

Abstract
Introduction
In Europe a disease is recognized as rare if less than 1 in 2000 people suffer from the specific disease. In patients with familial homozygous hypercholesterolemia (HoFH) the accumulation of low-density lipoprotein cholesterol (LDL-C) leads to generalized atherosclerosis due to an insufficient functioning of the LDL-C receptors. Patients die early sometimes even in the mid-30s, from myocardial infarction or stroke. For the German population, insufficient epidemiological evidence exists.
Methods
A systematic literature search in EMBASE and Medline was performed in conjunction with a targeted manual search for epidemiological HoFH studies. Additionally a nationwide survey was conducted in Germany in all identified apheresis- and lipid centers. The purpose of the survey was the validation of the systematic literature search results based on empirical (practice) data.
Results
In total 961 publications were found, 874 were excluded based on pre-defined exclusion criteria leaving only 87 for further review. After review of the identified abstracts (n = 87) 23 publications were identified as epidemiological studies. Only one publication was found which reported a prevalence of 1:1,000,000. The qualitative survey among 187 physicians in Germany also revealed a low prevalence: 95 HoFH patients were identified in 35 centers.
Conclusion
The estimated frequency of homozygous familial hypercholesterolemia patients in Germany is around 95 (1:860,000) and the disease should be recognized as rare according to the definition of the European Medical Agency.
Keywords: epidemiology, homozygous familial hypercholesterolemia (HoFH), Germany, survey
Introduction
The European Medicine Agency (EMA) defines a disease as orphan if the disease is life-threatening or a chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union (EU) at the time of submission of the designated drug application to the EMA. Additionally there must also be either no satisfactory method of diagnosis, prevention or treatment of the condition concerned being authorized, or, if such a method does exists, the treatment must be of significant benefit to those affected by the condition.1
Homozygous familiar hypercholesterolemia (HoFH) is a rare, inherited, genetic disorder affecting the low density lipoprotein cholesterol (LDL-C) metabolism, which results in greatly elevated blood cholesterol levels. It is most commonly caused by mutations in both alleles of the LDL receptor (LDLR) gene, but may potentially also be caused by mutations in the ApoB gene or proprotein convertase subtilisin/kexin type 9 gene. Mutations of the LDLR gene account for 85% to 90% of cases.2 Patients with HoFH may have two identical mutations (true homozygote) or two different mutations (compound heterozygote).3 HoFH is related to the more common disorder heterozygous familial hypercholesterolemia (HeFH). In HeFH, only one copy of the affected gene carries a mutation, preserving a variable level of normal LDL-C metabolism, which typically leads to less elevated LDL-C levels and a less severe disease course relative to HoFH.4
HoFH is an autosomal codominant disease,5 such that both parents of a homozygote patient must have at least one copy of the relevant mutation and are thus HeFH or HoFH patients themselves. The mutations which cause HoFH lead to failure of the removal of ApoB-containing lipoprotein particles and LDL-C from the blood by the LDLR5 resulting in an accumulation of LDL-C to levels far exceeding those in the general population; total cholesterol levels can range from 650 to 1000 mg/dL,3 with LDL-C levels > 500 mg/dL in untreated patients with HoFH. As HoFH is a disease affecting patients’ daily life it can be assumed that all patients receive treatment.
Exposure to chronically high cholesterol levels leave HoFH patients highly vulnerable to the development of atherosclerosis and cardiovascular disease (CVD).6 Rates of CVD are not only very high, but CVD can develop at a very early age in HoFH patients. For example, evidence of atherosclerosis, coronary heart disease and aortic stenosis may become detectable already during childhood.7,8 HoFH patients have a greatly reduced life expectancy, with a large proportion of patients suffering fatal coronary insufficiency or myocardial infarction before the age of 30 years.7,8
The excessively high levels of LDL-C associated with HoFH lead to visible signs of cholesterol build-up (xanthoma) in the skin and joints as well as around tendons, especially at the Achilles tendon and tendons of the fingers. Cholesterol depositions in the cornea may be present leading to a characteristic white-ringed appearance of the iris (arcus cornealis).4
HoFH is an extremely rare orphan disease, the exact prevalence of which is unknown. However it is thought to affect one person per million,3 which would lead to an estimated 82 patients in Germany, based on current estimates of the German population in 2012. However, the epidemiological data for Germany is insufficient and only little epidemiological evidence is known.
Methods
A systematic literature search was performed utilizing the MEDLINE and Embase databases. Before the search the following research frame using the PICOS (Patient, Intervention, Comparison, Outcome, Setting) criteria was defined:
Patient: familial homozygous hypercholesterolemia (HoFH).
Intervention: epidemiological studies.
Comparator: not applicable.
Outcome: epidemiology (prevalence and/or incidence of HoFH).
Setting: Germany.
The combined searches of MEDLINE and Embase yielded 961 unique abstracts; additional articles were identified upon evaluation of the bibliographies of review articles.
