

Abstract
In several neurodegenerative diseases, like Huntington's disease (HD), treatments are still lacking. To determine whether a treatment is effective, sensitive disease progression biomarkers are especially needed for the premanifest phase, since this allows the evaluation of neuroprotective treatments preventing, or delaying disease manifestation. On the basis of a longitudinal study we present a biomarker that was derived by integrating behavioural and neurophysiological data reflecting cognitive processes of action control. The measure identified is sensitive enough to track disease progression over a period of only 6 month. Changes tracked were predictive for a number of clinically relevant parameters and the sensitivity of the measure was higher than that of currently used parameters to track prodromal disease progression. The study provides a biomarker, which could change practice of progression diagnostics in a major basal ganglia disease and which may help to evaluate potential neuroprotective treatments in future clinical trials.
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by an extension of a CAG repeat at exon 1 of chromosome 4. The CAG repeat expansion is an established marker (trait biomarker) for the presence of HD. Currently, there is no cure for HD1. To determine whether a potential treatment is effective, we need definitive, quantitative, objective biomarkers for tracking disease progression2. Reliable disease state biomarkers that are sensitive enough to monitor the state and subtle progression of HD are lacking for the premanifest stage. During this stage, motor symptoms have not yet developed. To search for such a marker, several large-scale longitudinal studies, such as TRACK-HD3, PREDICT4, and REGISTRY5, have examined disease progression in the premanifest and/or the manifest stages of HD. These studies used neuropsychological tests, motor tests, biochemical parameters, and MRI neuroimaging techniques, the results of which were discussed as potential endpoints for clinical trials6. However, a single biomarker may not reflect the myriad aspects of the disease7. It has therefore been suggested that use and integration of multiple biomarkers might be more effective for tracking HD progression2.
Cross-sectional studies that integrate neuropsychological tests with electrophysiological (electroencephalographic, EEG) data show that these measures differ between controls and HD patients according to disease stage8,9 and reliably correlate with the estimated age of onset (eAO) and CAG repeat expansion10. Currently, no multicenter studies are using this approach, although use of a combination of electrophysiological measures seems promising as a way to evaluate progression2. By definition, a cognitive disease progression biomarker should vary according to disease progression in affected subjects but should not vary across longitudinal time points in control subjects2,11.
Here we present data from a longitudinal single centre study in premanifest HD gene mutation carriers (pre-HD subjects) that investigated the use of cognitive-neurophysiological measures as a biomarker of disease progression. To investigate whether these measures were sensitive to subtle changes that occurred over short time periods, the study employed two non-equidistant longitudinal endpoints after baseline measurement, 15 and 21 months (15-month and 6-month intervals). Hence, three time points were subjected to the analysis (i.e., baseline, 15-month, 21-month).
We examined processes related to action selection and control in a task that was a combination of a “Stroop Task” and a “Set-shifting Task.” Both tasks depend on fronto-striatal networks12,13, which play an important role in action selection14. These processes depend upon the integrity of striatal medium spiny neurons (MSNs)15, the genetically determined degeneration of which is a hallmark of HD16,17. The task used here requires that conflict monitoring18 and the flexible adaptation of actions13 be performed in parallel. Notably, this task can detect early deficits in premanifest HD compared to controls at the behavioural and neurophysiological level using event-related potentials (ERPs)19. Since changes in the fronto-striatal networks reflect disease progression6, it is possible that the parallel execution of conflict monitoring and flexible behavioural adaptation may be sensitive enough to detect even subtle changes in premanifest disease progression and may be used as a cognitive disease progression biomarker in premanifest HD.
Results
Behavioural data
Reaction times (RTs) and error rates were measured in each of the experimental condition (i.e., compatible switch, compatible non-switch, incompatible switch, incompatible non-switch). The RT data (means and SDs) in each of these conditions are given in Table 1 for the pre-HD group. For RT data, the highest interaction containing the factors “time point” and “group” was the interaction for “compatibility × switch/non-switch × time point × group” (F(2,110) = 49.83; P = 0.0001; η2 = 0.475). Subsequent analyses showed that there was an interaction for the pre-HD group for “compatibility × switch/non-switch × time point” (F(2,52) = 83.33; P = 3 × 10−6; η2 = 0.675), showing that task performance varied across the longitudinal time points in pre-HD subjects but not in controls. Further ANOVA analysis of data from the pre-HD subjects was performed separately for the compatible and incompatible trials and revealed that only for the incompatible trials, there was an interaction for “switch/non-switch × time point” (F(2,52) = 77.47; P = 1 × 10−6; η2 = 0.749). Bonferroni-corrected post-hoc tests revealed that RTs for the incompatible non-switch trials in the pre-HD group did not differ between time points (p > 0.4) (Figure 1). However, for the incompatible switch trials, RTs (in ms) were faster at baseline (831 ± 12) than at the 15-month time point (976 ± 15) (P = 0.0005, Cohen's d = 1.92). Interestingly, a further increase in RT was also evident between the 15-month (976 ± 15) and the 21-month endpoints (1023 ± 16) (P = 0.003, Cohen's d = 0.65) (Figure 1). In terms of error rates, there was no interaction for “compatibility × switch/nonswitch × time point × group” (F(2,110) = 0.98; P > 0.5) so this was not analysed further. As stated in the methods section, two of the examined participants revealed phenoconversion between the 15 and 21-month endpoint. When excluding these two manifest HD subjects from the data analyses, the results remained unchanged; i.e., the results are not biased with respect to these two patients. As to the effect sizes obtained for the reaction time data between the 15 and 21-month endpoint, the effects only decreased marginally (i.e., 0.03 in the effect size not normalized monthly, refer Figure 5) when excluding these two patients.
