

Abstract
Endoglin (CD105) is a type III auxiliary receptor for the transforming growth factor beta (TGFβ) superfamily. Several lines of evidence suggest that endoglin plays a critical role in maintaining cardiovascular homeostasis. Seemingly disparate disease conditions, including hereditary hemorrhagic telangiectasia, pre-eclampsia, and cardiac fibrosis, have now been associated with endoglin. Given the central role of the TGFβ superfamily in multiple disease conditions, this review provides a detailed update on endoglin as an evolving therapeutic target in the management of cardiovascular disease.
Keywords: endoglin, transforming growth factor beta, vascular, cardiac remodeling
Introduction
Endoglin (CD105) is a type III auxiliary receptor for the transforming growth factor beta (TGFβ) superfamily. Over the past three decades, several lines of evidence suggest that endoglin plays a critical role in maintaining cardiovascular homeostasis. First, mutations in human endoglin result in the autosomal dominant vascular dysplastic syndrome, hereditary hemorrhagic telangiectasia (HTT).1 Second, Endoglin null mice (Eng−/−) die at embryologic day 10–11.5 because of impaired cardiovascular development.2 Third, endoglin expression is increased in human atherosclerotic plaques3 and in models of balloon angioplasty-mediated vascular injury.4 Fourth, endoglin is abundantly expressed by neovascularized tumors in prostate, breast, and colon cancer.5 Finally, endoglin has recently been shown to regulate cardiac remodeling in heart failure6 and myocardial infarction.7 Given the central role of the TGFβ superfamily in multiple disease conditions, endoglin represents a potentially unique target of therapy for several debilitating conditions involving the cardiovascular system.
Endoglin structure, function, and distribution
Endoglin is a 180 kDa homodimeric integral membrane glycoprotein composed of disulfide-linked subunits. The receptor includes a large 561-amino acid extracellular domain with a single transmembrane domain and a serine/threonine-rich cytoplasmic region, with 47 amino acids in the long isoform (L-endoglin) and 14 residues in the short isoform (S-endoglin).8 The cytoplasmic domain of the predominantly expressed L-endoglin constitutes the region of the protein with the highest degree of conservation in mammalian species. A low-resolution structural analysis of the extracellular domain revealed a dome comprised of antiparallel-oriented monomers with a cavity at one end. Each endoglin monomer is comprised of three well defined domains. A large N-terminus domain extending from Glu26 to Ile359 forms a large flat area and does not show any degree of identity with known protein domains and is therefore referred to as the orphan domain. This domain mediates interactions with ligands of the TGFβ superfamily. Initial studies demonstrated that endoglin can be found in the receptor complex for the TGFβ1 and TGFβ3 isoforms in association with the type II receptor (TBR2).9 Subsequently, it was shown that endoglin also interacts with activin as well as bone morphogenetic proteins (BMP)-2 and BMP7 in association with the ligand binding receptor, which can be either a type I or type II receptor depending on the ligand involved.10 More recent studies have shown that endoglin may bind directly to BMP9 and BMP10.11,12
The orphan domain is followed by two zona pellucida subdomains, organized into an open U-shaped monomer and containing eight conserved cysteine residues localized to Lys362–Asp561.12–14 This zona pellucida domain is highly conserved among endoglin proteins from human, mouse, rat, pig, and dog models, and constitutes more than 30% of the extracellular region, suggesting an important functional role.11,15 The extracellular domain of endoglin also contains consensus motifs for several O-linked and N-linked oligosac-charide chains, with glycosylation accounting for about 30% of the molecular weight. The sequence of human endoglin originally revealed an arginine-glycine-aspartic acid (RGD) tripeptide at Arg399–Asp401, suggesting that endoglin may play a role in cell adhesion.14 However, that sequence was not conserved in other species. A recent study demonstrated that endoglin can bind to the α5β1 integrin via its RGD motif or a functionally equivalent murine TDD peptide that may facilitate leukocyte transmigration.16
The extracellular domain of endoglin can be proteolytically cleaved by matrix metalloproteinase 14 and circulate as soluble endoglin, which may serve as a naturally occurring antagonist for TGFβ signaling and therefore play an important role in cardiovascular disease.6,8,17 The cytoplasmic domain of endoglin is phosphorylated by serine-threonine kinase TBRI and TBR2 TGFβ receptors. Endoglin phosphorylation is thought to influence its subcellular localization by modulating its interaction with adhesive proteins such as zyxin and zyxin-related protein-1.18,19
Endoglin is present at high levels on the vascular endothelium in adults and is expressed early during development on vascular endothelium and on mesenchymal tissue derived from the endocardium.20 Stromal cells of mesenchymal origin,21 smooth muscle cells,22 the placental syncytiotrophoblast, mesenchymal and hematopoietic stem cells, pre-erythroblasts, leukemic cells of lymphoid and myeloid lineage, and activated blood monocytes also express endoglin. In cardiac tissue, endoglin is expressed by the endocardium and fibroblasts, with minimal expression by cardiomyocytes.8,21
Endoglin maintains vascular homeostasis
In quiescent nonproliferative endothelium, TGFβ signal transduction is predominantly mediated by the type I TGFβ receptor, activin receptor-like kinase-5,23 and phosphorylation of signaling mediators known as Smads-2/3. Alternatively, TGFβ-mediated activation of the activin receptor-like kinase-1 pathway phosphorylates Smads-1/5/8, which promotes endothelial proliferation and migration24 (Figure 1). Endoglin negatively regulates TGFβ-mediated activin receptor-like kinase-5 signaling in quiescent endothelium, while promoting activin receptor-like kinase-1 signaling in activated endothelium.24,25 Conversely, in the absence of endoglin, activin receptor-like kinase-1 signaling is attenuated, while activin receptor-like kinase-5-mediated growth arrest of endothelial cells is stimulated24 (Figure 1). Li et al determined that suppressed endoglin expression enhances the inhibitor effects and further that endoglin overexpression promotes endothelial proliferation.26,27 These pathways have important implications for the development of arteriovenous malformations associated with HHT, as described below.
Figure 1.
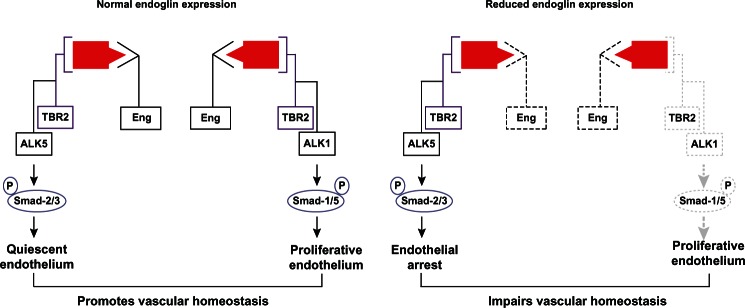
Endoglin signaling in endothelium.
Notes: Under normal conditions, endoglin promotes transforming growth factor beta-1 signaling via both ALK-1 and ALK-5, which balances endothelial quiescence and proliferation, thereby promoting vascular homeostasis. When endoglin expression is reduced, as in hereditary hemorrhagic telangiectasia 1, signaling via ALK-1 is attenuated, while ALK-5 activity is increased, thereby promoting arrest of endothelial proliferation and impairing vascular homeostasis.
Abbreviation: ALK, activin receptor-like kinase; P, phospho.
