hyperpolarized channel activation voltage, and slowed hyperpolarization- induced current activation (Fig. 1 A). GCK-3 also significantly (P < 0.008) increased both the rate and extent of MTSET inhibition (Fig. 1, B and C). These results demonstrate that channel inhibition induced by GCK-3 is associated with extracellular conformational changes that alter the MTS reagent reactivity of endogenous cysteine residues.
Figure 1.
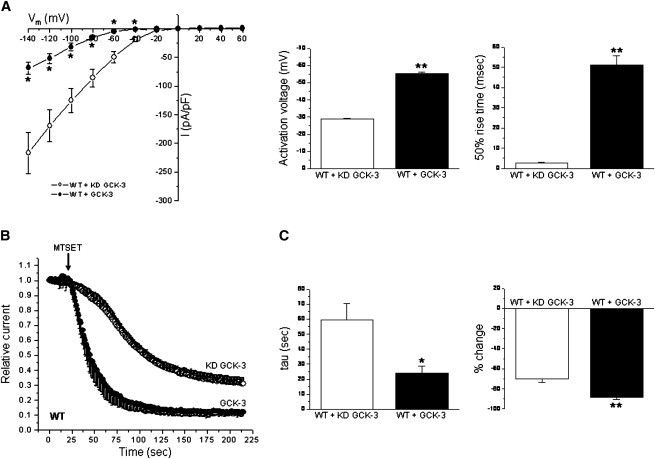
GCK-3 alters MTSET reactivity of WT CLH-3b. (A) Current-to-voltage relationships, activation voltages, and 50% rise times of CLH-3b coexpressed with KD or functional GCK-3. Values are means ± SE (n = 5–10). *P < 0.02 and **P < 0.0001 compared to KD GCK-3. (B) Time course of MTSET effects on CLH-3b. (C) Time constants (tau) of MTSET inhibitory effects and percent change in current amplitude. Values are means ± SE (n = 4–5). *P < 0.008 and **P < 0.005 compared to KD GCK-3.
CLH-3b contains 11 cysteines. To characterize the effects of phosphorylation on channel conformational changes in greater detail, we generated a cys-less CLH-3b mutant in which all 11 cysteine residues were replaced by alanine. The functional properties of the cys-less mutant were similar to those of WT CLH-3b. Cys-less CLH-3b exhibited strong inward rectification and a hyperpolarized activation voltage (Fig. 2 A). Hyperpolarization-induced activation was time dependent (Fig. 2 A). Channel activation voltage and 50% rise time were not significantly (P > 0.2) different from WT CLH-3b.
Figure 2.
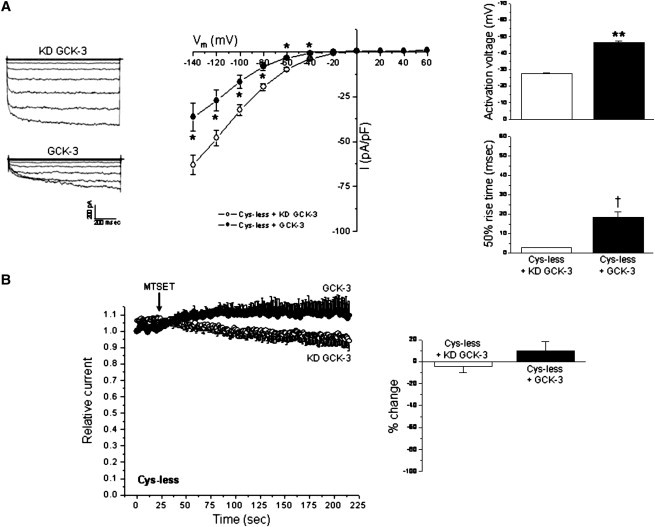
Cys-less CLH-3b is regulated by GCK-3 and insensitive to MTSET. (A) Whole cell current traces, current-to-voltage relationships, activation voltages, and 50% rise times of cys-less CLH-3b coexpressed with KD or functional GCK-3. Values are means ± SE (n = 10). *P < 0.01, **P < 0.0001, and †P < 0.0004 compared to KD GCK-3. (B) Effects of MTSET on current amplitude of cys-less CLH-3b. Values are means ± SE (n = 4–5). Relative changes in current amplitude induced by MTSET were not significantly (P > 0.3) different from 0 in the presence or absence of GCK-3 activity.
Whole cell current amplitudes for cys-less CLH-3b were generally smaller compared to those of WT channels suggesting lower expression levels and/or reduced single channel conductance. The mutant was responsive to GCK-3 as evidenced by significant (P < 0.01) kinase-dependent reductions in whole cell current levels, hyperpolarization of channel activation voltage, and slowing of voltage-dependent activation kinetics (Fig. 2 A). However, the GCK-3-dependent shifts in these parameters were typically smaller than those observed for WT CLH-3b.
In the absence of GCK-3, hyperpolarization-induced activation of WT CLH-3b is described by fast and slow time constants. However, the fast time constant is lost when the channel is inactivated by the kinase (31). The cys-less mutant behaved in an identical manner. When coexpressed with KD GCK-3, current activation at −140 mV was described by fast and slow time constants with mean ± SE values of 19 ± 3 ms and 131 ± 19 ms (n = 10), respectively. In contrast, activation at −140 mV of cys-less CLH-3b coexpressed with functional kinase was described by a single slow time constant with a mean ± SE value of 119 ± 20 ms (n = 10).
