

Abstract
Cancer-associated inflammation plays an important role in restraining anti-tumor immunity, particularly in pancreatic ductal adenocarcinoma (PDA) for which a massive infiltration of immunosuppressive leukocytes into the tumor stroma is an early and consistent event in oncogenesis. This pathophysiology is in contrast to many other solid tumors for which infiltration of effector T cells is often prominent, associated with improved clinical outcomes, and mechanistically contributes to tumor immunoediting that ultimately can mediate immune escape. In PDA, increasing evidence suggests that the ras oncogene drives an inflammatory program that establishes immune privilege in the tumor microenvironment. Indeed, PDA cells might remain intrinsically sensitive to T cell killing because they have never been exposed to T cell selective pressure in vivo. In support of this hypothesis, recent studies demonstrate that derailing immune suppressive pathways in the PDA microenvironment, such as tumor derived GM-CSF, facilitates T-cell mediated tumor rejection. These findings carry major implications for the development of novel, combination immunotherapies for pancreatic cancer.
Introduction
Leukocytes can be a major part of the tumor microenvironment, with evidence supporting both tumor-suppressing and tumor-promoting roles [1]. The host-protective and the tumor-sculpting actions of the immune system on a developing tumor are embodied in the prevailing model of cancer immunosurveillance, recently understood to occur in three phases – elimination, equilibrium, and escape [2,3]. In the elimination phase, the immune system acts as a powerful extrinsic tumor suppressor, and recent elegant studies underscore the large extent to which this process is T-cell dependent. [4–6]. In cases where the elimination phase fails to rid the host completely of transformed cells, a dynamic equilibrium ensues between residual malignant cells and the host immune system [7]. This phase is marked by genetic instability in the cancer cells, immune pressure, Darwinian selection, and in some model systems the outgrowth of tumor cells with reduced immunogenicity [2,3]. The selection of tumor cells with reduced immunogenicity (“immunoediting”) renders them more capable of surviving in an immunocompetent host. In the escape phase, immunoedited tumor cells can grow unrestrained by immune pressure and manifest as invasive tumors. In other model systems, active immune suppression mediated by the tumor enforces tolerance in tumor-specific T cells as a dominant mechanism of immune escape [8]. In so far as immunoediting has been documented in patients with cancer [9,10], a major implication of the “triple E” theory is that clinically apparent tumors in humans have escaped adaptive immunity and may therefore be inherently resistant to immune pressure. This may help explain why most therapeutic cancer vaccines and other immunotherapies have been ineffective, or only effective in a small fraction of patients, even in diseases such as melanoma which are considered to be immunogenic.
It is increasingly appreciated that tumor-associated leukocytes can promote tumor development [11–13]. Neoplastic cells are often capable of driving inflammatory pathways that recruit leukocytes to the tumor microenvironment. While the mechanisms underlying tumor-induced inflammatory responses remain incompletely understood, innate immune cells of the myeloid lineage such as tumor-associated macrophages (TAMs) and immature myeloid cells, are integrally involved [14]. These cell populations can promote tumor growth through the production of cytokines, chemokines, proteases, and growth factors; foster angiogenesis and tissue remodeling; and mediate local and systemic immune suppression [12].
Immunobiology of pancreatic cancer
Determining the balance between tumor-promoting innate immune responses and tumor-suppressing adaptive immune responses is critical both to understanding the pathophysiology of cancer and to developing new immunotherapies. A number of laboratories have investigated immunosurveillance and inflammation in the context of pancreatic ductal adenocarcinoma (PDA) [15–22]. PDA is a devastating tumor, highly refractory to standard therapies and almost universally lethal [23], owing in large part to a striking desmoplastic reaction and fibrosis in the stromal compartment [24]. PDA is one of only two cancers among 21 identified in the 2012 AACR Cancer Progress Report for which the death rate in the United States has actually risen since 1990 (the other is liver/intrahepatic bile duct cancer). Indeed, a recent report from the Pancreatic Cancer Action Network estimates that by 2020; PDA will become the second leading cause of cancer-related death in the US (behind lung cancer) [25]. For patients with metastatic PDA, only one new drug has been approved by the FDA in the last decade, and despite recent advances with combination chemotherapy [26,27], PDA remains a significant medical problem that requires an innovative, aggressive, and novel approach.