Further substantiating the role of endoglin in maintaining vascular homeostasis, cells from Endoglin heterozygous (Eng+/−) mice exhibit lower levels of endothelial nitric oxide synthase (eNOS), which results in less nitric oxide generation and more eNOS-derived superoxide production due to an uncoupling of eNOS activity.28 Endoglin may modulate vascular homeostasis via its colocalization to endothelial membrane-associated caveolae.26 Consistent with this observation, resistance arteries from Eng+/− mice demonstrate impaired myogenic responses and enhanced eNOS-dependent vasodilatation despite reduced eNOS levels. These findings may reflect increased stabilization of eNOS protein expression and enhanced calcium-induced activation of eNOS by endoglin.28,29 Subsequent data have also shown that TGFβ1 directly induces eNOS-dependent vasorelaxation, which is blocked by high circulating levels of soluble endoglin18 and may contribute to elevated vascular resistance.17 In addition to regulating eNOS activity, endoglin expression may correlate with myeloperoxidase activity and expression of vascular cell adhesion molecule 1, inducible nitric oxide synthase, and CD68 during ischemia-reperfusion injury.30
Endoglin modulates cardiac development
The importance of endoglin in hematopoiesis, angiogenesis, cardiovascular development, and vascular remodeling has been well established31 and is supported by the observation that Endoglin null mice (Eng−/−) die at embryonic day 10–11.5 as a result of cardiovascular abnormalities.2,20,32,33 The critical role of endoglin in cardiovascular development is further supported by high levels of endoglin expression in the endocardial cushion during valve formation and heart septation.34 Originating from the lateral plate mesoderm, both cardiac myocytes and cardiac endothelial progenitor cells ultimately constitute the primitive spongy heart tube. A subpopulation of endothelial cells in the region of the future cardiac valve and septal formation transforms into mesenchymal cells, which migrate into the cardiac jelly and begin to express α-actin.35 A critical aspect of this process is the ability of the developing myocardium to induce endothelial transformation via endocardial-myocardial signaling pathways, which is mediated in part by signaling through TGFβ1, TGFβ3, and endoglin.35–37 The role of endoglin in valve formation was recently highlighted by a systems biology approach which identified a haplotype within the endoglin (ENG) gene that is strongly associated with the presence of a bicuspid aortic valve in patients.38
Regulators of endoglin expression
Endoglin is localized to human chromosome 9 and contains 15 exons of which 13 code for the extracellular domain. The 5′ flanking region of the gene lacks consensus TATA and CAAT boxes, but contains two regions rich in G-C and consensus motifs for SP1, ETS, GATA, AP-2, NFkB, MAD, and response elements for TGFβ, glucocorticoids, vitamin D, and estrogen.39 Importantly, both endoglin and type I collagen promoters contain Smad-binding elements in close proximity to Sp1 binding sites, suggesting a possible mechanism by which TGFβ1 may coactivate endoglin and type I collagen expression.40 Factors known to induce endoglin expression in various cell types include angiotensin II,41 TGFβ,6,42 hypoxia-inducible factor-1,43 hypercholesterolemia,44 and balloon-mediated vascular injury via the Kruppel-like zinc finger transcription factor, KLF6.4 The inflammatory cytokine, tumor necrosis factor-alpha (TNFα), attenuates endoglin expression by endothelial cells in vitro.26
Several pharmacologic agents also influence endoglin expression. Tranexamic acid is a competitive inhibitor of plasminogen activation to plasmin, and at much higher concentrations, a noncompetitive inhibitor of plasmin. In endothelial cells, tranexamic acid promotes endoglin gene and protein expression and TGFβ1 signaling via activin receptor-like kinase-1.45 Conversely, inhibitors of angiotensin-converting enzyme, such as trandolapril,46 and angiotensin type I receptor inhibitors, such as valsartan, losartan, and candesartan, attenuate angiotensin II-induced endoglin expression.42 The 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor, atorvastatin, also decreases endoglin expression in mouse models of hyperlipidemia, which appears to correlate with the hypolipidemic effects of statin therapy.44,47
Acting as a biosensor for humoral and mechanical stimuli, the vascular endothelium plays a primary role in maintaining local and systemic homeostasis under conditions of mechanical stress. As a central marker of endothelial cell integrity and function, the effect of hemodynamic forces and mechanical deformation on endoglin activity remains largely uncharacterized. Several lines of evidence suggest that endoglin expression is regulated by mechanical forces such as stretch and strain. First, the evolutionarily conserved zona pellucida domain in the extracellular region of endoglin is mechanically responsive in fruit flies.15 Second, in vivo models of placental hypertension and balloon-induced arterial injury are characterized by increased endoglin expression in vascular tissue.4,17 Third, endoglin expression is increased in hemorrhoidal tissue and correlates with an increased propensity for thrombosis, suggesting that increases in venular wall tension may modulate endoglin expression and vascular thromboresistance.48 Recent data showed that mechanically stretching myoblasts increases endoglin expression; a process which appears to be regulated by microRNA-208A.49
As a central regulator of cardiovascular homeostasis, a functional role for endoglin has been identified in several important clinical disease conditions, including HHT, pre-eclampsia, cardiac remodeling, cancer, pulmonary hypertension, and other fibroproliferative disorders. Several of these conditions are discussed in detail below.
Hereditary hemorrhagic telangiectasia
HHT, known historically as Osler-Weber-Rendu syndrome, is a disorder of vascular homeostasis affecting small and large vessels. The prevalence of HHT varies depending on the population studied, from as low as about 1:39,000 in northern England to as high as about 1:1330 in the Afro-Caribbean population of Curaçao and Bonaire, with an average prevalence estimated at 1:5000–1:8000.50–53 The hallmark of HHT is the formation of arteriovenous malformations, which may occur in small vessels, leading to cutaneous telangiectases, nosebleeds, and gastrointestinal bleeds, or in large vessels of the lungs, brain, and liver. Important clinical sequelae of large-vessel arteriovenous malformations include hemorrhage, stroke, and brain abscess for pulmonary arteriovenous malformations, and pulmonary hypertension and high output heart failure arteriovenous malformations in the liver. Rupture of arteriovenous malformations, either spontaneous or associated with pregnancy or other physiological stressors, represents a significant source of morbidity in patients with HHT.51,54–57 The diagnosis of HHT is based on clinical observations that are summarized in consensus criteria known as the “Curaçao criteria”.56,58
Mutations in endoglin (ENG, OMIM#187300) and activin receptor-like kinase-1 (also known as ACVRL1, OMIM#600376) genes lead to HHT1 and HHT2, respectively, and account for >80% of cases.59 Mutations in Smad-4 (MADH4, OMIM #175050) are also associated with 2%–3% of cases with a combined juvenile polyposis-HHT syndrome.59 Two other loci have been shown to be in linkage disequilibrium with HHT; one on chromosome 5, defining HHT3, and the other on chromosome 7, defining HHT4.60,61 The precise genes involved remain to be identified and would give clues about the pathways deficient in HHT.