As expected, MTSET had no significant effect on cys-less CLH-3b. The mean steady-state MTSET induced changes in relative current amplitudes in cells expressing either KD or functional GCK-3 were <±10% (Fig. 2 B). These changes were not significantly (P > 0.3) different from 0.
MTS reagent reactivity of substituted cysteine mutants
Given that the functional properties of cys-less CLH-3b were similar to those of WT channels, we carried out a series of cysteine substitutions in this mutant. Our previous studies suggested that the CLC subunit interface may be an important site at which phosphorylation exerts its effects on CLH-3b structure and function (34). Several studies have suggested that the subunit interface plays an important role in common gating (6–8). We therefore focused our initial MTS reactivity studies in and around helices H, I, P, and Q, which comprise the interface (1–3). Cysteine mutations on the pore-forming helices D, F, N, and R, including the glutamate residue (E167) that forms the pore fast gate were also tested.
Fig. 3 shows ribbon diagrams of Escherichia coli CLC (EcCLC) and the location of homologous cysteine substitutions that were generated in cys-less CLH-3b. We generated a total of 22 cysteine substitution mutants (Table 1). All of the mutants except K166C and E167C showed gating behavior similar to WT CLH-3b including strong inward rectification, a hyperpolarized activation voltage, and time-dependent hyperpolarization-induced current activation (data not shown). The K166C and E167C mutants exhibited both inward and outward currents and lacked voltage- and time-dependent gating (data not shown). Of the 22 mutants, 13 expressed poorly or did not react with MTSET, and 3 showed no change in reactivity when the mutants were expressed with GCK-3 (Fig. 3 and Table 1). The remaining 6 mutants showed phosphorylation-dependent changes in MTS reagent reactivity (Fig. 3 and Table 1). These residues are highlighted in green in Fig. 3.
Figure 3.
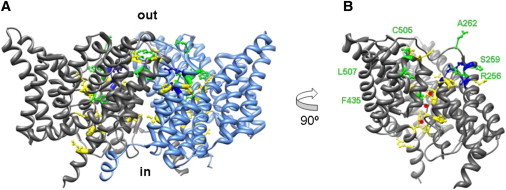
Location of cysteine substitutions. (A) Ribbon diagram of EcCLC dimer viewed from the side. Colored balls and sticks indicate homologous location of CLH-3b cysteine substitutions. Yellow, mutations that expressed poorly or did not react with MTS reagents. Blue, mutations that reacted with MTS reagents, but reactivity did not change with GCK-3 coexpression. Green, mutations that showed altered MTS reagent reactivity when channel was coexpressed with GCK-3. All cysteine substitutions were made in the cys-less CLH-3b background. See Table 1 for additional details. (B) Ribbon diagram of EcCLC monomer rotated 90°. Red spheres denote Cl− ions within the channel pore. Residues that exhibited GCK-3-dependent changes in MTS reagent reactivity are labeled and shown in green.
Table 1.
Location and MTS reagent reactivity of cysteine substitution mutants
| Residue | Location | Functional properties |
|---|---|---|
| S124C | D-helix | Poor expression |
| P127C | D-helix | Poor expression |
| K166C | F-helix | No reactivity |
| E167C | F-helix | No reactivity |
| S216C | G-H loop | MTS reactive; no GCK-3 effect |
| A217C | G-H loop | Poor expression |
| P218C | H-helix | Poor expression |
| I226C | H-helix | Poor expression |
| R253C | I-helix | MTS reactive; no GCK-3 effect |
| L255C | I-helix | Poor expression |
| R256C | I-helix | MTS reactive; reactivity altered by GCK-3 |
| M257C | I-J loop | MTS reactive; no GCK-3 effect |
| S259C | I-J loop | MTS reactive; reactivity altered by GCK-3 |
| A262C | I-J loop | MTS reactive; reactivity altered by GCK-3 |
| F435C | N-helix | MTS reactive; reactivity altered by GCK-3 |
| P437C | N-helix | Poor expression |
| G502C | P-helix | Poor expression |
| Q503C | P-Q loop | Poor expression |
| C505 | P-Q loop | MTS reactive; reactivity altered by GCK-3 |
| L507C | Q-helix | MTS reactive; reactivity altered by GCK-3 |
| Y529C | R-helix | No reactivity |
| K536C | R-helix | No reactivity |
Fig. 4, A and B, show details of the MTSET reactivity of two subunit interface cysteine mutants. R256 is located on the I-helix (Fig. 3). MTSET increased current amplitude of the R256C mutant coexpressed with KD GCK-3 ∼55%. Coexpression of R256C with functional kinase strikingly and significantly reduced both the extent (P < 0.01) and rate (P < 0.025) of MTSET-induced current activation (Fig. 4 A).
Figure 4.