In the first level of screening, each abstract was evaluated independently by one investigator for inclusion in the study according to the inclusion and exclusion criteria. Any decision to exclude a study was reviewed by a second investigator. Any discrepancies in these decisions were reviewed and resolved by a third investigator. For abstracts that were deemed relevant during the first level of review, full articles were retrieved and reviewed.
Inclusion criteria for abstracts selected for retrieval were studies published in English and German language evaluating humans, published since 1980, reporting outcomes relevant to the epidemiology for HoFH. Studies including different groups, such as patients with heterozygous familial hypercholesterolemia (HeFH), were allowed provided the data were reported separately for the HoFH population. Exclusion criteria included articles published prior to 1980 and studies without epidemiological outcomes. If studies only included HeFH patients they were excluded. In vitro studies; animal; fetal; molecular; genetic; and pharmacokinetics/pharmacodynamics studies; narrative reviews published prior to May 2009; expert opinions; case reports; meeting abstracts; and editorials were excluded from this review.
Information from the full text articles reporting clinical outcomes related to treatment of HoFH were extracted and abstracted by a single investigator, with a second senior investigator validating the extracted information independently; any differences were resolved by discussion with a third investigator.
In order to get a complete and supplementary epidemiological overview of HoFH patients in Germany, a survey was conducted in Germany in all identified apheresis- and lipid centers. The purpose of the survey was the validation of the systematic literature search results based on empirical data. As a starting point all centers treating HoFH patients were identified before the survey start. As a second step all apheresis centers and lipid ambulances for which contact details could be identified were contacted (one priority group (14 contacts), one apheresis center group (103 contacts), one lipid ambulance group (45 contacts), for a total 162 contacts). All contacts were contacted first by email, and followed up by phone if they did not respond to emails. This direct contact by telephone helped enlarge the contact database by 25 physicians to a total of 187.
Results
Of the 961 abstracts reviewed, 207 articles were selected for further review in full text, and 754 were excluded (see Figure 1). Of the 207 full text articles reviewed, 23 of the identified articles consisted of epidemiological information (overview of the 23 articles available on request).
Figure 1.
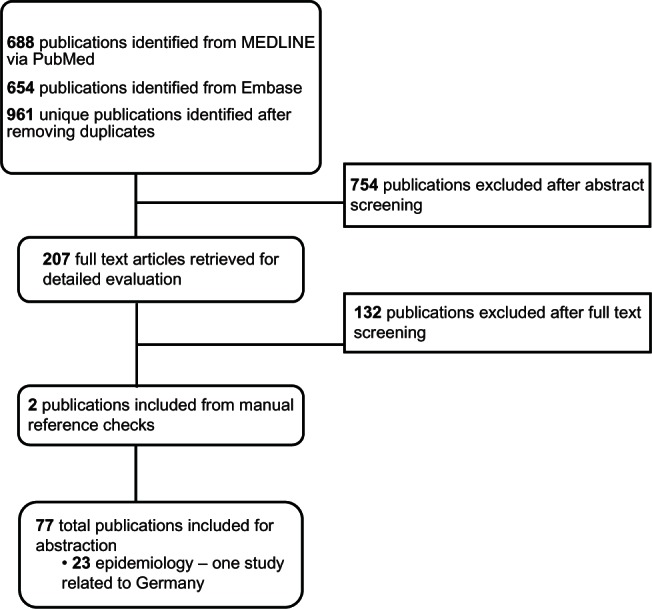
Literature search process for epidemiology evidence of homozygous familial hypercholesterolemia (HoFH) in Germany.
The prevalence of HoFH is often cited as 1:1,000,000 in the general population; however, our review identified only one citation with this figure, another citation supporting and citing this prevalence was found by the related hand search.3,9 Six other publications10–15 identified in this review, describe higher estimates for HoFH prevalence. All were cross-sectional surveys of HoFH patients, in varying regions of the world, with estimates of HoFH prevalence ranging from 1:30,000 in a cross-sectional survey of a South African population,14 to 1:640,000 from a survey of 10,889 Dutch individuals.12 The estimates in this review suggest a higher general prevalence of HoFH than the 1:1,000,000; however, the role of founder effects and observational bias cannot necessarily be discounted.
Our qualitative survey with 187 physicians in Germany also demonstrated a low prevalence: 95 HoFH patients were identified. The reported HoFH patient numbers for the defined four German regions are visualized within Figure 2.
Figure 2.