Transgenic mouse models of deficient expression of endoglin or ALK-1 recapitulate clinical features of HHT.62,63 In Eng−/− mice, complete loss of endoglin is embryonically lethal due to impaired angiogenesis, while heterozygosity leads to the clinical syndrome of HHT1 with variable penetrance.2,62 In humans with HHT1, endoglin expression in peripheral blood monocytes and newborn umbilical vein endothelial cells is half of control levels and mutant proteins are rarely detected.59 These and other data support the role of haploinsufficiency in the pathogenesis of HHT1 as opposed to reduced local expression of endoglin through a “second hit” process or interference by the mutated allele with the function of the normal allele.59,62
Mutations in ALK-1 and to a lesser degree ENG are associated with pulmonary arterial hypertension, which can occur in the absence of HHT.64–66 Both endoglin and ALK-1 can associate with the BMP type II receptor, which is primarily responsible for most familial forms of pulmonary arterial hypertension.23 The mechanistic link between pulmonary arterial hypertension and HHT was recently studied in adult ALK-1+/− and Eng+/− mice, which develop signs of pulmonary arterial hypertension that can be attenuated by treatment with the antioxidant, tempol.67,68 These findings highlight the importance of eNOS-derived superoxide in these mice due to uncoupling of eNOS, which contributes to impaired vascular tone in either HHT or pulmonary arterial hypertension. Furthermore, endoglin and ALK-1 are coexpressed in the terminal-most segments of the pulmonary vasculature, which is most affected in mouse models and where most HHT-associated arteriovenous malformations tend to form.69 However, physiologically, pulmonary arterial hypertension is associated with increased pulmonary vascular resistance, while arteriovenous malformations are associated with abnormal vasodilatation.70
An important treatment option for patients with HHT is embolization of visceral arteriovenous malformations, which can prevent major adverse clinical events, including stroke, high-output heart failure, pulmonary hypertension, and hemorrhage. Hormonal and antiangiogenic agents have also been explored as potential therapy for HHT. Clinical trials of estrogen preparations including the estrogen receptor antagonist, tamoxifen, and the selective estrogen receptor modulator, raloxifene, can reduce episodes of epistaxis and transfusion requirements.71,73,74 Inhibition of angiogenesis through the use of agents such as thalidomide, lenalidomide, and bevacizumab reduces the incidence of nasal and gastrointestinal bleeding in some patients.75–80 However, the lack of randomized controlled trial data for bevacizumab and the absence of long-term safety or efficacy data for antiangiogenesis therapy limits enthusiasm for these HHT treatment approaches, which remain experimental and limited to highly selected patients. The therapeutic action of thalidomide in HHT derives in part from increased platelet-derived growth factor expression at low doses and a direct antiangiogenic effect at high doses.77 Raloxifene and tranexamic acid increase in vitro mRNA and protein expression of endoglin and ALK-1 in endothelial cells.45,71 Whether future therapies such as gene transfer methods can target the underlying genetic defect associated with HHT remains unknown.81,82 Further insight into the role of endoglin and the TGFβ signaling pathway in the pathogenesis of HHT, together with evolving molecular approaches, may lead to novel therapeutic approaches for this debilitating condition.
Pre-eclampsia
Pre-eclampsia is a systemic disorder occurring in 3% of pregnant women after 20 weeks of gestation with symptoms of new-onset hypertension and proteinuria defined as a systolic blood pressure > 140 mmHg or diastolic blood pressure > 90 mmHg in a previously normotensive patient and at least 0.3 g of protein in a 24-hour urine specimen. Eclampsia is defined as pre-eclampsia with grand mal seizures without an identifiable cause.83,84 The incidence of maternal death associated with pre-eclampsia has been estimated at one in 100,000 live births.85 Maternal complications include central nervous system involvement, acute renal or liver failure, and hematological dysfunction.86 Incident pre-eclampsia is associated with increased risk of subsequent cardiovascular morbidity, including hypertension and ischemic heart disease, although it is not clear if pre-eclampsia is causal or reflects an underlying predisposition to cardiovascular disease.87 Fetal effects of pre-eclampsia include prematurity, fetal growth restriction, oligohydramnios, and placental abruption.88,89 Notable risk factors for the development of pre-eclampsia include a prior personal or family history of pre-eclampsia, advanced maternal age, obesity, and pregestational diabetes.90
The pathophysiology of pre-eclampsia remains poorly understood. Early changes in the placenta prior to the appearance of clinical manifestations of pre-eclampsia include failure of migrating placental cells to express endothelial surface adhesion markers, resulting in incomplete invasion of maternal arteries by the developing trophoblast.91–94 Pathological evidence of placental ischemia has been observed in some cases of pre-eclampsia,95 which is thought to trigger release of antiangiogenic factors, including soluble endoglin and soluble fms-like tyrosine kinase (sFlt1) into the maternal circulation.96,97 sFlt1 antagonizes the angiogenic factors, vascular endothelial growth (VEGF) factor and placental growth factor, while soluble endoglin antagonizes TGFβ1 and TGFβ3 activity.9,17,19,97 Furthermore, systemic disruption of vascular hemostasis caused by elaboration of these antiangiogenic factors promotes pre-eclampsia.86,98
The VEGF and TGFβ axes are the major signaling pathways implicated in the pathogenesis of pre-eclampsia (Figure 2). sFlt1, or the type 1 VEGF receptor, can be generated by either alternative splicing of the Flt1 gene or by cleavage of the Flt1 extracellular domain in response to hypoxia or thrombin.99,100 Elevated levels of sFlt1 in the maternal circulation correspond to a fall in VEGF and placental growth factor levels.97,101–103 A putative role for sFlt1 in pre-eclampsia is supported by studies showing that administration of exogenous sFlt1 to pregnant rats induces severe pre-eclampsia, while removal of sFlt1 by immunoprecipitation normalizes the angiogenic response of cells derived from placental villous explants.17,96 Similarly, pre-eclamptic placentae express increased levels of membrane-bound and soluble endoglin.17 A recent study demonstrated that oxysterol activation promotes matrix metalloproteinase 14-mediated cleavage of soluble endoglin in cells of trophoblast origin,104 which suggests that hypoxia and oxidative stress are important triggers for release of soluble endoglin. Once in the circulation, soluble endoglin antagonizes TGFβ1-induced vasodilation, thereby promoting hypertension.28,105,106 A rise in circulating maternal soluble endoglin heralds the onset of symptoms in patients with pre-eclampsia.102,107,108 Individually and in concert with sFlt1, soluble endoglin induces a pre-eclamptic state in pregnant rats.17 Investigation into the regulation of sFlt and soluble endoglin in pre-eclampsia is ongoing.109
Figure 2.
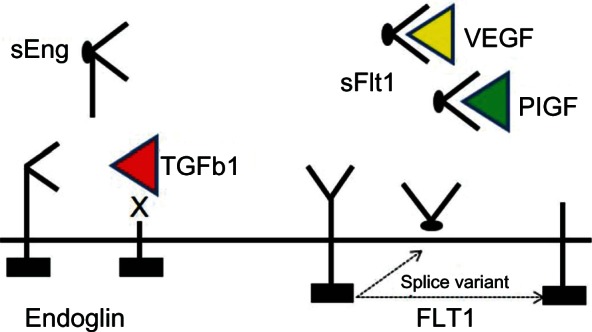
Circulating angiogenic peptides in pre-eclampsia.
Notes: Soluble endoglin is released by ectodomain cleavage of membrane-associated endoglin, thereby disrupting TGFβ1 signaling. Soluble Flt1 is generated by either ectodomain cleavage of the VEGF receptor type I or alternative splicing of FLT-1 pre-mRNA and interferes with VEGF signaling.
Abbreviations: TGFβ1, transforming growth factor beta-1; PIGF, placental growth factor; VEGF, vascular endothelial growth factor; sEng, soluble endoglin; mRNA, messenger RNA.