Table 1. Means and standard deviations (SD) for pre-HD subjects in the different experimental conditions at all time points (longitudinal, 15-month, 21-month).
| Baseline | 15-month | 21-month | |
|---|---|---|---|
| compatible switch | 654 (50) | 660 (53) | 646 (55) |
| incompatible switch | 834 (49) | 976 (65) | 1023 (65) |
| compatible non-switch | 601 (38) | 611 (38) | 598 (37) |
| incompatible non-switch | 666 (37) | 672 (35) | 658 (38) |
Figure 1.
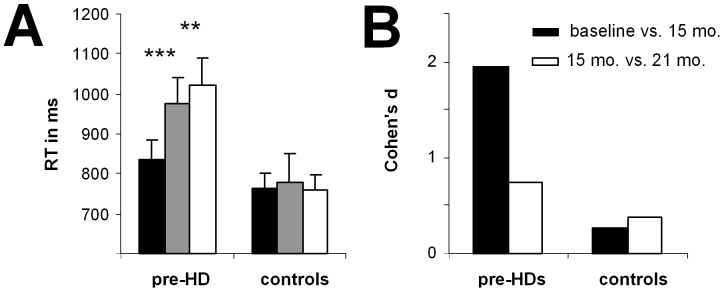
(A) Mean RTs ± SEM for the “incompatible” condition in pre-HD subjects and control subjects at baseline and at the two longitudinal endpoints. Black bars show the amplitudes at baseline, grey bars show amplitudes at the 15-month longitudinal endpoint, and white bars show amplitudes at the 21-month endpoint. (B) Cohen's d effect sizes in the pre-HD and control groups show the effects between baseline and 15 months and between the 15-month and 21-month endpoints.
Neurophysiological data
For the ERP data the amplitudes (in μV/m2) and latencies (in ms) were used as dependent variables. For the time-frequency decomposed data, the power in the different frequency band was used a dependent variable.
The N2 amplitudes for the subjects in this study are shown in Figure 2A at all three time points for the pre-HD subjects and control subjects. The grand average ERPs are shown in Figure 2B. The N2 ERP revealed an interaction for “compatibility × switch/non-switch × time point × group” (F(2,110) = 28.84; P = 0.0003; η2 = 0.344). As with the behavioural data, subsequent analyses show that only for the pre-HD group, there was an interaction “compatibility × switch/non-switch × time point” (F(2,52) = 26.06; P = 0.0001; η2 = 0.501). ANOVA tests of the pre-HD subjects, performed separately for the compatible and incompatible trials, revealed that only for the incompatible trials, there was an interaction “switch/non-switch × time point” (F(2,52) = 22.26; P = 0.0005; η2 = 0.461). Bonferroni-corrected post-hoc tests revealed that the N2 amplitude for incompatible non-switch trials in the pre-HD group did not differ between time points (P > 0.6). However, in line with the behavioural data, the N2 amplitude was larger (i.e. more negative) at baseline (−5.57 ± 0.3), compared to the 15-month time point (−5.09 ± 0.2) (P = 0.0008; d = 0.97). Importantly, the N2 amplitude declined further at the 21-month time point (−4.41 ± 0.3) compared to the 15-month time point (P = 0.002; d = 0.4).
Figure 2.

(A) ERP traces at electrode FCz in control subjects and pre-HD subjects at the three longitudinal time points. The stimulus was presented at time 0, the time scale (x-axis) is in milliseconds (ms). Red lines indicated results for “incompatible” switch trials, green lines “compatible” switch trials, black curves “compatible” non-switch trials, and brown curves “incompatible” non-switch trials. (B) Mean N2 amplitudes at electrode FCz for pre-HD subjects and control subjects at the three time points in the incompatible switch condition. (C) Cohen's d effect sizes in the pre-HD and control groups show the effects between baseline and 15 months and between the 15-month and 21-month endpoints. (D) Mean P3 amplitudes at electrode Pz for pre-HD subjects and control subjects at the three time points.
In terms of the N2 peak latency, there was no interaction for “compatibility × switch/non-switch × time point × group” (F(2,110) = 26.33; P = 0.0005; η2 = 0.312), and this was not analysed further. Similarly, there was no interaction for “compatibility × switch/non-switch × time point × group” for the P3 amplitudes and latencies measured at electrode Pz, P1, and P2 (all F < 0.9; P > 0.4). The P3 ERP components at electrode Pz are shown in Figure 2D.
In addition to the N2 time domain data, the baseline data showed that the time-frequency decomposed data was different for pre-HD subjects compared to controls (Beste et al., 2012). To test whether this parameter was sensitive enough to track premanifest disease progression over short time periods, we also quantified this parameter within the N2 time range (Figure 3A). The time-frequency plots of N2 for the pre-HD group and controls at all time points are shown in Figure 3B.
Figure 3.