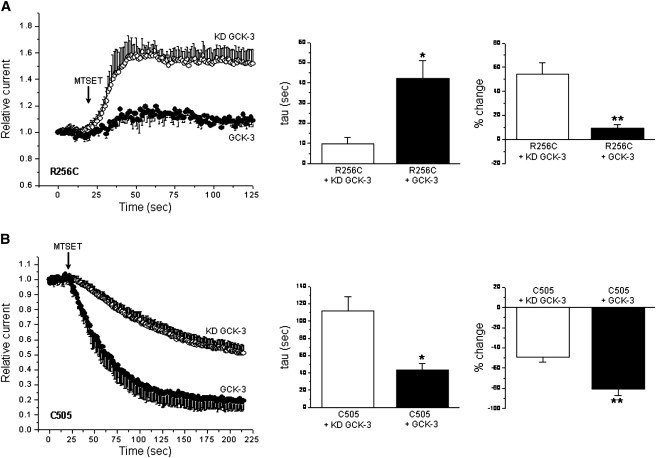
Characteristics of MTSET reactivity of the R256C and C505 mutants. (A) R256C mutant. Values are means ± SE (n = 3–4). *P < 0.025 and **P < 0.01 compared to KD GCK-3. (B) C505 mutant. Values are means ± SE (n = 3–5). *P < 0.02 and **P < 0.007 compared to KD GCK-3.
C505 is an endogenous cysteine residue located on an extracellular loop connecting helices P and Q (Fig. 3). MTSET inhibited the C505 channel both in the presence and absence of GCK-3 coexpression (Fig. 4 B). However, the extent and rate of MTSET inhibition were significantly (P < 0.02) increased by GCK-3-mediated phosphorylation.
In addition to C505, eight other endogenous cysteine residues are located in membrane helices or extracellular facing loops, and two cysteines are located on the N- and C-terminal cytoplasmic domains. We generated a mutant channel in which the first seven membrane domain associated cysteine residues (C113–C380) were replaced with alanine. This mutant showed MTSET reactivity similar to the WT and C505 channels (data not shown). Replacement of 10 of the 11 endogenous cysteines except for C484 gave rise to a channel that was insensitive to MTSET (data not shown). Taken together, these results indicate that C505 is most likely responsible for the MTSET-induced inhibition of WT CLH-3b (Fig. 1, B and C), and suggest that the other 10 endogenous cysteine residues do not react with MTS reagents in the absence or presence of GCK-3.
Fig. 5 A summarizes the effects of MTS reagents on interface and pore mutants exhibiting reactivity. S216C (G-H loop), R253C (I helix), and M257C (I-J loop) mutant channels showed similar MTSET reactivity in the presence and absence of GCK-3 coexpression. S259C (I-J loop) mutant channels were inhibited ∼30% by MTSET and coexpression with GCK-3 induced a complete loss of reactivity. The A262C (I-J loop) mutant was inhibited 15–20% by MTSET. Interestingly, in the presence of GCK-3, MTSET became stimulatory and activated A262C channels ∼15%. L507C (Q helix) mutants showed similar degrees of MTSET inhibition with or without kinase coexpression. However, GCK-3 coexpression significantly (P < 0.04) increased the rate of MTSET inhibition. A number of other interface cysteine mutants tested (A217C, G-H loop; P218C, and I226C, H helix; L255C, I helix; G502C, P helix, and Q503C, P-Q loop) either expressed poorly or did not react with MTSET. Overall, data shown in Figs. 4 and 5 show that GCK-3 induces conformational changes in extracellular-facing domains associated with the subunit interface.
Figure 5.
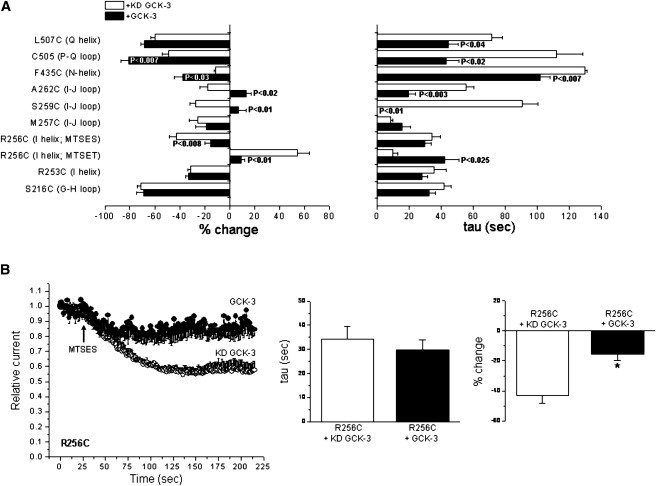
MTS reagent reactivity of subunit interface and pore amino acid residues. (A) Summary of MTS reagent effects on various pore and subunit interface cysteine substitution mutants. Values are means ± SE (n = 3–6). P-values shown are compared to channels expressed with KD GCK-3. (B) Characteristics of MTSES inhibition of the R256C mutant. Values are means ± SE (n = 4). *P < 0.008 compared to KD GCK-3.
Most cysteine mutations in helices D, F, N, and R comprising the channel pore expressed poorly or did not react with MTSET (Fig. 3 and Table 1). However, the N helix mutant F435C showed significantly (P < 0.03) enhanced MTSET reactivity when it was coexpressed with GCK-3 (Fig. 5 A) indicating that phosphorylation of the C-terminus activation domain also induces conformational changes in extracellular domains associated with the channel pores.
As shown in Fig. 4 A, MTSET had a stimulatory effect on the R256C mutant. The crystal structure of EcCLC (1,2) suggests that R256 is located near the outer mouth of the CLH-3b pore. Charged residues located close to the intracellular pore opening of CLC-0 modulate conductance and fast gating (46–49). MTSET is positively charged. Its stimulatory effect could thus reflect an important channel regulatory role for the positively charged arginine residue at position 256. Therefore, to determine if the effect of MTSET was charge dependent, we treated R256C expressing cells with negatively charged MTSES. R256C expressed with KD GCK-3 was inhibited ∼40% by MTSES (Fig. 5 B). The extent of inhibition was reduced to ∼15% (P < 0.01) by GCK-3 coexpression, but the rate constant for inhibition was not significantly (P > 0.7) altered. The stimulatory and inhibitory effects of MTSET and MTSES, respectively, are consistent with a role for R256 in modulation of channel gating and conductance (see Discussion).