To understand the immune reaction to PDA, we and others have evaluated genetically defined mouse models of PDA [15–20,28]. In one model, mutations in Kras and p53 alleles are targeted to the pancreas via a Cre-lox approach [29]. These “KPC” animals develop PDA with reproducible kinetics and with features that closely resemble the human disease (histologically and molecularly), including progression from preinvasive pancreatic intraepithelial neoplasia (PanIN) to invasive and metastatic PDA (Figure 1). Furthermore, the dense desmoplasia and leukocytic infiltration classically observed in the tumor stroma of patients with PDA is reproduced with high fidelity in the KPC murine model [16,29,30]. Immunosuppressive cells, including TAMs, Gr-1+ CD11b+ myeloid cells, and regulatory T cells (Treg), are prominent at even the earliest stages of neoplasia and persist through invasive cancer [16,17]. Effector T cells are scarce in preinvasive and invasive lesions, and most T cells show a naïve phenotype without evidence of activation. In some model systems, tumor specific (non-Treg) T cells have been noted, but these are typically dysfunctional [28,31]. These findings are consistent with the immune infiltrate observed in human PDA which involves a marked macrophage and myeloid cell infiltration associated with few effector T cells (unless there is underlying chronic pancreatitis) [21,32,33]. Identifiable T cells are skewed toward a Th2 phenotype [32]. Autoantibody production to pancreatic tumor cells is weak relative to antibody responses seen in other tumors [34]. Thus, suppressive cells of the immune system appear early during pancreatic tumorigenesis – preceding and outweighing antitumor cellular immunity. The absence of effector T cell surveillance in PDA may indicate that immunoediting is not a major outcome of immune surveillance in this cancer [16]. Rather, we have hypothesized that T cell evasion in PDA manifests in in four stages (the “four I” hypothesis): induction of cancer by an oncogene (i.e. mutant ras), inflammation linked to downstream pathways of the oncogene, immune suppression imposed early and persistently by the inflammatory reaction, and ultimately, immune privilege [16]. Potential mechanisms driving this pathophysiology range from immunological ignorance to defects in immune effector function, both of which might contribute to the failure of immune surveillance in pancreatic cancer. This scenario of tumor-induced immune privilege, if true, creates an apparent paradox as well as an opportunity for intervention: PDA cells intrinsically might be sensitive to T cell killing because they have never been exposed to T cell selective pressure in vivo (Figure 2).
Figure 1.

The PDA microenvironment is characterized by a dense desmoplastic reaction and prominent infiltration of leukocytes. (A) H&E stain and (B) immunohistochemistry for CD45 of a representative primary PDA lesion from a tumor-bearing KPC mouse. Scale bars, 100 μM.
Figure 2.
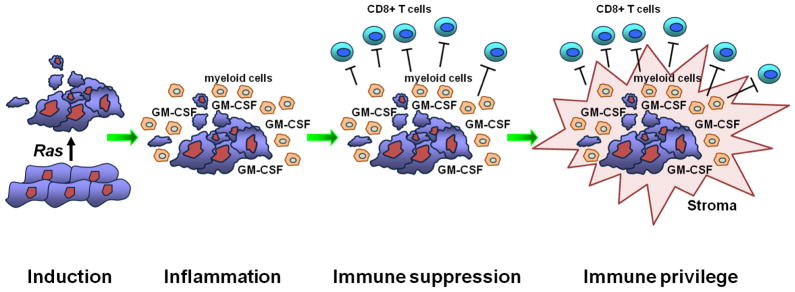
In pancreatic ductal adenocarcinoma, the ras oncogene induces an inflammatory program that establishes immune suppression and ultimately immune privilege in the tumor microenvironment. One critical mediator is tumor-derived GM-CSF which drives the accumulation of immature myeloid cells that then directly suppress infiltrating T cells.