(A) Time-frequency plots show the evoked wavelet power at electrode FCz in pre-HD subjects and control subjects at the three time points. (B) Mean N2 evoked power at electrode FCz for pre-HD subjects and control subjects at the three time points. (C) Cohen's d effect sizes in the pre-HD and control groups show the effects between baseline and 15 months and between the 15-month and 21-month endpoints.
For the N2 evoked wavelet power the interaction of “compatibility × switch/non-switch × time point × group” was significant (F(2,110) = 37.24; P = 0.00009; η2 = 0.404). Subsequent ANOVAs revealed that the interaction of “compatibility × switch/non-switch × time point” (F(2,52) = 98.84; P = 1 × 10−6; η2 = 0.641) was only significant for the pre-HD group. Further ANOVA tests of data from pre-HD subjects, which were performed separately for the compatible and incompatible trials, revealed that there was an interaction of “switch/non-switch × time point” (F(2,52) = 21.06; P = 0.0004; η2 = 0.447) only for the incompatible trials. Bonferroni-corrected post-hoc tests revealed that the evoked wavelet power was highest at baseline (3.48 ± 0.01) and had declined at the 15-month endpoint (3.44 ± 0.03) (P = 0.0003; d = 1.17). The evoked wavelet power was even lower at the 21-month endpoint (3.40 ± 0.02) than at the 15-month endpoint (P = 0.0001; d = 1.42) (Figure 3B). To underline the validity of the results obtained we performed non-parametric test using Monte-Carlo simulations (5000 permutations, 95% confidence interval). The Friedman test revealed differences between the longitudinal endpoints (χ2 = 48.22; df = 2; lower bound p = .00001; upper bound p = .001). A Wilcoxon test (5000 permutations, 95% confidence interval) revealed differences between baseline and the 15-month endpoint (Z = −4.51; lower bound p = .00001; upper bound p = .001), as well as between the 15-month and 21-month endpoint (Z = −4.22; lower bound p = .00001; upper bound p = .001).
As the P3 data did not reveal an interaction for the factors “compatibility × switch/non-switch × time point × group” in the time domain data, the time-frequency decomposed data was not analysed. When excluding these two manifest HD subjects from the data analyses, the results remained unchanged; i.e., the results are not biased with respect to these two patients. As to the effect sizes obtained for the reaction time data between the 15 and 21-month endpoint, the effects only decreased marginally (i.e., 0.08 in the effect size not normalized monthly) when excluding these two patients.
Regression analyses using the behavioural and time-frequency decomposed data were performed using the disease burden score (DBS) giving a measure of toxic load6 and the probability of disease manifestation in the next 5 years as dependent variables20. There were substantial correlations between both dependent variables and behavioural and neurophysiological parameters (Figure 4).
Figure 4.

(A) Correlations between changes in the 15-month longitudinal period for the behavioural (y1-axis; blue crosses) and neurophysiological data (y2-axis; red crosses). The left plots show correlations with the disease burden score (DBS; x-axis), and the right plots show correlations with 5-year onset probability (x-axis). The y-axes indicate the degree of change in the behavioural parameter (i.e. the difference in reaction time between baseline and the 15-month endpoint) and the neurophysiological parameter (i.e. the difference in evoked wavelet power in the N2 range between baseline and the 15-month endpoint). (B) Correlations between the degree of change in the 6-month longitudinal period (between the 15-month and 21-month endpoints).
Comparison of the effects in this study with those from other studies
To put the current results in the context of those from large-scale multi-centre studies, we compared the effect sizes for the behavioural and neurophysiological parameters in this study with the scores obtained in the cognitive battery of the TRACK-HD study. The TRACK-HD study is one of the largest longitudinal, multi-centre, observational studies of HD6. To conduct a comparison, we used the data shown in Table 3 from the analysis by Stout et al.21 to calculate Cohen's d values. The results of this analysis are shown in Figure 5A, together with Cohen's d as derived from the behavioural and neurophysiological data in the current study. To correct for the different endpoints in the current study and in the TRACK-HD study, we provide Cohen's d estimations for monthly changes (Figure 5B).
Figure 5.

(A) Histograms of the Cohen's d effect sizes. Blue bars indicate the effect sizes obtained in the 15-month period; orange bars indicate effect sizes obtained in the current study in the 6-month period between the 15-month and 21-month endpoints. Green bars indicate effect sizes that were calculated based on the neuropsychological data in Stout et al.21. Note that for the Stout et al.21 data, the direction of longitudinal performance development is not coded. (B) Cohen's d effect sizes were normalized monthly. Normalization was calculated on the basis of the 15-month longitudinal data (this study) and the 12-month longitudinal data from the Stout et al.21 study.
Discussion
In this longitudinal study, we evaluate the sensitivity of cognitive-neurophysiological parameters for documenting disease progression in pre-HD. The results show that parallel monitoring of conflict and flexible adaptation of actions became increasingly compromised in pre-HD subjects during a 15-month period from baseline as well as during the subsequent 6-month period. To the best of our knowledge, this is the first study to show declines in premanifest disease progression over a period of just 6 months. Other studies on cognition have not shown changes in cognitive function in pre-HD subjects22 or, alternatively, the pre-HD subjects did not differ from controls23. The effect sizes show the high sensitivity of the measures we use (Fig. 6). Two pre-HD subjects revealed phenoconversion between the 15 and 21-month endpoint. Excluding these subjects did not affect the pattern of results or the effect sizes obtained. The effect sizes are greater than those obtained from longitudinal neuropsychological data in the TRACK-HD study21.