At present, the primary mode of therapy for pre-eclampsia is accelerated delivery of the placenta. The available evidence strongly implicates an imbalance of the antiangiogenic factors, sFlt1 and soluble endoglin, and the proangiogenic factors, placental growth factor, TGFβ, and VEGF, in the pathogenesis of pre-eclampsia. Strategies targeting maternal depletion of soluble endoglin or sFlt1 would plausibly de-escalate the disease process. Given the growing appreciation for the role of sFlt1 and soluble endoglin in the pathophysiology of pre-eclampsia, novel therapeutic approaches include the use of antibodies and small molecules to sequester or limit synthesis of antiangiogenic molecules. Alternatively, modulating the balance of angiogenic factor levels has been recently explored in a study that showed improvement in blood pressure and renal function after administration of exogenous VEGF in a preclinical model of pre-eclampsia.110–112 More recently, directly removing sFlt1 from the maternal circulation by extracorporeal apheresis using a dextran sulfate column has been introduced as a potential therapeutic approach for pre-eclampsia.113 Other approaches include induction of hemoxygenase-1 with cobalt protoporphyrin in a rat model of pre-eclampsia114 and direct inhibition of matrix metalloproteinase 14 to prevent the release of soluble endoglin.115,116 Limiting fetal consequences of any treatment approach is mandatory and remains a challenging limitation to the development of new treatments.
Cardiac remodeling
Heart failure is a major cause of global morbidity and mortality that affects nearly 23 million individuals worldwide.117,118 Heart failure commonly occurs secondary to hypertensive heart disease, myocardial infarction, infection, and inherited cardiomyocyte dysfunction. A decline in cardiac function activates several signaling cascades that promote cardiomyocyte hypertrophy and cardiac fibrosis, a process known as cardiac remodeling.119 At each phase of cardiac remodeling from acute load to compensatory hypertrophy, various signaling cascades are implicated. Among these, TGFβ1 promotes the cardiac hypertrophy and fibrosis associated with heart failure. In response to angiotensin II, TGFβ1 is increased, converts fibroblasts into myofibroblasts, and generates extracellular matrix proteins, such as type I collagen.120 Excess collagen deposition exaggerates mechanical stiffness of the left ventricle, impairs myocyte contractility, disrupts electrical coupling, and worsens tissue hypoxia,119 thereby promoting heart failure.
TGFβ1-induced fibrosis involves both canonical and noncanonical signaling (Figure 3). Among the canonical pathways, Smad-3 plays a central role.120 First, Smad-3 mediates TGFβ1-induced activation of a TGFβ-response element57 located in the promoter region of type I collagen.121 Second, Smads-2/3 and Smad-4 form a complex with activated transcription factor-2 to stimulate collagen synthesis. Activated transcription factor-2 phosphorylation requires activation of TGFβ-activated kinase.122 Third, Smad-3 mediates TGFβ1-induced expression of connective tissue growth factor via a TGFβ1 response element located in the connective tissue growth factor promoter.123 Connective tissue growth factor is a critical downstream mediator of tissue fibrosis. Noncanonical pathways contributing to TGFβ1-induced collagen synthesis involve activation of mitogen-activated protein kinases including: extracellular regulated kinases, jun-n-terminal kinases, and p38 kinase.120 While all three cascades are activated in advanced heart failure, the extracellular regulated kinase pathway is required for TGFβ1-induced connective tissue growth factor expression and collagen synthesis.124 In cardiac fibroblasts, connective tissue growth factor is an important downstream mediator of TGFβ1-induced fibrosis because it promotes binding of TGFβ1 to TBR2 and blocks inhibition of the pathway by Smad-7.124,125 Given its central role in stimulating fibrosis, TGFβ1 has been nonselectively targeted in heart failure models using multiple approaches, none of which have had clearly beneficial therapeutic effects.126,127
Figure 3.
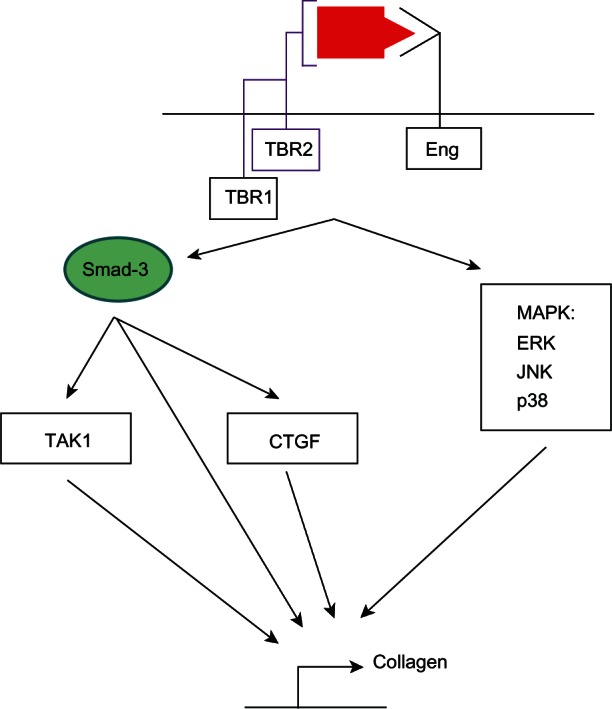
Canonical and noncanonical transforming growth factor beta-1/endoglin signaling in fibrosis.
Notes: On binding of transforming growth factor beta-1 to a heteromeric complex of receptors including type I, type II, and endoglin, downstream signaling via Smad-dependent or Smad-independent pathways occurs. Smad-3 promotes collagen synthesis by activating transforming growth factor beta-1 activated kinase or CTGF. Smad-3 can also directly activate collagen synthesis by binding a Smad-binding element within the collagen promoter. Noncanonical signaling can stimulate collagen synthesis via several mitogen-activated protein kinases including ERK, JNK, or p38 kinase.
Abbreviations: CTGF, connective tissue growth factor; ERK, extracellular regulated kinase; JNK, jun-n-terminal kinase; Eng, endoglin; MAPK, mitogen-activated protein kinase.
Recently, endoglin expression was found to be increased in left ventricular tissue from patients with end-stage heart failure and in a murine model of thoracic aortic constriction-induced heart failure.6 Further analysis showed that endoglin is highly expressed by cardiac fibroblasts and endothelium, but poorly on cardiomyocytes. Compared with Eng+/+ wild-type mice, Eng+/− mice demonstrated improved survival, limited cardiac fibrosis, and enhanced myocardial capillarity after thoracic aortic constriction (Figure 4). Loss-of-function studies in vitro confirmed the dependence of TGFβ1 activity on endoglin expression in human cardiac fibroblasts. Paradoxically, adenoviral-mediated overexpression of full-length endoglin also blocked TGFβ1-induced collagen synthesis. Further study showed that levels of soluble endoglin were elevated in the conditioned medium after treatment with the adenovirus, thereby implicating soluble endoglin as a negative regulator of TGFβ1 activity. This observation was confirmed by adenoviral-mediated overexpression of human soluble endoglin or treatment with recombinant human soluble endoglin in vitro. Treatment with adenoviral-mediated overexpression of human soluble endoglin attenuated cardiac fibrosis in wild-type mice after thoracic aortic constriction. These findings suggest that endoglin is required for TGFβ1 signaling in cardiac fibroblasts and that selectively inhibiting TGFβ1 signaling by reducing endoglin activity attenuates cardiac fibrosis and improves survival in a mouse model of heart failure. In contrast with the functional role of endoglin in promoting TGFβ1 signaling, soluble endoglin limits TGFβ1 signaling, type I collagen synthesis, and ultimately cardiac fibrosis.