GCK-3-induced extracellular conformational changes are mediated by the intracellular H-I loop/CBS2 α1 interface
The inhibitory effect of GCK-3 on CLH-3b is prevented by alanine mutagenesis of a conserved tyrosine residue, Y232, on the intracellular H-I loop or a conserved histidine residue, H805, on the first α-helix (α1) of CBS2 (34). These two residues are closely apposed in CmCLC (3) and may therefore functionally interact. We have proposed that conformational information associated with phosphorylation of the cytoplasmic C-terminus is transduced to extracellular domains via the H-I loop/CBS2 α1 interface (34). If GCK-3-dependent extracellular conformational changes are required for channel regulation, these changes should then be prevented by mutations that disrupt this putative signal transduction domain. We therefore mutated Y232 to alanine in the R256C and C505 mutants and characterized the effect of GCK-3 on extracellular conformation. As shown in Fig. 6, A and B, the Y232A mutation fully blocked the effect of GCK-3 on R256C and C505 MTSET reactivity.
Figure 6.
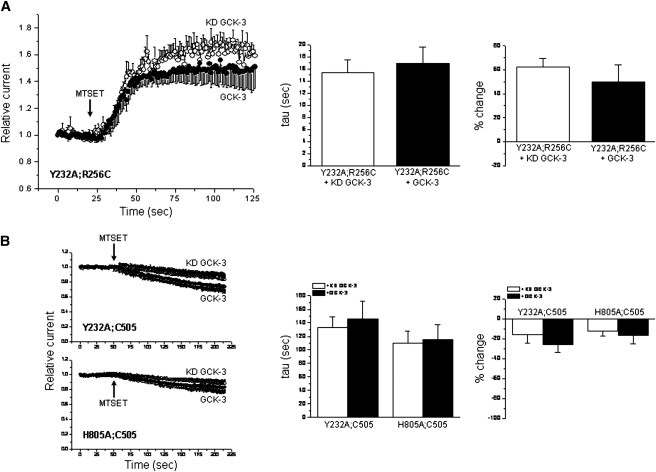
Effect of mutation of the H-I loop/CBS2 α1 interface on GCK-3-induced changes in MTSET reactivity. Y232 and H805 are located on the H-I loop and α1 of CBS2, respectively. Mutation of these residues to alanine fully blocks the inhibitory effect of GCK-3 on channel activity (34). These mutations also block the effect of GCK-3 on MTSET reactivity of the R256C (A) and C505 (B) CLH-3b mutants. The Y232A and H805A mutations additionally reduce the inhibitory effect of MTSET on C505 in the presence and absence of GCK-3 (compare to Fig. 4, A and B). Values are means ± SE (n = 3–4).
We also mutated H805 on CBS2 α1 to alanine in the C505 mutant. This mutation fully blocked the effect of GCK-3 on MTSET reactivity. Both the Y232A and H805A mutation also reduced the inhibitory effect of MTSET on the C505 mutant in the presence and absence of GCK-3 (Fig. 6 B). These and our previous results (34) demonstrate that the H-I loop/CBS2 α1 interface plays a critical role in intraprotein signaling that mediates phosphorylation-dependent channel conformational changes and regulation.
GCK-3-mediated phosphorylation alters Zn2+ inhibition
The subunit interface is the site of multiple disease causing mutations (50,51) and plays an important role in common gating (6,7). Zinc inhibits CLCs and it is widely accepted that its inhibitory action is mediated by the common gate (41–43). Given the striking effect GCK-3 has on the MTS reagent reactivity of amino acid residues associated with the subunit interface (Figs. 4 and 5), we examined the Zn2+ sensitivity of WT CLH-3b coexpressed with or without functional kinase.
When exposed to 5 mM Zn2+, WT channels coexpressed with KD GCK-3 exhibited slower and less extensive inhibition compared to channels coexpressed with functional kinase (Fig. 7 A). We determined time constants for Zn2+ inhibition using mono- or multiexponential fits. Zinc inhibition of channels expressed with KD GCK-3 was described by fast and slow time constants (Fig. 7 B). In contrast, a single fast time constant described Zn2+ inhibition of CLH-3b coexpressed with functional GCK-3 (Fig. 7 B). Reversal of Zn2+ inhibition showed similar kinetics in the presence and absence of the kinase (Fig. 7 B).
Figure 7.