Figure 6. Schematical overview of the experimental paradigm to assess parallel execution of response selection and conflict monitoring.

In particular, the results show increases in RTs in incompatible switch trials that are paralleled by reduction of the N2 ERP component and evoked wavelet power in the delta frequency band. The N2 ERP reflects conflict monitoring, response selection processes, and inhibition of responses24,25. The N2 ERP data, together with the behavioural effects, most likely reflect an increasing inability to inhibit processes related to the irrelevant task set from the previous trial26 and to select the appropriate response in the current trial19,24. In contrast, the P3 ERP component is related to working memory processes, and in particular to the updating, organization, and implementation processes involved in a new task set27. Since this neurophysiological parameter does not show longitudinal changes in pre-HD subjects, working memory processes may not contribute to the longitudinal changes we observed.
N2-related processes are mediated via the fronto-striatal networks, including the anterior cingulate cortex (ACC)25. Processes reflected by the P3 component are mediated largely via parietal cortical networks28. As opposed to the parietal areas, the ACC is closely connected to the striatum. It is possible that differences in the strength of connectivity between the striatal areas and the frontal or parietal areas underlie the differences we observed during premanifest disease progression. The dependence on N2-related processes in fronto-striatal circuits, together with the fact that the parallel execution of cognitive processes (i.e., conflict monitoring and flexible adaptation) depends on the fronto-striatal networks, may explain the high sensitivity across the longitudinal endpoints that we observed in this study. This is underlined by the finding that no longitudinal changes were observed in other trials in which conflict processing and set-shifting did not coincide.
A disease progression biomarker should be sensitive enough to vary with disease progression in pre-HD subjects, should not vary in controls, and should correlate with clinically important parameters2,11. The measures presented here fulfil all these requirements. In both longitudinal periods, disease progression, as tracked by behavioural and neurophysiological parameters, reveal substantial correlations with clinically relevant parameters such as “disease burden score” (DBS), also called the “toxic load,” and with the probability of disease manifestation in the next 5 years. Systematic changes were not detected at the longitudinal endpoints in controls, but were detected in pre-HD subjects and showed considerable sensitivity as indicated by the Cohen's d effect sizes in pre-HD subjects over a 6-month period of premanifest disease progression. As shown in Figure 5A and 5B, the effect sizes as estimated by Cohen's d for the behavioural and neurophysiological parameters in the current study were considerably greater than the effect sizes obtained from standard neuropsychological tests. However, when we used neuropsychological tests similar to those used in the TRACK-HD study21 and looked at effect sizes obtained from structural MRI data, the effect sizes were similar to those obtained in the current study. Compared to these measures, effect sizes obtained from the time-frequency decomposed neurophysiological data were higher. This is likely due to the fact that the way the test was applied allowed us to conduct a series of trials, and this considerably increased the reliability of the behavioural and neurophysiological measures. The measures we used have been shown to be sensitive to disease-modifying therapy in Parkinson's disease29 and may also be sensitive enough to monitor the effects of a potential disease-modifying therapy for HD. In contrast to the TRACK-HD study, the current study was not a multi-centre study, which is clearly a limitation. Further studies are needed to evaluate whether the parameters identified here are suitable for use in larger multi-centre studies and can be used as outcome parameters in clinical trials that assess potential neuroprotective treatments for HD.
In summary, this study showed that behavioural and neurophysiological measures of cognitive response selection processes are sensitive enough to detect changes in premanifest disease progression over a short 6-month period. The effect sizes in pre-HD subjects, correlations with clinically relevant parameters, and a lack of similar changes in control subjects suggest that the measures have potential as a novel cognitive-neurophysiological state biomarker and merit further evaluation in larger multi-centre studies.
Methods
Participants
At baseline, a group of 30 right-handed pre-HD subjects were enrolled in the study. After 15 months, three pre-HD subjects dropped out due to personal reasons. There were no further dropouts at 21 months. At each time point, all pre-HD subjects were scored according to the Unified Huntington's Disease Rating Scale (UHDRS) motor score (MS), total functional capacity scale (TFC), and independence scale (IS). Each HD subject completed the verbal fluency test, symbol digit test, and stroop colour naming, stroop word reading, and stroop interference tests; these were summarised in a single cognitive score (CS)30. The pre-HD subjects also completed several motor tests. A rating of “absence of clinical motor symptoms” was based on experts' assessments of motor signs with the finding that the motor signs were not sufficient for a diagnosis of HD (Diagnostic Confidence Level < 4)30. For each pre-HD participant, the probability of estimated disease onset (eAO) within five years was calculated according to Langbehn's parametric model20. In addition, we calculated the disease burden score, or DBS, for each subject [(CAG repeat - 35.5 × age)3. MRI was performed to assess caudate size.
The study also included 30 right-handed, healthy control subjects that were matched to the pre-HD group in terms of age, sex, educational status, and socio-economic background. All participants gave written informed consent before the study began. The control subjects were examined at all three time points (baseline, 15 months, and 21 months). The Ethics Committee at the Ruhr-University Bochum (Germany) approved the study. Detailed sample characteristics are shown in the supplementary information (Table 2) for 27 pre-HD subjects completing all time points. Statistical analysis was carried out on the basis of these 27 pre-HD subjects. Clinical values reported for the pre-HD subjects were in the normal range and are comparable to the clinical characteristics of pre-HD subjects in other studies.