Figure 4.

Functional role of endoglin in cardiac fibrosis.
Notes: In heart failure, increased ventricular pressure overload stimulates TGFβ1 expression. In mice with normal endoglin expression (Eng+/+), endoglin promotes TGFβ1-induced type I collagen synthesis and cardiac fibrosis. Reduced endoglin expression in Eng+/− mice attenuates TGFβ1-induced pSmad-2/3, type I collagen expression, and cardiac fibrosis.
Abbreviation: TGFβ1, transforming growth factor beta-1.
In a separate study, circulating levels of soluble endoglin were significantly increased in patients with suspected left ventricular dysfunction and were strongly correlated with predictors of mortality in heart failure, such as elevated left ventricular end-diastolic pressure, reduced left ventricular ejection fraction, and worsening New York Heart Association class symptoms.128 Soluble endoglin levels demonstrated superior sensitivity, specificity, accuracy, and predictive value compared with atrial and brain natriuretic peptides for identifying subjects with increased left ventricular end-diastolic pressure. Furthermore, levels were reduced in patients receiving medical therapy for congestive heart failure and correlated with reduced cardiac filling pressures. These findings identify elevated soluble endoglin levels as a biomarker of increased cardiac filling pressure and perhaps a potential biomarker of endoglin activity in heart failure.129 These findings also represent an important paradox in endoglin biology. Expression of membrane-associated endoglin is increased in heart failure and correlates with a direct increase in circulating soluble endoglin. However, membrane-associated endoglin promotes adverse cardiac remodeling by driving cardiac fibrosis, while soluble endoglin may serve as a negative feedback loop to limit TGFβ1/endoglin signaling. The exact functional role of soluble endoglin remains to be determined, but appears to serve as a potentially important diagnostic marker of heart failure as well as a participant in the cardiac remodeling associated with heart failure.
The role of soluble endoglin in heart failure remains poorly understood. First, the mechanism underlying increased expression of soluble endoglin in heart failure is unknown. Proteolytic cleavage of soluble endoglin from membrane endoglin may occur both locally in cardiac tissue or systemically because levels of matrix metalloproteinase 14 are known to be elevated in heart failure.130 Second, the mechanism by which higher soluble endoglin levels interrupt TGFβ1 signaling remains poorly characterized. First, endoglin may modulate signaling via several TGFβ-family ligands and soluble endoglin could serve as a ligand trap for ligands of the TGFβ superfamily.10 However, recent studies indicate that BMP9 and BMP10 may be the only ligands that bind to soluble endoglin with high affinity.12 Second, soluble endoglin may promote alternate signaling pathways that indirectly inhibit TGFβ1 signaling, such as BMP7.131 Finally, release of soluble endoglin by ectodomain shedding may render the receptor nonfunctional by removing the primary active component of endoglin, the extracellular domain, and leaving behind a nonfunctional cytoplasmic tail that cannot interact with TBR2.
Further supporting the role of endoglin in cardiac remodeling, studies of acute myocardial infarction have shown that Eng+/− mice exhibit showed neoangiogenesis in the infarct zone, progressive left ventricular dilatation, worsening cardiac function, and elevated cardiac filling pressures compared with wild-type controls. In this model, adverse cardiac remodeling in Eng+/− mice was partially rescued by intravenous injection of mononuclear cells from healthy human donors with normal endoglin expression but not by those from patients with HHT1 harboring mutant endoglin.7
In contrast with heart failure, circulating levels of soluble endoglin in patients presenting with acute myocardial infarction were significantly lower than in healthy controls, and decreased further in the first 48 hours of admission.132 Reduced levels of soluble endoglin were an independent predictor of short-term and long-term cardiovascular mortality. Unlike other known circulating angiogenic proteins, including sFlt1 and VEGF, levels of soluble endoglin are not altered by treatment with the anticoagulants commonly used to manage patients with myocardial infarction.133 Whether alterations in soluble endoglin levels correlate with atherosclerotic burden remains under investigation.134 Collectively, these data suggest that both membrane-associated and soluble endoglin serve important roles in cardiac remodeling due to heart failure or acute myocardial infarction, and may serve as potential targets of therapy for these debilitating conditions.
In summary, the role of endoglin as a critical mediator of cardiovascular homeostasis has been well established over the past two decades. Studies have now begun to explore the potential utility of targeting endoglin and the signaling program it regulates as a novel therapeutic approach for patients suffering from cardiovascular diseases, including HHT1, pre-eclampsia, pulmonary hypertension, heart failure, myocardial infarction, atrial fibrillation, pulmonary hypertension, and atherosclerosis.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemor-rhagic telangiectasia type 1. Nat Genet. 1994;8(4):345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 2.Li DY, Sorensen LK, Brooke BS, et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284(5419):1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 3.Conley BA, Smith JD, Guerrero-Esteo M, Bernabeu C, Vary CP. Endoglin, a TGF-beta receptor-associated protein, is expressed by smooth muscle cells in human atherosclerotic plaques. Atherosclerosis. 2000;153(2):323–335. doi: 10.1016/s0021-9150(00)00422-6. [DOI] [PubMed] [Google Scholar]
- 4.Botella LM, Sanchez-Elsner T, Sanz-Rodriguez F, et al. Transcriptional activation of endoglin and transforming growth factor-beta signaling components by cooperative interaction between Sp1 and KLF6: their potential role in the response to vascular injury. Blood. 2002;100(12):4001–4010. doi: 10.1182/blood.V100.12.4001. [DOI] [PubMed] [Google Scholar]
- 5.Li C, Guo B, Bernabeu C, Kumar S. Angiogenesis in breast cancer: the role of transforming growth factor beta and CD105. Microsc Res Tech. 2001;52(4):437–449. doi: 10.1002/1097-0029(20010215)52:4<437::AID-JEMT1029>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 6.Kapur NK, Wilson S, Yunis AA, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;125(22):2728–2738. doi: 10.1161/CIRCULATIONAHA.111.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Laake LW, van den Driesche S, Post S, et al. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation. 2006;114(21):2288–2297. doi: 10.1161/CIRCULATIONAHA.106.639161. [DOI] [PubMed] [Google Scholar]
- 8.Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem. 2007;102(6):1375–1388. doi: 10.1002/jcb.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheifetz S, Bellon T, Cales C, et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267(27):19027–19030. [PubMed] [Google Scholar]
- 10.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274(2):584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 11.Alt A, Miguel-Romero L, Donderis J, et al. Structural and functional insights into endoglin ligand recognition and binding. PLoS One. 2012;7(2):e29948. doi: 10.1371/journal.pone.0029948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castonguay R, Werner ED, Matthews RG, et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem. 2011;286(34):30034–30046. doi: 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gougos A, Letarte M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J Immunol. 1988;141(6):1925–1933. [PubMed] [Google Scholar]