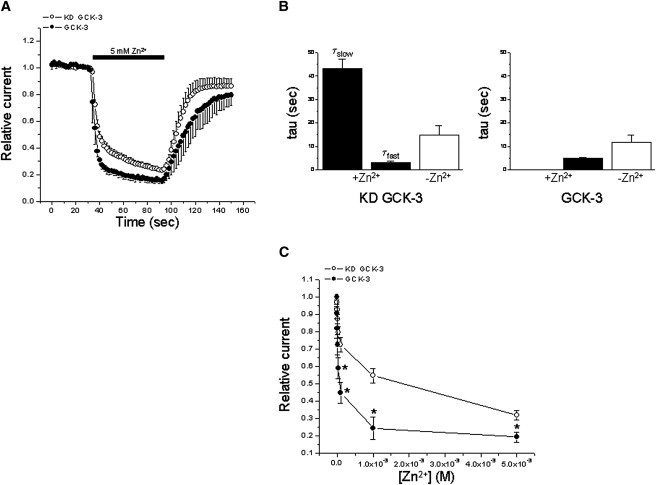
GCK-3 alters the kinetics and concentration dependence of Zn2+ inhibition. (A) Time course of 5 mM Zn2+ inhibition and washout. (B) Time constants for 5 mM Zn2+ inhibition and washout. In the absence of functional GCK-3, the kinetics of Zn2+ inhibition are described by fast and slow time constants. A single time constant describes Zn2+ inhibition of CLH-3b coexpressed with functional kinase. (C) Concentration dependence of Zn2+ inhibition. Values are means ± SE (n = 3–5). *P < 0.03 compared to KD GCK-3.
We also examined the concentration dependence of Zn2+ inhibition in the presence and absence of GCK-3. As shown in Fig. 7 C, GCK-3 inhibited channels exhibited increased sensitivity to Zn2+. Taken together, data in Figs. 4–7 suggest that GCK-3-mediated phosphorylation may modulate the common gate.
Pore fast gate mutations alter subunit interface cysteine reactivity
E167 comprises the pore fast gate of CLH-3b. We previously demonstrated that replacement of E167 with cysteine in WT CLH-3b alters MTSET reactivity, and that this effect is reversed by mutations in the cytoplasmic C-terminus. Our interpretation of these results was that E167C MTSET reactivity was modulated by C-terminus conformational changes suggesting that cytoplasmic domains may regulate pore fast gating (45). However, results of the current studies suggest an alternative hypothesis. Given that E167C does not react with MTSET in a cys-less background, and that C505 is likely the only reactive cysteine residue in WT CLH-3b, it is possible that pore fast gate mutations (i.e., E167C) may alter C505 MTSET reactivity. If this is the case, it suggests that conformational changes in the pore fast gate alter the conformation of the subunit interface and vice versa. Conformational interaction between these two domains has implications for understanding CLC gating.
To assess the effect of pore fast gate conformation on subunit interface conformation, we mutated E167 in the single cysteine C505 background and quantified C505 MTSET reactivity. C505 is located on the extracellular loop connecting subunit interface helices P and Q (Fig. 3 and Table 1). On the basis of our previous findings with the E167C mutant (45), we mutated E167 to either alanine or leucine. Cysteine and alanine are both slightly hydrophobic amino acids with side-chain volumes of ∼45 Å3 and 25 Å3, respectively (52). Leucine has a much larger side-chain volume of ∼110 Å3 (52) and is strongly hydrophobic. As shown in Fig. 8 A, E167A;C505 channels showed little MTSET reactivity. The mean MTSET-induced change in current amplitude was not significantly (P > 0.2) different from 0 either in the presence or absence of GCK-3. E167L;C505 channels exhibited increased (P < 0.04) MTSET reactivity when they were coexpressed with GCK-3. However, the extent of MTSET-induced channel inhibition was significantly (P < 0.02) less than that observed in the C505 mutant coexpressed with or without functional kinase. These results suggest that the conformation of the pore fast gate and possibly other channel domains influence the conformation of the subunit interface.
Figure 8.
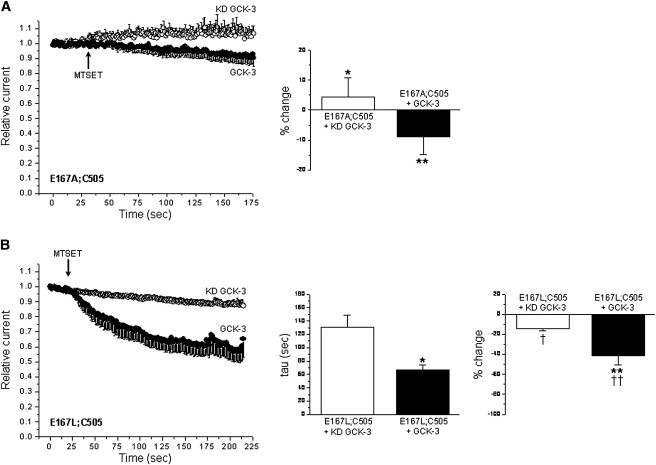
Mutation of the E167 pore fast gate alters the MTSET reactivity of the subunit interface cysteine residue C505. (A) E167A;C505 mutant. Values are means ± SE (n = 4–5). Current amplitude changes induced by MTSET were not significantly (P > 0.2) different from 0 in the presence or absence of GCK-3 activity. *P < 0.0002 and **P < 0.0004 compared to C505 mutant (see Fig. 5A). (B) E167L;C505 mutant. Values are means ± SE (n = 3–5). *P < 0.04 and **P < 0.008 compared to KD GCK-3. †P < 0.0006 and ††P < 0.02 compared to C505 mutant (see Fig. 5A).
Discussion
Hyperpolarization-induced activation of CLH-3b is described by fast and slow time constants. However, when the channel is inactivated by GCK-3, a single, slow process dominates voltage-dependent gating (31). Fast and slow time constants have been derived from exponential fits of gating events in CLC-1 and CLC-2 (53–55). Fast time constants are thought to reflect opening and closing of pore gates, whereas slow time constants have been ascribed to the common gating mechanism.