Impact of Gr-1+ CD11b+ cells on T cell responses in PDA
To understand the potential potency of T cell responses against PDA, immune reactions have been monitored in tumor-bearing KPC mice in which key microenvironmental regulators of immune suppression have been abrogated genetically or pharmacologically [18,19,30,35]. Targets evaluated include TAMs, Tregs, B cells, and mesenchymal cells, including cells expressing fibroblast activation protein. In recent work, we [18] and others [19] have also tested the immunological consequence of blocking the in vivo recruitment of Gr-1+ CD11b+ immature myeloid cells. These cells infiltrate both PanIN and PDA lesions in KPC mice, as well as metastatic foci. Cells of this phenotype have been well-recognized in multiple other tumor models (with an equivalent myeloid cell also being appreciated in cancer patients, including those with PDA) [36–38]. In mice, these immature myeloid cells, often termed myeloid-derived suppressor cells (MDSCs), are a heterogeneous population of cells that co-express CD11b and the myeloid-cell lineage differentiation antigen Gr-1, and represent precursors to macrophages, dendritic cells, and granulocytes [36–38]. Numerous studies have reported the expansion of MDSCs in a variety of tumor models and have demonstrated the ability of these cells to impair T cell responses in vitro in a cell contact-dependent manner [39–41].
In the KPC and other mouse models of PDA, Gr-1+ CD11b+ cells potently suppress both antigen-specific and polyclonal T cell responses in vitro (consistent with the nomenclature MDSCs) [15,17,18]. In vivo, there is an interesting mutual exclusion observed between Gr-1+ CD11b+ cells and CD8+ T cells infiltrating PDA tumors [17]. Among the many cytokines and chemokines produced by PDA tumors, GM-CSF appears both necessary and sufficient for the generation of functional MDSCs from c-kit+ Gr-1neg CD11bneg precursors in tumor-bearing mice [18]. In vivo, abrogation of GM-CSF in KPC PDA tumor cell lines with either neutralizing antibodies or shRNA inhibits the recruitment of Gr-1+ CD11b+ cells to the tumor microenvironment and blocks tumor development. Experimental depletion of CD8+ T cells fully rescues tumor growth, even though Gr-1+ CD11b+ cell infiltration is not completely restored [18]. Likewise, GM-CSF knockdown PDA cells grow progressively and establish tumors in Rag2 deficient or perforin-deficient mice (Bayne and Vonderheide, unpublished). CD8+ T cell-dependent rejection in vivo also occurs for pancreatic epithelial lines harboring a ras mutation and injected orthotopically, provided GM-CSF is abrogated [19]. In patients, based on immunohistochemistry, more than 90% of both primary PDA and PanIN samples prominently express GM-CSF, suggesting that GM-CSF-mediated recruitment of immature myeloid cells is an early event in the clinical disease [18,19]. Interestingly, expression of GM-CSF has been linked to activity of mutant ras [19,42]. Thus, suppressive cells of the host immune system appear early during pancreatic tumorigenesis and effectively shield developing tumors from immune pressure, thereby preserving the underlying susceptibility of PDA cells to T cell attack (Figure 2). Elaboration of a productive anti-tumor T cell response, therefore, requires uncoupling of the ras-driven pathway of immunosuppressive myeloid inflammation.
Interestingly, modulation of the PDA tumor microenvironment can have other consequences on immune surveillance that are independent of T cells. Although CD40 agonistic antibodies have been shown to drive anti-tumor T cell responses in vivo (a notion being aggressively tested in patients) [43,44], we found that in tumor-bearing KPC mice, agonistic CD40 mAb re-educates macrophages in the tumor microenvironment, transforming immunosuppressive macrophages into tumoricidal macrophages that facilitate the depletion of tumor stroma [30]. Similar tumor regressions are observed equally in T cell replete and T cell depleted mice. In the clinic, gemcitabine and the agonistic CD40 mAb CP-870,893 produces objective responses in 24% of patients with metastatic PDA [30]. Thus, productive immune surveillance of PDA is not necessarily T cell dependent. CD40-activation of macrophages appears to be a major mechanism of tumor rejection with implications beyond PDA immunobiology [30,45,46].