- 14.Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265(15):8361–8364. [PubMed] [Google Scholar]
- 15.Jovine L, Qi H, Williams Z, Litscher E, Wassarman PM. The ZP domain is a conserved module for polymerization of extracellular proteins. Nat Cell Biol. 2002;4(6):457–461. doi: 10.1038/ncb802. [DOI] [PubMed] [Google Scholar]
- 16.Rossi E, Sanz-Rodriguez F, Eleno N, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. 2013;121(2):403–415. doi: 10.1182/blood-2012-06-435347. [DOI] [PubMed] [Google Scholar]
- 17.Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 18.Conley BA, Koleva R, Smith JD, et al. Endoglin controls cell migration and composition of focal adhesions: function of the cytosolic domain. J Biol Chem. 2004;279(26):27440–27449. doi: 10.1074/jbc.M312561200. [DOI] [PubMed] [Google Scholar]
- 19.Sanz-Rodriguez F, Guerrero-Esteo M, Botella LM, Banville D, Vary CP, Bernabeu C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the Lim family of proteins. J Biol Chem. 2004;279(31):32858–32868. doi: 10.1074/jbc.M400843200. [DOI] [PubMed] [Google Scholar]
- 20.Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104(10):1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gougos A, St Jacques S, Greaves A, et al. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int Immunol. 1992;4(1):83–92. doi: 10.1093/intimm/4.1.83. [DOI] [PubMed] [Google Scholar]
- 22.Adam PJ, Clesham GJ, Weissberg PL. Expression of endoglin mRNA and protein in human vascular smooth muscle cells. Biochem Biophys Res Commun. 1998;247(1):33–37. doi: 10.1006/bbrc.1998.8734. [DOI] [PubMed] [Google Scholar]
- 23.Machado RD, Aldred MA, James V, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27(2):121–132. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 24.Lebrin F, Goumans MJ, Jonker L, et al. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23(20):4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65(3):599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Hampson IN, Hampson L, Kumar P, Bernabeu C, Kumar S. CD105 antagonizes the inhibitory signaling of transforming growth factor beta1 on human vascular endothelial cells. FASEB J. 2000;14(1):55–64. doi: 10.1096/fasebj.14.1.55. [DOI] [PubMed] [Google Scholar]
- 27.Pece-Barbara N, Vera S, Kathirkamathamby K, et al. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem. 2005;280(30):27800–27808. doi: 10.1074/jbc.M503471200. [DOI] [PubMed] [Google Scholar]
- 28.Toporsian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96(6):684–692. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 29.Jerkic M, Rivas-Elena JV, Santibanez JF, et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ Res. 2006;99(3):248–256. doi: 10.1161/01.RES.0000236755.98627.69. [DOI] [PubMed] [Google Scholar]
- 30.Docherty NG, Lopez-Novoa JM, Arevalo M, et al. Endoglin regulates renal ischaemia-reperfusion injury. Nephrol Dial Transplant. 2006;21(8):2106–2119. doi: 10.1093/ndt/gfl179. [DOI] [PubMed] [Google Scholar]
- 31.Chen CZ, Li M, de Graaf D, et al. Identification of endoglin as a functional marker that defines long-term repopulating hematopoietic stem cells. Proc Natl Acad Sci USA. 2002;99(24):15468–15473. doi: 10.1073/pnas.202614899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho SK, Bourdeau A, Letarte M, Zuniga-Pflucker JC. Expression and function of CD105 during the onset of hematopoiesis from Flk1(+) precursors. Blood. 2001;98(13):3635–3642. doi: 10.1182/blood.v98.13.3635. [DOI] [PubMed] [Google Scholar]
- 33.Borges L, Iacovino M, Mayerhofer T, et al. A critical role for endoglin in the emergence of blood during embryonic development. Blood. 2012;119(23):5417–5428. doi: 10.1182/blood-2011-11-391896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu R, Silver MM, Letarte M. Distribution of endoglin in early human development reveals high levels on endocardial cushion tissue mesen-chyme during valve formation. Cell Tissue Res. 1998;292(2):333–343. doi: 10.1007/s004410051064. [DOI] [PubMed] [Google Scholar]
- 35.Molin DG, Bartram U, Van der Heiden K, et al. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev Dyn. 2003;227(3):431–444. doi: 10.1002/dvdy.10314. [DOI] [PubMed] [Google Scholar]
- 36.Nomura-Kitabayashi A, Anderson GA, Sleep G, et al. Endoglin is dispensable for angiogenesis, but required for endocardial cushion formation in the midgestation mouse embryo. Dev Biol. 2009;335(1):66–77. doi: 10.1016/j.ydbio.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322(1):208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wooten EC, Iyer LK, Montefusco MC, et al. Application of gene network analysis techniques identifies AXIN1/PDIA2 and endoglin haplotypes associated with bicuspid aortic valve. PLoS One. 2010;5(1):e8830. doi: 10.1371/journal.pone.0008830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rius C, Smith JD, Almendro N, et al. Cloning of the promoter region of human endoglin, the target gene for hereditary hemorrhagic telangiectasia type 1. Blood. 1998;92(12):4677–4690. [PubMed] [Google Scholar]
- 40.Botella LM, Sanchez-Elsner T, Rius C, Corbi A, Bernabeu C. Identification of a critical Sp1 site within the endoglin promoter and its involvement in the transforming growth factor-beta stimulation. J Biol Chem. 2001;276(37):34486–34494. doi: 10.1074/jbc.M011611200. [DOI] [PubMed] [Google Scholar]
- 41.Li D, Chen H, Mehta JL. Angiotensin II via activation of type 1 receptor upregulates expression of endoglin in human coronary artery endothelial cells. Hypertension. 2001;38(5):1062–1067. doi: 10.1161/hy1101.092971. [DOI] [PubMed] [Google Scholar]
- 42.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95(12):1167–1173. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem. 2002;277(46):43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 44.Pospisilova N, Semecky V, Jamborova G, et al. Endoglin expression in hyper-cholesterolemia and after atorvastatin treatment in apoE-deficient mice. J Pharm Pharm Sci. 2006;9(3):388–397. [PubMed] [Google Scholar]
- 45.Fernandez LA, Garrido-Martin EM, Sanz-Rodriguez F, et al. Therapeutic action of tranexamic acid in hereditary haemorrhagic telangiectasia (HHT): regulation of ALK-1/endoglin pathway in endothelial cells. Thromb Haemost. 2007;97(2):254–262. [PubMed] [Google Scholar]
- 46.Prieto M, Rodriguez-Pena A, Arevalo M, et al. Effect of the long-term treatment with trandolapril on endoglin expression in rats with experimental renal fibrosis induced by renal mass reduction. Kidney Blood Press Res. 2005;28(1):32–40. doi: 10.1159/000081439. [DOI] [PubMed] [Google Scholar]
- 47.Shyu KG, Wang BW, Chen WJ, Kuan P, Hung CR. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-beta1 in cultured cardiac fibroblasts. Eur J Heart Fail. 2010;12(3):219–226. doi: 10.1093/eurjhf/hfq011. [DOI] [PubMed] [Google Scholar]
- 48.Chung YC, Hou YC, Pan AC. Endoglin (CD105) expression in the development of haemorrhoids. Eur J Clin Invest. 2004;34(2):107–112. doi: 10.1111/j.1365-2362.2004.01305.x. [DOI] [PubMed] [Google Scholar]
- 49.Shyu KG, Wang BW, Wu GJ, Lin CM, Chang H. Mechanical stretch via transforming growth factor-beta1 activates microRNA208a to regulate endoglin expression in cultured rat cardiac myoblasts. Eur J Heart Fail. 2013;15(1):36–45. doi: 10.1093/eurjhf/hfs143. [DOI] [PubMed] [Google Scholar]
- 50.Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19(2):140–148. doi: 10.1002/humu.10026. [DOI] [PubMed] [Google Scholar]