The loss of the fast time constant induced by GCK-3 (31) suggests that phosphorylation of the channel inhibits the pore fast gate and/or activates the common gating mechanism. A number of recent studies have demonstrated that intracellular nucleotide binding, extracellular Ca2+ binding, and the interacting protein barttin likely mediate their regulatory effects on various CLC proteins via the common gate (15,17,21,24,26,27,56,57). GCK-3 induces significant changes in MTS reagent reactivity at amino acid residues associated with the subunit interface (Table 1 and Figs. 4 and 5). Conformational changes at the subunit interface have been proposed to underlie common gating in CLC-1 (6–8). In addition, GCK-3 alters the inhibitory effects of Zn2+ (Fig. 7), which is thought to act on the common gate (41–43). On the basis of these findings and studies in other CLC proteins, we propose that phosphorylation-dependent inhibition of CLH-3b is mediated by activation of common gating.
GCK-3-induced MTSET reactivity changes (Fig. 6) and channel inhibition (34) are both blocked by alanine mutation of Y232 or H805. Y232 and H805 are conserved residues located on an intracellular loop that connects membrane helices H and I, which form part of the subunit interface (1–3), and the first α-helix of CBS2, respectively. The H-I loop interfaces with CBS2 α1 in CmCLC (3). We have proposed recently that this interface functions as a conserved signal transduction module that mediates long-range intraprotein signaling in CLC channels (34). The linkage between channel activity and subunit interface conformation changes mediated by the H-I loop/CBS2 α1 interface further supports our hypothesis that activation of the common gate underlies GCK-3-induced channel inhibition.
At least one amino acid residue associated with the channel pore also exhibits GCK-3-induced changes in MTSET reactivity (Fig. 5 A) indicating that GCK-3 also modifies pore conformation. It is not clear, however, whether this conformational change is direct or a result of changes at the subunit interface. The membrane helices that form the CLC pores are closely apposed to the helices that form the subunit interface in EcCLC (1,2) and CmCLC (3) and studies from several laboratories suggest that there is functional coupling between common and pore gating (6,8,53,58). Thus, it is possible that conformational changes at the subunit interface induce conformational changes in the pore and vice versa. Consistent with this idea, we observed that mutation of the glutamate residue (E167) that forms the pore fast gate alters the MTSET reactivity of the subunit interface amino acid residue C505 (Fig. 8).
Our working model raises an interesting and important question. Does activation of common gating represent a conformational change that blocks both pores simultaneously without affecting the function of the pore fast gate? Or does activation of the common gate induce conformational changes that function to close the fast gates of both pores simultaneously as suggested by Ma et al. (8)? We have observed that GCK-3 inhibits the activity of various CLH-3b pore fast gate glutamate (i.e., E167) mutants (T. Yamada and K. Strange, unpublished observations). This suggests that common gating may inactivate CLH-3b independently of the pore fast gates. However, it is not possible to rule out pore gating in E167 mutants. Although neutralization of the glutamate residue comprising the pore fast gate is expected to increase channel open probability and alter voltage- and Cl−-dependent pore gating, it is still possible for substituted amino acids to modulate pore conductance. In addition, mutagenesis and electrophysiological studies in eukaryotic CLC channels have suggested that pore fast gating may involve conformational changes beyond those occurring at the fast gate glutamate residue (59). CLC-K channels provide an example of these possibilities. These channels exhibit both fast and common gating processes, but lack the glutamate residue comprising the pore fast gate (24). Clearly, additional electrophysiological and structural studies are needed to define how phosphorylation-induced conformational changes at the subunit interface regulate CLH-3b activity.
It is interesting to note the stimulatory effect of positively charged MTSET on the whole cell current of the R256C mutant (Fig. 4 A). Studies on CLC-0 have shown that a lysine residue located near the inner mouth of the pore, K519, controls channel conductance and pore gating and that the effects of charge at this position can be mimicked by positively and negatively charged MTS reagents in a K519C mutant (46–49). R256 is located on the I-helix and is thus part of the subunit interface (Fig. 3). Examination of the crystal structure of EcCLC (1,2) suggests that this residue is located near the outer mouth of the CLH-3b pore ∼13–14 Å away from the pore gate. As with CLC-0 then, charged residues near the pore may exert electrostatic control over channel conductance and/or fast gating. Consistent with this idea, we have found that replacement of the positively charged arginine residue at position 256 with glutamate hyperpolarizes CLH-3b activation voltage by 10–15 mV (T. Yamada and K. Strange, unpublished observations).
Importantly, the MTSET-induced stimulation of R256C current is strikingly suppressed by GCK-3 (Fig. 4 A). This suggests that phosphorylation induces a conformational change that reduces the putative electrostatic effect of R256 on pore conductance and/or gating. Such a change could reflect at least one mechanism by which GCK-3 inhibits CLH-3b activity. Clearly, additional structure/function studies will be needed to test this hypothesis directly.
In conclusion, our studies have provided the first, to our knowledge, detailed insights into structure/function relationships that mediate phosphorylation-dependent regulation of a CLC anion transport protein. We propose that phosphorylation of the C-terminus of CLH-3b is transduced into a subunit interface conformational change and activation of the common gating mechanism via the interaction between α1 of CBS2 and the intracellular H-I loop. Activation of the common gate directly blocks pore conduction and/or inhibits pore fast gating. The current studies together with our previous work (34) provide an important foundation for understanding the mechanistic basis of regulation of other CLC proteins by phosphorylation, intracellular nucleotide binding, extracellular Ca2+, and accessory proteins.