The good and bad of GM-CSF
Further experiments are underway to understand the function of tumor-derived GM-CSF as a potential mechanism of immune suppression in PDA, i.e. that GM-CSF drives an early accumulation of MDSCs even around PanIN lesions and thereby blocks tumor-specific recognition by T cells. Immunosuppressive effects of tumor-derived GM-CSF have been well appreciated in other model systems [47,48], but it should be further emphasized that GM-CSF is neither the only tumor-derived cytokine implicated in MDSC biology [49–54] nor necessary in all tumor models in which MDSC accumulate [55]. Ironically, GM-CSF is also well-described as a key immunological activator (rather than suppressor) when tumor cells are transduced to express GM-CSF, irradiated and injected subcutaneously as therapy in tumor-bearing mice or humans. Moreover, the potency of such vaccines (often called “G-VAX”) has been clearly demonstrated in patients with PDA [56]. The determinants driving immune suppression in the PDA microenvironment on one hand vs. immune activation via G-VAX therapy on the other remain to be fully understood, but are likely linked to differences in GM-CSF dose and location, as well as the immunogenicity of the tumor antigen released. In any event, it is hypothesized that G-VAX vaccines in patients with PDA will require concomitant therapeutic down-regulation of the relevant immunosuppressive factors in the tumor microenvironment to achieve full potency and ultimate clinical success.
Conclusions
PDA is a highly inflammatory and lethal tumor for which infiltration of immune suppressive myeloid and other cells is an early event. In genetically engineered mouse models, myeloid inflammation occurs concomitantly with neoplastic induction, and T cell infiltration is scarce throughout the natural history of pancreatic cancer. These results raise the hypothesis that the consequence of immune surveillance in pancreatic cancer is more akin to immune privilege than immunoediting, although further studies are needed to understand mechanism. Pancreatic tumor cells therefore might be paradoxically sensitive to T cell attack, and an emerging body of experimental evidence suggests this is the case. Clinically, it will be important in pancreatic cancer to test novel cancer immunotherapies in combination with agents that inhibit or alter oncogene-driven immunosuppressive mechanisms in the tumor microenvironment.
Highlights for Vonderheide and Bayne submission.
Immune suppressive leukocytes are prominent in pancreatic cancer
T cell infiltration is rare throughout disease progression
Pancreatic tumors may therefore remain intrinsically sensitive to T cell killing
Blockade of tumor GM-CSF blocks myeloid inflammation and mediates T cell immunity
Combination immunotherapies in pancreatic cancer are warranted
Acknowledgments
We thank Drs. Ben Stanger and Gregory Beatty for helpful discussions. This work was supported by NIH grant R01 CA169123.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
** of outstanding interest
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- **2.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. Outstanding analysis of the prevailing “triple E” hypothesis of cancer immunosurveillance. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- *4.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. Results highlight the importance of tumor-specific-antigen expression in immune surveillance, and potentially, immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *5.Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, Wang Z, Derudder E, Li S, Chakraborty T, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell. 2012;148:739–751. doi: 10.1016/j.cell.2011.12.031. T cells function as potent extrinic immune suppressors in hematological malignancies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *6.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. T cells function as potent extrinic immune suppressors in solid tumors. Mutation in tumor associated antigens drives immune escape. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–907. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 8.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 9.Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, Kenyon K, Davis MM, Riddell SR, Greenberg PD. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of T cell-mediated vitiligo. J Exp Med. 2000;192:1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sterman DH, Gillespie CT, Carroll RG, Coughlin CM, Lord EM, Sun J, Haas A, Recio A, Kaiser LR, Coukos G, et al. Interferon beta adenoviral gene therapy in a patient with ovarian cancer. Nat Clin Pract Oncol. 2006;3:633–639. doi: 10.1038/ncponc0658. [DOI] [PubMed] [Google Scholar]
- 11.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantovani A, Garlanda C, Allavena P. Molecular pathways and targets in cancer-related inflammation. Ann Med. 2010;42:161–170. doi: 10.3109/07853890903405753. [DOI] [PubMed] [Google Scholar]