Table 2. Baseline group statistics of premanifest HD mutation carriers at baseline, 15 and 21 month follow up and controls.
| Age [yr] | 39.3 ± 10.6 (23–63) | 40.6 ± 10.7 (24–64) | 41.1 ± 10.5 (25–64) |
|---|---|---|---|
| Gender (male/female) | 12/15 | 12/15 | 12/15 |
| CAG expanded | 42.1 ± 2.2 (38–48) | 42.1 ± 2.2 (38–48) | 42.1 ± 2.2 (38–48) |
| Disease burden score | 250.1 ± 79.3 (108.5–412.5) | 258.3 ± 81.9 (115.5–427.5) | 262.1 ± 82.4 (115.5–427.5) |
| 5 year OP [%] | 17.2 ± 17.4 (0.6–60.1) | 16.9 ± 16.7 (0.7–52.9)# | 18 ± 17.3 (0.7–52.9)# |
| YTO Langbehn [yr] | 16.1 ± 8.7 (5.3–38.4) | 16.4 ± 8.9 (5.9–36.4) # | 16 ± 8.8 (5.9–36.4) # |
| YTO Ranen [yr] | 6.6 ± 8 (−12.5–19.8) | 6.8 ± 6.6 (−7.7–17.8) # | 6.2 ± 6.5 (−7.7–17.8)# |
| UHDRS MS | 2.9 ± 2.6 (0–8) | 4.4 ± 4.8 (0–15) | 4.4 ± 4.7 (0–17) |
| UHDRS TFC | 12.9 ± 0.2 (12–13) | 12.8 ± 0.3 (12–13) | 12.8 ± 0.4 (12–13) |
| UHDRS IS | 99.4 ± 1.6 (95–100) | 98.7 ± 3.6 (85–100) | 98.7 ± 3.3 (90–100) |
| UHDRS CS | 332.4 ± 41.4 (238–423) | 329.7 ± 44 (234–425) | 342 ± 41.7 (242–433) |
| Verbal fluency | 42.4 ± 10.9 (17–64) | 41 ± 10.4 (21–64) | 43.9 ± 14.8 (15–83) |
| SDMT | 54.3 ± 9.9 (36–78) | 53.4 ± 10.6 (36–76) | 56.1 ± 10.5 (37–74) |
| Stroop interference | 51.4 ± 8.9 (31–66) | 50.4 ± 9.1 (31–78) | 51 ± 9.2 (33–77) |
| Stroopcolor | 77.5 ± 10.5 (59–101) | 79.9 ± 11.8 (61–107) | 81.7 ± 10.3 (58–104) |
| Stroop word | 106.8 ± 14.2 (81–136) | 105.1 ± 15.6 (76–137) | 109.1 ± 13 (81–138) |
| Pegboard dominant | 42.8 ±9.7 (32–56) % | 43.7 ± 4.7 (33.7–52.6)% | 42.4 ± 4.7 (33.8–52.8)% |
| Pegboard non dominant | 45.8 ± 11.3 (33.4–63.5)% | 46.8 ± 6.2 (32.7–56.3)% | 45.2 ± 6.4 (35.9–59.1)% |
| Tapping dominant37,38 | 195.2 ± 21.6 (143–226) | 188.9 ± 24.2 (140–230) | 192.1 ± 22.8 (147–238) |
| Tapping non dominant | 176.4 ± 24.1 (127–239) | 170.6 ± 23.7 (128–221) | 175 ± 23.2 (122–224) |
| Hamilton Depression Inventory | 6.6 ± 6.2 (0–22) | 6.4 ± 8.1 (0–28) | 6.2 ± 5.5 (0–22) |
| Caudatum volume [ml] | 3.02 ± 0.76 (1.3–4.15) | 2.92 ± 0.75 (1.31 – 4.04)+ | 2.84 ± 0.75 (1.16–4.14)+ |
Data of matched controls are only shown at baseline. Of the 30 participants enrolled at baseline, 27 (90%) completed the 15 month and 27 (90%) completed the 21 month follow-up assessment. We included 27 participants who contributed to all three datasets in the statistical analysis. Values are given as mean ± SD; range (min-max) in brackets; $ = including two mutation carriers with a pheno-conversion to manifest HD, # = two manifest HD patients excluded, % = complete datasets for 26 participants; + = complete datasets for 25 participants (Cohen’s d effect size for absolute MRI measures in ml: 15 month interval 0.14, for 6 month interval 0.11).
Abbreviations: yr – years, OP – onset probability, YTO – years to onset, UHDRS – unified Huntington's disease rating scale, MS – motor score TFC – total functional capacity, IS – independence scale, CS – cognitive sum score; SDMT – symbol digit modality test.
Task
The task is identical to the task described in Beste et al.19 presenting the baseline data. The outline of the paradigm is depicted below (Figure 6).