Acknowledgments
The authors thank Jerod Denton and Michael Pusch for helpful discussions and Angela Parton and Rebecca Morrison for technical assistance.
This work was supported by National Institutes of Health (NIH) grant R01 DK51610 to K.S. and by NIH Institutional Development Award (IDeA) grant P20 GM103423.
Experiments described in this work were proposed and designed by T.Y., M.B., and K.S. T.Y. and M.B. performed the experimental studies. All authors participated in the analysis and interpretation of the data, in the writing of the manuscript, and in the approval of the final version of the manuscript for publication.
References
- 1.Dutzler R., Campbell E.B., MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 2.Dutzler R., Campbell E.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 3.Feng L., Campbell E.B., MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duran C., Thompson C.H., Hartzell H.C. Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jentsch T.J. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 6.Cederholm J.M., Rychkov G.Y., Bretag A.H. Inter-subunit communication and fast gate integrity are important for common gating in hClC-1. Int. J. Biochem. Cell Biol. 2010;42:1182–1188. doi: 10.1016/j.biocel.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Duffield M., Rychkov G., Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. J. Gen. Physiol. 2003;121:149–161. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma L., Rychkov G.Y., Bretag A.H. Movement of hClC-1 C-termini during common gating and limits on their cytoplasmic location. Biochem. J. 2011;436:415–428. doi: 10.1042/BJ20102153. [DOI] [PubMed] [Google Scholar]
- 9.Bykova E.A., Zhang X.D., Zheng J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat. Struct. Mol. Biol. 2006;13:1115–1119. doi: 10.1038/nsmb1176. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa T., Ogura T., Inagaki N. Phosphorylation and functional regulation of ClC-2 chloride channels expressed in Xenopus oocytes by M cyclin-dependent protein kinase. J. Physiol. 2002;540:883–893. doi: 10.1113/jphysiol.2001.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsiao K.M., Huang R.Y., Lin M.J. Functional study of CLC-1 mutants expressed in Xenopus oocytes reveals that a C-terminal region Thr891-Ser892-Thr893 is responsible for the effects of protein kinase C activator. Cell. Physiol. Biochem. 2010;25:687–694. doi: 10.1159/000315088. [DOI] [PubMed] [Google Scholar]
- 12.Cuppoletti J., Tewari K.P., Malinowska D.H. Sites of protein kinase A activation of the human ClC-2 Cl(−) channel. J. Biol. Chem. 2004;279:21849–21856. doi: 10.1074/jbc.M312567200. [DOI] [PubMed] [Google Scholar]
- 13.Duan D., Cowley S., Hume J.R. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J. Gen. Physiol. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson N.C., Huang P., Nelson D.J. Identification of an N-terminal amino acid of the CLC-3 chloride channel critical in phosphorylation-dependent activation of a CaMKII-activated chloride current. J. Physiol. 2004;556:353–368. doi: 10.1113/jphysiol.2003.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennetts B., Parker M.W., Cromer B.A. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 2007;282:32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- 16.De Angeli A., Moran O., Gambale F. ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J. Biol. Chem. 2009;284:26526–26532. doi: 10.1074/jbc.M109.005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tseng P.Y., Yu W.P., Chen T.Y. Binding of ATP to the CBS domains in the C-terminal region of CLC-1. J. Gen. Physiol. 2011;137:357–368. doi: 10.1085/jgp.201010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott J.W., Hawley S.A., Hardie D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer S., Savaresi S., Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007;14:60–67. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 20.Wellhauser L., Luna-Chavez C., Bear C.E. ATP binding induces conformational changes in the carboxy terminal region of ClC-5. J. Biol. Chem. 2011;268:6733–6741. doi: 10.1074/jbc.M110.175877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennetts B., Yu Y., Parker M.W. Intracellular β-nicotinamide adenine dinucleotide inhibits the skeletal muscle ClC-1 chloride channel. J. Biol. Chem. 2012;287:25808–25820. doi: 10.1074/jbc.M111.327551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeworutzki E., López-Hernández T., Estévez R. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(−) channel auxiliary subunit. Neuron. 2012;73:951–961. doi: 10.1016/j.neuron.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leisle L., Ludwig C.F., Stauber T. ClC-7 is a slowly voltage-gated 2Cl(−)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011;30:2140–2152. doi: 10.1038/emboj.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer M., Janssen A.G., Fahlke C. Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 2010;21:1281–1289. doi: 10.1681/ASN.2009121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholl U., Hebeisen S., Fahlke C. Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. USA. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gradogna A., Babini E., Pusch M. A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 2010;136:311–323. doi: 10.1085/jgp.201010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gradogna A., Fenollar-Ferrer C., Pusch M. Dissecting a regulatory calcium-binding site of CLC-K kidney chloride channels. J. Gen. Physiol. 2012;140:681–696. doi: 10.1085/jgp.201210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutledge E., Bianchi L., Strange K. CLH-3, a ClC-2 anion channel ortholog activated during meiotic maturation in C. elegans oocytes. Curr. Biol. 2001;11:161–170. doi: 10.1016/s0960-9822(01)00051-3. [DOI] [PubMed] [Google Scholar]
