

Abstract
During an analysis of the virome of bats from Myanmar, a large number of reads were annotated to orthohepadnaviruses. We present the full genome sequence and a morphological analysis of an orthohepadnavirus circulating in bats. This virus is substantially different from currently known members of the genus Orthohepadnavirus and represents a new species.
Keywords: hepatitis, bats, bat hepatitis virus, full genome, viral morphology, prevalence, viruses, Myanmar
The family Hepadnaviridae comprises 2 genera (Orthohepadnavirus and Avihepadnavirus), and viruses classified within these genera have a narrow host range. The genus Orthohepadnavirus consists of pathogens that infect mammals, and it currently contains 4 species: hepatitis B virus, woodchuck hepatitis virus, ground squirrel hepatitis virus, and woolly monkey hepatitis B virus. The genus Avihepadnavirus contains 2 avian species: duck hepatitis B virus and heron hepatitis B virus (1). Hepadnaviruses mainly infect the liver cells of their hosts and, in humans, cause hepatitis B, cirrhosis, and hepatocellular carcinoma (2). Approximately 2 billion persons worldwide are infected with hepatitis B virus (HBV), and 600,000 persons die every year from the consequences of hepatitis B (3).
Bats are associated with an increasing number of emerging and reemerging viruses, many of which pose major threats to public health (4). We conducted a viral metagenomic analysis of 6 species of bats from Myanmar. The analysis revealed a large number of viral contigs annotated to orthohepadnavirus with <70% nt identity (B. He, unpub. data), suggesting the presence of orthohepadnaviruses in these animals. We describe the virus by full genomic analysis and morphologic observation.
The Study
We purchased 853 freshly killed insectivorous bats in Sedon and Wutao Counties in southeastern Kachin State, Myanmar; the counties are adjacent to Yunnan Province, People’s Republic of China. The bats covered 6 species: Miniopterus fuliginosus (n = 640), Hipposideros armiger (n = 8), Rhinolophus ferrumequinum (n = 176), Myotis chinensis (n = 11), Megaderma lyra (n = 6), and Hipposideros fulvus (n = 12). All bat tissue samples were subjected to viral metagenomic analysis (unpublished data). The sampling of bats for this study was approved by the Administrative Committee on Animal Welfare of the Institute of Military Veterinary, Academy of Military Medical Sciences, China.
We used PCR to further study the prevalence of orthohepadnavirus in the 6 bat species; the condition of the samples made serologic assay and pathology impracticable. Viral DNA was extracted from liver tissue of each of the 853 bats by using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). To detect virus in the samples, we conducted PCR by using the TaKaRa PCR Kit (TaKaRa, Dalian, China) with a pair of degenerate pan-orthohepadnavirus primers (sequences available upon request). The PCR reaction was as follows: 45 cycles of denaturation at 94°C for 30 s, annealing at 54°C for 30 s, extension at 72°C for 40 s, and a final extension at 72°C for 7 min. Positive results were obtained for 22 long-fingered bats (Miniopterus fuliginosus). Of these bats, 2.19% (7/320) were from Sedon County and 4.69% (15/320) from Wutao County; the viruses they harbored shared >98% nt identity. No other species had positive amplification results, indicating that M. fuliginosus was the most likely species to harbor orthohepadnaviruses.
Of the 22 positive samples, 3 were randomly selected for full genome amplification: M086 from Sedon County and 776 and M005 from Wutao County. PCR was conducted by using the PCR protocol defined above with high-fidelity Pfu DNA polymerase (Promega, Madison, WI, USA) and 4 pairs of specific primers (sequences available upon request). Four overlapping amplicons were obtained, sequenced in both directions, and assembled into the full genomic sequence by using SeqMan, version 7.1.0 (DNASTAR, Madison, WI, USA). All 3 full genomes (GenBank accession nos. JX941466– JX941468) were 3,230 nt in length, which is close to the size of primate hepatitis viruses (≈3,200 nt) but smaller than rodent hepatitis viruses (≈3,300 nt). We analyzed the genome structure by using Vector NTI Advance 10 (Invitrogen, Carlsbad, CA). The results showed that the bat hepatitis viruses (BtHVs) contained the same circular and compact genomic structure as other orthohepadnaviruses, comprising 4 open reading frames encoding the multifunctional Pol, preS1/preS2/S, preC/C, and X proteins in the same direction (Figure 1, panel A).
Figure 1.