Briefly, the task combines a stroop paradigm with a switching paradigm. The stimuli were four colour words (i.e., RED, BLUE, YELLOW and GREEN) presented at the centre of the screen. These colour words are presented either in a rhomb or in a square. These shapes serve as cue stimuli denoting the task rule. Cue and target stimulus are separated by a short delay of 150 ms. When a rhomb is presented, subjects are instructed to respond according to the ‘colour rule’; when a square is presented the subjects respond according to the ‘word rule’. The subjects respond using their index fingers to BLUE (left key press) and YELLOW (right key press). The middle fingers are used to respond to the RED (left middle finger) and GREEN colour (right middle finger). For the ‘colour rule’ the subjects respond according to the print-colour of the word and ignore the meaning of the word (e.g. BLUE printed in green, subjects respond with the left index finger). For the ‘word rule’ subjects respond according to the meaning of the word and ignore the print-colour of the word. In the following sections, colour rule trials and word rule trials are referred to as ‘incompatible’ and ‘compatible’ trials respectively. The paradigm contains four different trial types: (i) non-switch, compatible [i.e., on two consecutive trials the font colour of the word corresponds to its meaning]; (ii) switch, compatible [i.e., on two consecutive trials the rule changes, with the font colour of the word corresponding to its meaning]; (iii) non-switch, incompatible trials [i.e., on two consecutive the font colour of the word does not correspond to its meaning]; and (iv) switch, incompatible trials [i.e., on two consecutive trials the rule changes and the font colour of the word does not correspond to its meaning]. The latter condition is the most difficult condition, since conflict monitoring and switching processes are required in parallel.
EEG recording and analysis
In both experiments conducted, the EEG was recorded from 65 Ag–AgCl electrodes at standard scalp positions against a reference electrode located at Cz. The sampling rate was 500 Hz. All electrode impedances were kept below 5 kΩ. Data processing involved a manual inspection of the data to remove technical artefacts. After manual inspection, a band-pass filter ranging from 0.5 to 20 Hz (48 db/oct) was applied. After filtering, the raw data were inspected a second time. To correct for periodically recurring artefacts (pulse artefacts, horizontal and vertical eye movements) an independent component analysis (ICA; Infomax algorithm) was applied to the un-epoched data set. Afterwards, the EEG data was segmented according to the four different conditions. Segmentation was applied with respect to the occurrence of the stimuli (i.e., stimulus-locked). Automated artefact rejection procedures were applied after epoching: rejection criteria included a maximum voltage step of more than 50 μV/ms, a maximal value difference of 200 μV in a 200 ms interval or activity below 0.1 μV. Then the data was CSD-transformed (current source density transformation31) in order to eliminate the reference potential from the data. Moreoever, the CSD-transformation serves as a spatial filter32. As a consequence activity is confined to only a few electrodes, and analysis of amplitudes and latencies can be confined to these electrodes. After the CSD-transformation, data were corrected relative to a baseline extending from 200 ms before stimulus presentation until stimulus onset and averaged. The data pre-processing is identical to the study by Beste et al.19. The N2 at electrode FCz and the P3 at electrode Pz were quantified in the longitudinal data, since these electrodes revealed strongest effects in the baseline data19. The scalp topography plots for the other longitudinal endpoints suggest that these electrode positions were still the most relevant ones. Baseline correction was performed in the time interval between −200 ms until stimulus presentation. The N2 was quantified relative to the pre-stimulus baseline and defined as the most negative peak occurring within the time interval of 250 till 320 ms. The P3 was defined as the most positive peak within a time range from 350 to 500 ms. Both components were quantified in amplitude and latency on single subject level. The whole procedure is comparable to the analysis of the baseline data19.
Time-frequency decomposition (TF-decomposition)
Time-frequency (TF) analysis of stimulus-related potentials was performed by means of a continuous wavelet transform (CWT), applying Morlet wavelets (w) in the time domain to different frequencies (f):
t is time, 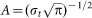 ,
, 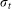 is the wavelet duration, and
is the wavelet duration, and 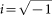 . For analysis and TF-plots, a ratio of f0/σf = 5.5 was used, where f0 is the central frequency and σf is the width of the Gaussian shape in the frequency domain. The analysis was performed in the frequency range 0.5–20 Hz with a central frequency at 0.5 Hz intervals. The ‘evoked wavelet power’ was calculated, which refers to event-related changes in EEG power that are phase-locked with respect to the event onset across trials33. The segments used for the wavelet analysis were 4000 ms long; starting 2000 ms before stimulus onset and ending 2000 ms after stimulus onset. This epoch length was chosen to allow a reliable estimation of the evoked power of low frequent oscillations34,35. Maximal TF power and corresponding peak power latencies were measured in time intervals used for ERP quantification and was quantified in the delta and theta frequency band. Their central frequencies were 3 and 5 Hz. We used these a-priori defined frequencies in the analyses of the longitudinal time points to have the same data compared between pre-HD and controls, as for the baseline data already presented19. The delta and the theta frequency band were analyzed in separate ANOVAs. Evoked power was quantified at the same electrode positions, as the ERP data. A time window from 600 to 800 ms prior to the response was used to estimate background noise. Wavelet power in the time range of interest was measured normalized to wavelet power at this baseline. Data quantification was performed on single subject level. TF power was log10-transformed to normalize the distributions for statistical analyses. The whole procedure is comparable to the analysis of the baseline data19.