- 51.Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245(1):31–39. doi: 10.1046/j.1365-2796.1999.00398.x. [DOI] [PubMed] [Google Scholar]
- 52.Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992;29(8):527–530. doi: 10.1136/jmg.29.8.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westermann CJ, Rosina AF, De Vries V, de Coteau PA. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: a family screening. Am J Med Genet A. 2003;116A(4):324–328. doi: 10.1002/ajmg.a.10002. [DOI] [PubMed] [Google Scholar]
- 54.Ference BA, Shannon TM, White RI, Jr, Zawin M, Burdge CM. Life-threatening pulmonary hemorrhage with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Chest. 1994;106(5):1387–1390. doi: 10.1378/chest.106.5.1387. [DOI] [PubMed] [Google Scholar]
- 55.Sabba C, Pasculli G, Cirulli A, et al. Hereditary hemorrhagic teleangiectasia (Rendu-Osler-Weber disease) Minerva Cardioangiol. 2002;50(3):221–238. [PubMed] [Google Scholar]
- 56.Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000;91(1):66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 57.Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 58.Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87. doi: 10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- 59.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43(2):97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140(20):2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 61.Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42(7):577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourdeau A, Cymerman U, Paquet ME, et al. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformations of patients with hereditary hemorrhagic telangiectasia type 1. Am J Pathol. 2000;156(3):911–923. doi: 10.1016/S0002-9440(10)64960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srinivasan S, Hanes MA, Dickens T, et al. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. 2003;12(5):473–482. doi: 10.1093/hmg/ddg050. [DOI] [PubMed] [Google Scholar]
- 64.Cottin V, Dupuis-Girod S, Lesca G, Cordier JF. Pulmonary vascular manifestations of hereditary hemorrhagic telangiectasia (Rendu-Osler disease) Respiration. 2007;74(4):361–378. doi: 10.1159/000103205. [DOI] [PubMed] [Google Scholar]
- 65.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–861. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- 66.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(Suppl 1):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 67.Jerkic M, Kabir MG, Davies A, et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc Res. 2011;92(3):375–384. doi: 10.1093/cvr/cvr232. [DOI] [PubMed] [Google Scholar]
- 68.Toporsian M, Jerkic M, Zhou YQ, et al. Spontaneous adult-onset pulmonary arterial hypertension attributable to increased endothelial oxidative stress in a murine model of hereditary hemorrhagic telangiectasia. Arterioscler Thromb Vasc Biol. 2010;30(3):509–517. doi: 10.1161/ATVBAHA.109.200121. [DOI] [PubMed] [Google Scholar]
- 69.Mahmoud M, Borthwick GM, Hislop AA, Arthur HM. Endoglin and activin receptor-like-kinase 1 are co-expressed in the distal vessels of the lung: implications for two familial vascular dysplasias, HHT and PAH. Lab Invest. 2009;89(1):15–25. doi: 10.1038/labinvest.2008.112. [DOI] [PubMed] [Google Scholar]
- 70.Mache CJ, Gamillscheg A, Popper HH, Haworth SG. Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax. 2008;63(1):85–86. doi: 10.1136/thx.2007.076109. [DOI] [PubMed] [Google Scholar]
- 71.Albinana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabeu C, Botella LM. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): effects of raloxifene on Endoglin and ALK1 expression in endothelial cells. Thromb Haemost. 2010;103(3):525–534. doi: 10.1160/TH09-07-0425. [DOI] [PubMed] [Google Scholar]
- 72.van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet. 1990;335(8695):953–955. doi: 10.1016/0140-6736(90)91010-8. [DOI] [PubMed] [Google Scholar]
- 73.Yaniv E, Preis M, Shevro J, Nageris B, Hadar T. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia – a long-term clinical trial. Rhinology. 2011;49(2):214–216. doi: 10.4193/Rhino09.201. [DOI] [PubMed] [Google Scholar]
- 74.Shovlin CL, Sulaiman NL, Govani FS, Jackson JE, Begbie ME. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): association with venous thromboembolism. Thromb Haemost. 2007;98(5):1031–1039. [PubMed] [Google Scholar]
- 75.Bowcock SJ, Patrick HE. Lenalidomide to control gastrointestinal bleeding in hereditary haemorrhagic telangiectasia: potential implications for angiodysplasias? Br J Haematol. 2009;146(2):220–222. doi: 10.1111/j.1365-2141.2009.07730.x. [DOI] [PubMed] [Google Scholar]
- 76.Karnezis TT, Davidson TM. Efficacy of intranasal bevacizumab (Avastin) treatment in patients with hereditary hemorrhagic telangiectasia-associated epistaxis. Laryngoscope. 2011;121(3):636–638. doi: 10.1002/lary.21415. [DOI] [PubMed] [Google Scholar]
- 77.Lebrin F, Srun S, Raymond K, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16(4):420–428. doi: 10.1038/nm.2131. [DOI] [PubMed] [Google Scholar]
- 78.Simonds J, Miller F, Mandel J, Davidson TM. The effect of bevacizumab (Avastin) treatment on epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 2009;119(5):988–992. doi: 10.1002/lary.20159. [DOI] [PubMed] [Google Scholar]
- 79.Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948–955. doi: 10.1001/jama.2012.250. [DOI] [PubMed] [Google Scholar]
- 80.Mitchell A, Adams LA, MacQuillan G, Tibballs J, van den Driesen R, Delriviere L. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl. 2008;14(2):210–213. doi: 10.1002/lt.21417. [DOI] [PubMed] [Google Scholar]
- 81.High KA. The gene therapy journey for hemophilia: are we there yet? Blood. 2012;120(23):4482–4448. doi: 10.1182/blood-2012-05-423210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.ACOG Committee on Practice Bulletins–Obstetrics ACOG Practice Bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, Jan 2002. Obstet Gynecol. 2002;99(1):159–167. doi: 10.1016/s0029-7844(01)01747-1. [DOI] [PubMed] [Google Scholar]
- 84.Hutcheon JA, Lisonkova S, Joseph KS. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol. 2011;25(4):391–403. doi: 10.1016/j.bpobgyn.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 85.MacKay AP, Berg CJ, Atrash HK. Pregnancy-related mortality from preeclampsia and eclampsia. Obstet Gynecol. 2001;97(4):533–538. doi: 10.1016/s0029-7844(00)01223-0. [DOI] [PubMed] [Google Scholar]
- 86.Maynard SE, Karumanchi SA. Angiogenic factors and preeclampsia. Semin Nephrol. 2011;31(1):33–46. doi: 10.1016/j.semnephrol.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. 2007;335(7627):974. doi: 10.1136/bmj.39335.385301.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Odegard RA, Vatten LJ, Nilsen ST, Salvesen KA, Austgulen R. Risk factors and clinical manifestations of pre-eclampsia. BJOG. 2000;107(11):1410–1416. doi: 10.1111/j.1471-0528.2000.tb11657.x. [DOI] [PubMed] [Google Scholar]
- 89.Sibai BM, Ramadan MK, Usta I, Salama M, Mercer BM, Friedman SA. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome) Am J Obstet Gynecol. 1993;169(4):1000–1006. doi: 10.1016/0002-9378(93)90043-i. [DOI] [PubMed] [Google Scholar]
- 90.Duckitt K, Harrington D. Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. BMJ. 2005;330(7491):565. doi: 10.1136/bmj.38380.674340.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Redman CW. Platelets and the beginnings of preeclampsia. N Engl J Med. 1990;323(7):478–480. doi: 10.1056/NEJM199008163230710. [DOI] [PubMed] [Google Scholar]
- 92.Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, van Asshe A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101(8):669–674. doi: 10.1111/j.1471-0528.1994.tb13182.x. [DOI] [PubMed] [Google Scholar]