- 14.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao F, Obermann S, von Wasielewski R, Haile L, Manns MP, Korangy F, Greten TF. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. 2009;128:141–149. doi: 10.1111/j.1365-2567.2009.03105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 17.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- **18.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophagecolony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;2021(21):822–835. doi: 10.1016/j.ccr.2012.04.025. Tumor-derived GM-CSF drives the accumulation of Gr-1+ CD11b+ myeloid cells as part of the cancer-associated inflammatory reaction in pancreatic cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **19.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. Mutational activation of Kras in pancreatic ductal cells triggers production of GM-CSF and restrains antitumor immune responses in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukherjee P, Basu GD, Tinder TL, Subramani DB, Bradley JM, Arefayene M, Skaar T, De Petris G. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J Immunol. 2009;182:216–224. [PMC free article] [PubMed] [Google Scholar]
- 21.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8 T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Transl Sci. 2008;1:228–239. doi: 10.1111/j.1752-8062.2008.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 24.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matrisian LM, Aizenberg R, Rosenzweig A. Newsletter of the Pancreatic Cancer Action Network. 2012. The alarming rise of pancreatic cancer deaths in the United States: Why we need to stem the tide today. [Google Scholar]
- 26.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 28.Garbe AI, Vermeer B, Gamrekelashvili J, von Wasielewski R, Greten FR, Westendorf AM, Buer J, Schmid RM, Manns MP, Korangy F, et al. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. 2006;66:508–516. doi: 10.1158/0008-5472.CAN-05-2383. [DOI] [PubMed] [Google Scholar]
- 29.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 30.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee P, Ginardi AR, Madsen CS, Tinder TL, Jacobs F, Parker J, Agrawal B, Longenecker BM, Gendler SJ. MUC1-specific CTLs are non-functional within a pancreatic tumor microenvironment. Glycoconj J. 2001;18:931–942. doi: 10.1023/a:1022260711583. [DOI] [PubMed] [Google Scholar]
- 32.Tassi E, Gavazzi F, Albarello L, Senyukov V, Longhi R, Dellabona P, Doglioni C, Braga M, Di Carlo V, Protti MP. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. 2008;181:6595–6603. doi: 10.4049/jimmunol.181.9.6595. [DOI] [PubMed] [Google Scholar]
- 33.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gnjatic S, Ritter E, Buchler MW, Giese NA, Brors B, Frei C, Murray A, Halama N, Zornig I, Chen YT, et al. Seromic profiling of ovarian and pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:5088–5093. doi: 10.1073/pnas.0914213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *35.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2011;330:827–830. doi: 10.1126/science.1195300. Fibroblast activation protein-expressing cells are a nonredundant, immune-suppressive component of the tumor microenvironment. [DOI] [PubMed] [Google Scholar]
- 36.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid- derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 39.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 40.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 41.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 42.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-12-2064. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13:1083–1088. doi: 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- 45.O’Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. 2012 doi: 10.1084/jem.20112738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rakhmilevich AL, Buhtoiarov IN, Malkovsky M, Sondel PM. CD40 ligation in vivo can induce T cell independent antitumor effects even against immunogenic tumors. Cancer Immunol Immunother. 2008;57:1151–1160. doi: 10.1007/s00262-007-0447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 48.Sotomayor EM, Fu YX, Lopez-Cepero M, Herbert L, Jimenez JJ, Albarracin C, Lopez DM. Role of tumor-derived cytokines on the immune system of mice bearing a mammary adenocarcinoma. II. Down-regulation of macrophage-mediated cytotoxicity by tumor-derived granulocyte-macrophage colony-stimulating factor. J Immunol. 1991;147:2816–2823. [PubMed] [Google Scholar]
- 49.Melani C, Chiodoni C, Forni G, Colombo MP. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145. doi: 10.1182/blood-2003-01-0190. [DOI] [PubMed] [Google Scholar]
- 50.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 51.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005;175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 53.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz- Oesterreicher M, Bjorkdahl O, Fox JG, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *54.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328:749–752. doi: 10.1126/science.1185837. CCL21-secreting tumors shift the host immune response from immunogenic to tolerogenic, which facilitates tumor progression. [DOI] [PubMed] [Google Scholar]
- 55.Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, Dranoff G. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest. 2011;121:2436–2446. doi: 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor- secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]