. For analysis and TF-plots, a ratio of f0/σf = 5.5 was used, where f0 is the central frequency and σf is the width of the Gaussian shape in the frequency domain. The analysis was performed in the frequency range 0.5–20 Hz with a central frequency at 0.5 Hz intervals. The ‘evoked wavelet power’ was calculated, which refers to event-related changes in EEG power that are phase-locked with respect to the event onset across trials33. The segments used for the wavelet analysis were 4000 ms long; starting 2000 ms before stimulus onset and ending 2000 ms after stimulus onset. This epoch length was chosen to allow a reliable estimation of the evoked power of low frequent oscillations34,35. Maximal TF power and corresponding peak power latencies were measured in time intervals used for ERP quantification and was quantified in the delta and theta frequency band. Their central frequencies were 3 and 5 Hz. We used these a-priori defined frequencies in the analyses of the longitudinal time points to have the same data compared between pre-HD and controls, as for the baseline data already presented19. The delta and the theta frequency band were analyzed in separate ANOVAs. Evoked power was quantified at the same electrode positions, as the ERP data. A time window from 600 to 800 ms prior to the response was used to estimate background noise. Wavelet power in the time range of interest was measured normalized to wavelet power at this baseline. Data quantification was performed on single subject level. TF power was log10-transformed to normalize the distributions for statistical analyses. The whole procedure is comparable to the analysis of the baseline data19.
MRI scanning and analyses
At each time point (baseline, 15 months, 21 months) structural MRI scanning was conducted to assess caudate head volume. Scanning data was available for N = 25 of the 27 pre-HD participants completing all three visits (2 pre-HDs had to be excluded due to claustrophobia). MR-imaging was performed on a 1.5 T scanner (Magnetom SymphonyTM, Siemens, Erlangen, Germany) using a standard head coil and a Turbo FLASH 3D sequence with the following parameters: TE (echo time): 3.93 ms, TR (repetition time): 1900 ms, TI (inversion time): 1100 ms, FA: 15°, NA: 1, resolution: 1 mm × 1 mm, 128 sagittal slices, voxel-size in slice selected direction 1.0 mm. Subjects were positioned within the head coil using a standard procedure according to outer anatomical markers. Caudate volume was calculated using the manual tracing method described by Aylward et al.36. The whole procedure is comparable to the analysis of the baseline data19.
Statistics
The data were analysed by univariate or mixed ANOVA. The mixed measures ANOVA tests contained the factors “trial type” (switch vs. non-switch), “context” (compatible vs. incompatible), and "time point" (baseline, 15 months, and 21 months) as the within-subject factors. The factor “group” (pre-HD vs. controls) was used as the between-subject factor. The degrees of freedom were adjusted using the Greenhouse-Geisser correction for all ANOVAs and for all experiments. All post-hoc tests were adjusted using the Bonferroni correction as necessary. The standard error of the mean (SEM) is reported as a measure of variability. Data were normally distributed in all of the tests in this study and at all time points as indicated by the results of the Kolmogorov-Smirnov Tests (all z < 0.5; P > 0.4; one-tailed).
Author Contributions
C.B. and C.S. conceived and designed the study. A.K.S., V.S., R.H., C.L. performed data collection, C.B. performed data analysis, C.B. and C.S. wrote the manuscript and compiled the figures.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by Grant from the Deutsche Forschungsgemeinschaft BE4045/10-1 and a Grant by the CHDI Foundation to C.B.
References
- Ross C. A. & Tabrizi S. J. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–88 (2011). [DOI] [PubMed] [Google Scholar]
- Weir D. W., Sturrock A. & Leavitt B. R., Development of biomarkers for Huntington's disease. Lancet Neurol. 10, 573–590 (2011). [DOI] [PubMed] [Google Scholar]
- Tabrizi S. J. et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 8, 791–801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J. S. et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J. Neurol. Neurosurg. Psychiatry 79, 874–880 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M. et al. Observing Huntington's disease: the European Huntington's disease Networks REGISTRY. J. Neurol. Neurosurg. Psychiatry 82, 1409–1412 (2011). [DOI] [PubMed] [Google Scholar]
- Tabrizi S. J. et al. Potential endpoints for clinical trails in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 11, 42–53 (2012). [DOI] [PubMed] [Google Scholar]
- O'Keefe G. C., Michell A. W. & Marker R. A. Biomarkers in Huntington's and Parkinson's disease. Ann. N. Y. Acad. Sci. 1180, 97–110 (2009). [DOI] [PubMed] [Google Scholar]
- Beste C. et al. Levels of error processing in Huntington' disease: a combined study using event-related potentials and voxel-based morphometry. Hum. Brain Mapp. 29, 121–130 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste C. et al. Functional compensation or pathology in cortico-subcortical interactions in preclinical Huntington's disease. Neuropsychologia 45, 2922–2930 (2007). [DOI] [PubMed] [Google Scholar]
- Nguyen L., Bradshaw J. L., Stout J. C., Croft R. J. & Georgiou-Karistianis N. Electrophysiological measures as potential biomarkers in Huntington's disease: review and future directions. Brain. Res. Rev. 64, 177–194 (2010). [DOI] [PubMed] [Google Scholar]