- 29.Rutledge E., Denton J., Strange K. Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7α/β phosphatases. J. Cell Biol. 2002;158:435–444. doi: 10.1083/jcb.200204142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falin R.A., Morrison R., Strange K. Identification of regulatory phosphorylation sites in a cell volume- and Ste20 kinase-dependent ClC anion channel. J. Gen. Physiol. 2009;133:29–42. doi: 10.1085/jgp.200810080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denton J., Nehrke K., Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J. Gen. Physiol. 2005;125:113–125. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falin R.A., Miyazaki H., Strange K. C. elegans STK39/SPAK ortholog-mediated inhibition of ClC anion channel activity is regulated by WNK-independent ERK kinase signaling. Am. J. Physiol. Cell Physiol. 2011;300:C624–C635. doi: 10.1152/ajpcell.00343.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delpire E., Gagnon K.B. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem. J. 2008;409:321–331. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 34.Miyazaki H., Yamada T., Strange K. CLC anion channel regulatory phosphorylation and conserved signal transduction domains. Biophys. J. 2012;103:1706–1718. doi: 10.1016/j.bpj.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ignoul S., Eggermont J. CBS domains: structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005;289:C1369–C1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 36.Baykov A.A., Tuominen H.K., Lahti R. The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem. Biol. 2011;6:1156–1163. doi: 10.1021/cb200231c. [DOI] [PubMed] [Google Scholar]
- 37.Townley R., Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315:1726–1729. doi: 10.1126/science.1137503. [DOI] [PubMed] [Google Scholar]
- 38.Jin X., Townley R., Shapiro L. Structural insight into AMPK regulation: ADP comes into play. Structure. 2007;15:1285–1295. doi: 10.1016/j.str.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 39.Hattori M., Tanaka Y., Nureki O. Crystal structure of the MgtE Mg2+ transporter. Nature. 2007;448:1072–1075. doi: 10.1038/nature06093. [DOI] [PubMed] [Google Scholar]
- 40.Mahmood N.A., Biemans-Oldehinkel E., Poolman B. Engineering of ion sensing by the cystathionine beta-synthase module of the ABC transporter OpuA. J. Biol. Chem. 2009;284:14368–14376. doi: 10.1074/jbc.M901238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duffield M.D., Rychkov G.Y., Roberts M.L. Zinc inhibits human ClC-1 muscle chloride channel by interacting with its common gating mechanism. J. Physiol. 2005;568:5–12. doi: 10.1113/jphysiol.2005.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen T.Y. Extracellular zinc ion inhibits ClC-0 chloride channels by facilitating slow gating. J. Gen. Physiol. 1998;112:715–726. doi: 10.1085/jgp.112.6.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y.W., Lin C.W., Chen T.Y. Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J. Gen. Physiol. 1999;114:1–12. doi: 10.1085/jgp.114.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyazaki H., Strange K. Differential regulation of a CLC anion channel by SPAK kinase ortholog-mediated multisite phosphorylation. Am. J. Physiol. Cell Physiol. 2012;302:C1702–C1712. doi: 10.1152/ajpcell.00419.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He L., Denton J., Strange K. Carboxy terminus splice variation alters ClC channel gating and extracellular cysteine reactivity. Biophys. J. 2006;90:3570–3581. doi: 10.1529/biophysj.105.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pusch M., Ludewig U., Jentsch T.J. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature. 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- 47.Chen M.F., Chen T.Y. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 2003;122:133–145. doi: 10.1085/jgp.200308844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen T.Y., Chen M.F., Lin C.W. Electrostatic control and chloride regulation of the fast gating of ClC-0 chloride channels. J. Gen. Physiol. 2003;122:641–651. doi: 10.1085/jgp.200308846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Middleton R.E., Pheasant D.J., Miller C. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- 50.Lourdel S., Grand T., Teulon J. ClC-5 mutations associated with Dent’s disease: a major role of the dimer interface. Pflugers Arch. 2012;463:247–256. doi: 10.1007/s00424-011-1052-0. [DOI] [PubMed] [Google Scholar]
- 51.Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- 52.Zamyatnin A.A. Protein volume in solution. Prog. Biophys. Mol. Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
- 53.Zúñiga L., Niemeyer M.I., Sepúlveda F.V. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J. Physiol. 2004;555:671–682. doi: 10.1113/jphysiol.2003.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennetts B., Roberts M.L., Rychkov G.Y. Temperature dependence of human muscle ClC-1 chloride channel. J. Physiol. 2001;535:83–93. doi: 10.1111/j.1469-7793.2001.t01-1-00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saviane C., Conti F., Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999;113:457–468. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennetts B., Rychkov G.Y., Cromer B.A. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J. Biol. Chem. 2005;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- 57.Tseng P.Y., Bennetts B., Chen T.Y. Cytoplasmic ATP inhibition of CLC-1 is enhanced by low pH. J. Gen. Physiol. 2007;130:217–221. doi: 10.1085/jgp.200709817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yusef Y.R., Zúñiga L., Sepúlveda F.V. Removal of gating in voltage-dependent ClC-2 chloride channel by point mutations affecting the pore and C-terminus CBS-2 domain. J. Physiol. 2006;572:173–181. doi: 10.1113/jphysiol.2005.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Accardi A., Pusch M. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 2003;122:277–293. doi: 10.1085/jgp.200308834. [DOI] [PMC free article] [PubMed] [Google Scholar]