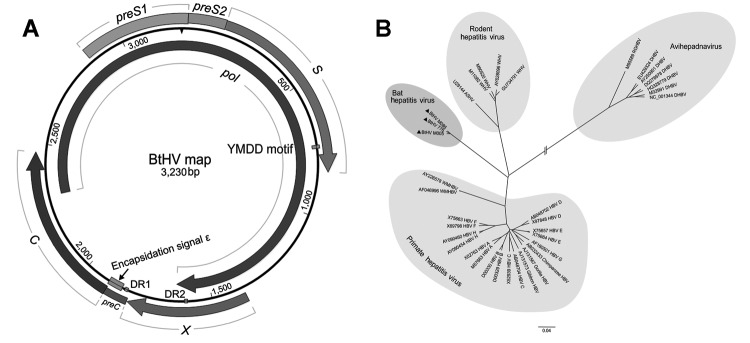
Predicted schematic representation of the bat hepatitis virus (BtHV) genome and its phylogenetic relationship with other hepadnaviruses. A) Genomic structural map of BtHV. Boxes and arrows represent the open reading frames encoding the main proteins: pol gene (2,305–1,636), preS1/S2 and S gene (2,864–833), preC/C gene (1,815–2,468) and X gene (1,378–1,812). Two 12-nt direct repeat sequences (DR1 from 1,825 to 1,836 and DR2 from 1,594 to 1,605), the encapsidation signal ε (1,848–1,903), and YMDD domain (734–745) are also depicted in the map. B) Phylogenetic analysis of BtHVs and other hepadnaviruses based on amino acid sequences of pol genes. Representatives of hepadnavirus species belonging to Orthohepadnavirus and Avihepadnavirus genera were used; their GenBank accession nos. are shown in the trees. The different genotypes of human hepatitis B virus are also included. The 3 BtHV isolates are identified by black triangles. Scale bar indicates nucleotide substitutions per site.
Genomic sequence comparison and phylogenetic analysis based on amino acids of the pol gene (2,562 bp) were constructed with ClustalW version 2.0 (www.clustal.org/) and MEGA5 (5). Phylogenetic tree analysis showed that previously described orthohepadnaviruses formed 2 clusters, primate hepatitis viruses and rodent hepatitis viruses, whereas the 3 newly identified BtHVs formed an independent cluster within the Orthohepadnavirus genus (Figure 1, panel B). Sequence comparison showed that the full genomes of the BtHVs were 63.1%–65.3% and 33.9%–34.8% identical to members of the Orthohepadnavirus and Avihepadnavirus genera, respectively. Similar low identities were also observed separately in the 4 genes of the BtHVs (Table). These results support the classification of the BtHVs within the Orthohepadnavirus genus, being distantly related to current species and likely to form a new species designated as BtHV.
Table. Gene lengths and percentage identity between bat orthohepadnavirus and other hepadnaviruses*.
| Virus† |
pol gene |
preS1/preS2/S gene |
preC/C gene |
X gene |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nt | % ID | aa | % ID | nt | % ID | aa | % ID | nt | % ID | aa | % ID | nt | % ID | aa | % ID | ||||
| BtHV776 | 2562 | – | 853 | – | 1200 | – | 399 | – | 654 | – | 217 | – | 435 | – | 144 | – | |||
| HBV | 2532 | 63 | 843 | 57 | 1203 | 63 | 400 | 59 | 639 | 65 | 212 | 66 | 465 | 61 | 154 | 49 | |||
| WMHBV | 2508 | 63 | 835 | 55 | 1176 | 64 | 391 | 60 | 636 | 65 | 211 | 63 | 459 | 66 | 152 | 50 | |||
| WHV | 2640 | 66 | 879 | 56 | 1281 | 66 | 426 | 51 | 678 | 69 | 225 | 71 | 426 | 67 | 141 | 44 | |||
| ASHV | 2634 | 67 | 877 | 53 | 1284 | 67 | 427 | 52 | 654 | 68 | 217 | 71 | 417 | 69 | 138 | 52 | |||
| DHBV | 2526 | 41 | 841 | 30 | 1104 | 43 | 367 | 30 | 888 | 42 | 295 | 22 | NA | – | NA | – | |||
*nt, nucleotide length; % ID, percentage identity of nt and amino acid sequence between BtHV and other viruses; aa, amino acid length; BtHV, bat hepatitis virus; –, not applicable; HBV, hepatitis B virus; WMHBV, woolly monkey HBV; WHV, woodchuck hepatitis virus; ASHV, arctic squirrel hepatitis virus; DHBV, duck HBV; NA, not available. †GenBank accession nos. for HBV, WMHBV, WHV, ASHV, and DHBV are D00329, AF046996, AY344076, U29144, and EU429324, respectively.