- Henley S. M., Bates G. P. & Tabrizi S. J. Biomarkers for neurodegenerative diseases. Curr. Opin. Neurol. 18, 698–705 (2005). [DOI] [PubMed] [Google Scholar]
- Aron A. R. et al. Taskset switching deficits in early-stage Huntington's disease: implications for basal ganglia function. J. Cogn. Neurosci. 15, 629–642 (2003). [DOI] [PubMed] [Google Scholar]
- Kehagia A. A., Cools R., Barker R. & Robbins T. W. Switching between abstract rules reflects disease severity but not dopaminergic status in Parkinson's disease. Neuropsychologia 47, 1117–1127 (2009). [DOI] [PubMed] [Google Scholar]
- Redgrave P. et al. Goal-directed and habitual control in the basal ganglia: implications for Parkinson's disease. Nat. Rev. Neurosci. 11, 760–772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenz D. When inhibition goes incognito: feedback interaction between spiny projection neurons in striatal function. Trends Neurosci. 26, 436–443 (2003). [DOI] [PubMed] [Google Scholar]
- Thomas E. A. et al. In vivo cell-autonomous transcriptional abnormalities revealed in mice expressing mutant huntingtin in striatal but not cortical neurons. Hum. Mol. Genet. 20, 1049–1060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C., Wu N., Andre V. M., Cummings D. M. & Levine M. S. The corticostriatal pathway in Huntington's disease. Prog. Neurobiol. 81, 253–271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botvinick M. M., Cohen J. D. & Carter C. S. Conflict monitoring and anterior cingulate cortex: an update. Trends Cogn. Sci. 8, 539–546 (2004). [DOI] [PubMed] [Google Scholar]
- Beste C. et al. Mechanisms mediating parallel action monitoring in fronto-striatal circuits. Neuroimage 62, 137–146 (2012). [DOI] [PubMed] [Google Scholar]
- Langbehn D. R., Brinkman R. R., Falush D., Paulsen J. S. & Hayden M. R. International Huntington's disease collaborative group. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin. Genet. 65, 267–277. [DOI] [PubMed] [Google Scholar]
- Stout J. C. et al. Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington's disease. J. Neurol. Neurosurg. Psychiatry. 83, 687–694 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campodonico J. R., Codori A. M. & Brandt J. Neuropsychological stability over two years in asymptomatic carriers of the Huntington's disease mutation. J. Neurol. Neurosurg. Psychiatry 61, 621–624 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjes-Ane M. N. et al. Longitudinal evaluation of presymptomatic carriers of Huntington's disease. J. Neuropsychiatry Clin. Neurosci. 19, 310–317 (2007). [DOI] [PubMed] [Google Scholar]
- Gajewski P. D., Kleinsorge T. & Falkenstein M. Electrophysiological correlates of residual switch costs. Cortex 46, 1138–1148 (2010). [DOI] [PubMed] [Google Scholar]
- Folstein J. R. & Van Petten C. Influence of cognitive control and mismatch on the N2 component of the ERP: a review. Psychophysiology 45, 152–170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr U. Age differences in the selection of mental sets: the role of inhibition, stimulus ambiguity, and response-set overlap. Psychol. Aging 16, 96–109 (2000). [DOI] [PubMed] [Google Scholar]
- Karayanidis F., Coltheart M., Michie P. T. & Murphy K. Electrophysiological correlates of anticipatory and poststimulus components of task switching. Psychophysiology 40, 329–348 (2003). [DOI] [PubMed] [Google Scholar]
- Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol 118, 2128–2148 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemssen R., Falkenstein M., Schwarz M., Müller T. & Beste C. Effects of aging, Parkinson's disease, and dopaminergic medication on response selection and control. Neurobiol. Aging 32, 327–335 (2011). [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Unified Huntington's disease rating scale: reliability and consistency. Mov. Disord. 11, 136–142 (1996). [DOI] [PubMed] [Google Scholar]
- Perrin F., Pernier J., Bertrand O. & Echallier J. F. Spherical splines for scalp potential and current density mapping. Electroencephalogr. Clin. Neurophysiol. 72, 184–187 (1989). [DOI] [PubMed] [Google Scholar]
- Nunez P. L. & Pilgreen K. L. The spline-laplacian in clinical neurophysiology: a method to improve EEG spatial resolution. J. Clin. Neurophysiol. 8, 397–413. [PubMed] [Google Scholar]
- Roach B. J. & Mathalon D. H. Event-related EEG time-frequency analysis: an overview of measures and an analysis of early gamma band phase locking in schizophrenia. Schizophr. Bull. 34, 907–926 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocklenburg S., Güntürkün O. & Beste C. Lateralized neural mechanisms underlying the modulation of response inhibition processes. Neuroimage 55, 1771–1778 (2011). [DOI] [PubMed] [Google Scholar]
- Beste C. et al. Functional 5-HT1a receptor polymorphism selectively modulates error-specific subprocesses of performance monitoring. Hum. Brain Mapp. 31, 621–630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward E. H. et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 63, 66–72 (2004). [DOI] [PubMed] [Google Scholar]
- Saft C., Andrich J., Meisel N. M., Przuntek H. & Müller T. Congruent deterioration of complex and simple movements in patients with Huntington's disease. J. Neural. Transm. Suppl. 68, 97–104 (2004). [DOI] [PubMed] [Google Scholar]
- Andrich J., Saft C., Ostholt N. & Müller T. Assessment of simple movements and progression of Huntington's disease. J. Neurol. Neurosurg. Psychiatry. 78, 405–407 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information