- 93.Zhou Y, Damsky CH, Fisher SJ. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest. 1997;99(9):2152–2164. doi: 10.1172/JCI119388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Y, Fisher SJ, Janatpour M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99(9):2139–2151. doi: 10.1172/JCI119387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salafia CM, Pezzullo JC, Ghidini A, Lopez-Zeno JA, Whittington SS. Clinical correlations of patterns of placental pathology in preterm pre-eclampsia. Placenta. 1998;19(1):67–72. doi: 10.1016/s0143-4004(98)90100-x. [DOI] [PubMed] [Google Scholar]
- 96.Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res. 2004;95(9):884–891. doi: 10.1161/01.RES.0000147365.86159.f5. [DOI] [PubMed] [Google Scholar]
- 97.Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;294(2):H541–H550. doi: 10.1152/ajpheart.01113.2007. [DOI] [PubMed] [Google Scholar]
- 99.Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA. 1993;90(22):10705–10709. doi: 10.1073/pnas.90.22.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kapur NK, Heffernan KS, Yunis AA, et al. Elevated soluble fms-like tyrosine kinase-1 levels in acute coronary occlusion. Arterioscler Thromb Vasc Biol. 2011;31(2):443–450. doi: 10.1161/ATVBAHA.110.215897. [DOI] [PubMed] [Google Scholar]
- 101.Clark DE, Smith SK, He Y, et al. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol Reprod. 1998;59(6):1540–1548. doi: 10.1095/biolreprod59.6.1540. [DOI] [PubMed] [Google Scholar]
- 102.Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355(10):992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 103.Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350(7):672–683. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 104.Valbuena-Diez AC, Blanco FJ, Oujo B, et al. Oxysterol-induced soluble endoglin release and its involvement in hypertension. Circulation. 2012;126(22):2612–2624. doi: 10.1161/CIRCULATIONAHA.112.101261. [DOI] [PubMed] [Google Scholar]
- 105.Jerkic M, Rivas-Elena JV, Prieto M, et al. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004;18(3):609–611. doi: 10.1096/fj.03-0197fje. [DOI] [PubMed] [Google Scholar]
- 106.Lopez-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2010;299(4):H959–H974. doi: 10.1152/ajpheart.01251.2009. [DOI] [PubMed] [Google Scholar]
- 107.Gu Y, Lewis DF, Wang Y. Placental productions and expressions of soluble endoglin, soluble fms-like tyrosine kinase receptor-1, and placental growth factor in normal and preeclamptic pregnancies. J Clin Endocrinol Metab. 2008;93(1):260–266. doi: 10.1210/jc.2007-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension. 2009;53(2):399–403. doi: 10.1161/HYPERTENSIONAHA.108.123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cudmore M, Ahmad S, Al-Ani B, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115(13):1789–1797. doi: 10.1161/CIRCULATIONAHA.106.660134. [DOI] [PubMed] [Google Scholar]
- 110.Bergmann A, Ahmad S, Cudmore M, et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J Cell Mol Med. 2010;14(6B):1857–1867. doi: 10.1111/j.1582-4934.2009.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Z, Zhang Y, Ying Ma J, et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension. 2007;50(4):686–692. doi: 10.1161/HYPERTENSIONAHA.107.092098. [DOI] [PubMed] [Google Scholar]
- 112.Suzuki H, Ohkuchi A, Matsubara S, et al. Effect of recombinant placental growth factor 2 on hypertension induced by full-length mouse soluble fms-like tyrosine kinase 1 adenoviral vector in pregnant mice. Hypertension. 2009;54(5):1129–1135. doi: 10.1161/HYPERTENSIONAHA.109.134668. [DOI] [PubMed] [Google Scholar]
- 113.Thadhani R, Kisner T, Hagmann H, et al. Pilot study of extracorpo-real removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124(8):940–950. doi: 10.1161/CIRCULATIONAHA.111.034793. [DOI] [PubMed] [Google Scholar]
- 114.George EM, Cockrell K, Aranay M, Csongradi E, Stec DE, Granger JP. Induction of heme oxygenase 1 attenuates placental ischemia-induced hypertension. Hypertension. 2011;57(5):941–948. doi: 10.1161/HYPERTENSIONAHA.111.169755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69(4):1517–1526. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 116.Overall CM, Kleifeld O. Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 117.Cowie MR, Mosterd A, Wood DA, et al. The epidemiology of heart failure. Eur Heart J. 1997;18(2):208–225. doi: 10.1093/oxfordjournals.eurheartj.a015223. [DOI] [PubMed] [Google Scholar]
- 118.Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics – 2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):e21–e181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 119.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117(3):568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74(2):207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 121.Ritzenthaler JD, Goldstein RH, Fine A, Smith BD. Regulation of the alpha 1(I) collagen promoter via a transforming growth factor-beta activation element. J Biol Chem. 1993;268(18):13625–13631. [PubMed] [Google Scholar]
- 122.Qiao B, Padilla SR, Benya PD. Transforming growth factor (TGF)-beta-activated kinase 1 mimics and mediates TGF-beta-induced stimulation of type II collagen synthesis in chondrocytes independent of Col2a1 transcription and Smad3 signaling. J Biol Chem. 2005;280(17):17562–17571. doi: 10.1074/jbc.M500646200. [DOI] [PubMed] [Google Scholar]
- 123.Leask A, Holmes A, Black CM, Abraham DJ. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem. 2003;278(15):13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- 124.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32(10):1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 125.Chuva de Sousa Lopes SM, Feijen A, Korving J, et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231(3):542–550. doi: 10.1002/dvdy.20162. [DOI] [PubMed] [Google Scholar]
- 126.Frantz S, Hu K, Adamek A, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008;103(5):485–492. doi: 10.1007/s00395-008-0739-7. [DOI] [PubMed] [Google Scholar]
- 127.Kuwahara F, Kai H, Tokuda K, et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106(1):130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 128.Kapur NK, Heffernan KS, Yunis AA, et al. Usefulness of soluble endoglin as a noninvasive measure of left ventricular filling pressure in heart failure. Am J Cardiol. 2010;106(12):1770–1776. doi: 10.1016/j.amjcard.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yanavitski M, Givertz MM. Novel biomarkers in acute heart failure. Curr Heart Fail Rep. 2011;8(3):206–211. doi: 10.1007/s11897-011-0065-5. [DOI] [PubMed] [Google Scholar]
- 130.Peterson JT, Li H, Dillon L, Bryant JW. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res. 2000;46(2):307–315. doi: 10.1016/s0008-6363(00)00029-8. [DOI] [PubMed] [Google Scholar]
- 131.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 132.Cruz-Gonzalez I, Pabon P, Rodriguez-Barbero A, et al. Identification of serum endoglin as a novel prognostic marker after acute myocardial infarction. J Cell Mol Med. 2008;12(3):955–961. doi: 10.1111/j.1582-4934.2008.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kapur NK, Shenoy C, Yunis AA, et al. Distinct effects of unfractionated heparin versus bivalirudin on circulating angiogenic peptides. PLoS One. 2012;7(4):e34344. doi: 10.1371/journal.pone.0034344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nachtigal P, Zemankova Vecerova L, Rathouska J, Strasky Z. The role of endoglin in atherosclerosis. Atherosclerosis. 2012;224(1):4–11. doi: 10.1016/j.atherosclerosis.2012.03.001. [DOI] [PubMed] [Google Scholar]