Hepadnaviruses have not been grown in any available in vitro cell system; thus, we did not attempt to isolate BtHV in cell culture. To detect the presence of virus particles, we used pooled liver tissues from the 3 bats that were randomly selected for full genome amplification. We homogenized the pooled tissues in SM buffer (50 mM Tris, 10 mM MgSO4, 0.1M NaCl; pH7.5), followed by clarification by low-speed centrifugation to remove cell debris. We then passed the pooled sample through a 0.22-μm syringe filter (Millipore, Carrigtwohill, Ireland). Polyethylene glycol 6000 was added, and the resulting precipitate was sedimented at 12,000 × g in a desktop centrifuge (Eppendorf, Hamburg, Germany) for 40 min at 4°C. The pellet was resuspended and examined after negative staining in a JEM-1200 EXII transmission electron microscope (JEOL, Tokyo, Japan). Numerous spherical particles of ≈20 nm diameter were observed (Figure 2). The particles were morphologically similar to the Australia antigens of HBV, the most abundant viral component found in HBV-infected humans and animals and also known as surface protein or S antigen (6,7). PCR amplification of DNA extracted from the virus pellet revealed the full genome of the BtHV, with the expected size of ≈3200 bp (image not shown).
Figure 2.

Electron microscopy of negative-stained orthohepadnavirus particles from a bat (arrow). Clumps of Australia antigen–like particles are seen.
Conclusions
Our observations provide strong evidence for the circulation of orthohepadnaviruses in at least 1 species of bats, M. fuliginosus, in Myanmar. These bats have a wide distribution (8), and increasing numbers of viruses, including coronaviruses and betaherpesviruses, are being isolated from them (9,10). Of the 6 bat species we sampled, only M. fuliginosus was positive for BtHV. The prevalence of BtHV-positive bats in the 2 counties from which we obtained bats, was 2.2% and 4.7%, respectively, indicating that this species is likely a natural reservoir host of BtHV. The lack of detection of BtHV in bats from the other 5 species may be due to the limited numbers of bats sampled (although no evidence of hepadnavirus was found in any of the 176 R. ferrumequinum bats) or to a narrow host range of the virus. Further study is required to determine the tropism and prevalence of BtHVs in other bat species.
Acknowledgments
This study was supported by the National Natural Science Foundation of China–Yunnan Province Joint Fund (U1036601) and the National “973” Program (grant no. 2012CB722501) to C.T.
Biography
Mr He is a doctoral candidate at Institute of Military Veterinary, Academy of Military Medical Sciences. He is majoring in animal virology, with research interests focusing on the discovery of bat emerging viruses.
Footnotes
Suggested citation for this article: He B, Fan Q, Yang F, Hu T, Qiu W, Feng Y, et al. Hepatitis virus in long-fingered bats, Myanmar. Emerg Infect Dis [Internet]. 2013 April [date cited]. http://dx.soi.org/10.3201/eid1904.121655
These authors contributed equally to this article.
References
- 1.International Committee on Taxonomy of Viruses. ICTV 2011. master species list v2 [cited 2012 Aug 20]. http://talk.ictvonline.org/files/ictv_documents/m/msl/4090.aspx
- 2.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. 10.1128/MMBR.64.1.51-68.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Hepatitis B, fact sheet no.204, July 2012. [cited 2012 Jul 16]. http://www.who.int/mediacentre/factsheets/fs204/en/index.html
- 4.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–45. 10.1128/CMR.00017-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen–associated hepatitis. Lancet. 1970;295:695–8. 10.1016/S0140-6736(70)90926-8 [DOI] [PubMed] [Google Scholar]
- 7.Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large surface proteins of hepatitis B virus containing the pre-s sequence. J Virol. 1984;52:396–402 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu KL, Wei L, Zhu TT, Wang XZ, Zhang LB. Dietary composition, echolocation pulses and morphological measurements of the long-fingered bat Miniopterus fuliginosus (Chiroptera: Vespertilioninae) [in Chinese]. Dongwuxue Yanjiu Dongwuxue Yanjiu. 2011;32:163–7. [DOI] [PubMed]
- 9.Shirato K, Maeda K, Tsuda S, Suzuki K, Watanabe S, Shimoda H, et al. Detection of bat coronaviruses from Miniopterus fuliginosus in Japan. Virus Genes. 2012;44:40–4. 10.1007/s11262-011-0661-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe S, Maeda K, Suzuki K, Ueda N, Iha K, Taniguchi S, et al. Novel betaherpesvirus in bats. Emerg Infect Dis. 2010;16:986–8. 10.3201/eid1606.091567 [DOI] [PMC free article] [PubMed] [Google Scholar]