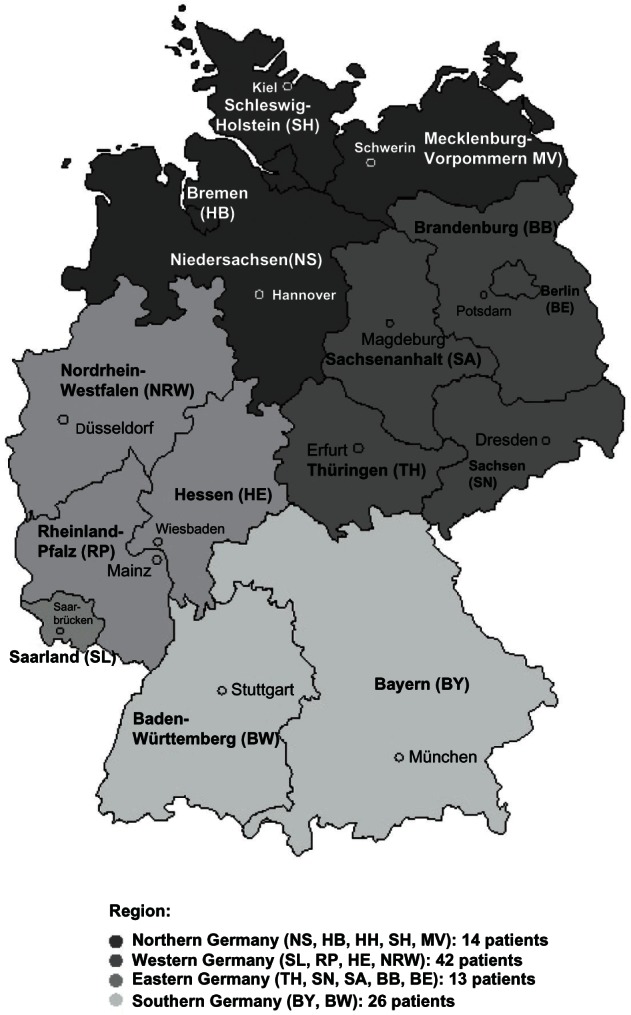
German HoFH patients in the different regions according to the qualitative survey.
Discussion
Patients with this rare and severe disease have only limited treatment options available. Besides very limited effective therapies (lipid-lowering drugs such as statins, diet) LDL-apheresis in conjunction with a maximum lipid-lowering drug therapy and diet is accepted in Germany as the standard of care for those patients. Based on this treatment paradigm the qualitative survey was developed around the German apheresis and lipid-ambulance centers in order to recruit and contact physicians to define a thorough epidemiological basis of HoFH in Germany. This approach has had the advantage of optimizing the likelihood of catching the majority of HoFH patients in Germany for the survey. As potential limitations the normal drawbacks of any qualitative approach apply, especially the potential reporting bias by physicians or the possibility of patient duplicate counting due to the possible treatment of one patient in an apheresis center and a lipid-ambulance around the same time.
Specifically for this research the following limitations apply: There might be double-counts among the 95 identified HoFH patients as no validation or formal check was being done. Hence this bias could have an effect in decreasing the actual number of patients. Additionally a few centers did not respond to the survey which may result in a potential increase in the estimated number of patients, if they did in-effect have patients with HoFH. Furthermore some physicians did not respond consistently to the survey as they responded as having HoFH patients in the screening phase of the project but did not provide patient data during the data collection phase.
Otherwise, to the best of our knowledge this is the first scientific attempt to define the epidemiological basis of homozygous familial hypercholesterolemia patients in Germany.
Conclusion
The estimated frequency of homozygous familial hypercholesterolemia patients in Germany is around 95 (1:860,000) and the disease should hence be recognized as a rare one according to the definition of the European Medical Agency. Further research in order to identify the correct number of HoFH patients in Germany is warranted.
Acknowledgment
The study was supported by an unrestricted grant from Aegerion Pharmaceuticals Inc. The company played no role in the execution of the study or in data collection, data management, data analysis, and interpretation of the data.
Footnotes
Disclosure
Walzer, Trevers, Rieder and Erazo-Fischer received funding for consultancy from Aegerion Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
- 1.European Medicines Agency (EMA) Definition of orphan diseases. [webpage on the Internet] Available from: http://www.emea.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000029.jsp&mid=WC0b01ac05800240ceAccessed August 6, 2012
- 2.Goldberg AC, Hopkins PN, Toth PP, et al. Familial Hypercholesterolemia: Screening, diagnosis and management of pediatric and adult patients. J Clin Lipidol. 2011;5:S1–S8. doi: 10.1016/j.jacl.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Marais AD. Familial hypercholesterolaemia. Clin Biochem Rev. 2004;25(1):49–68. [PMC free article] [PubMed] [Google Scholar]
- 4.Raal FJ, Pilcher GJ, Veller MG, Kotze MJ, Joffe BI. Efficacy of vitamin E compared with either simvastatin or atorvastatin in preventing the progression of atherosclerosis in homozygous familial hypercholesterolemia. Am J Cardiol. 1999;84(11):1344–1346. doi: 10.1016/s0002-9149(99)00570-6. [DOI] [PubMed] [Google Scholar]
- 5.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111(12):1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeg JM, Feuerstein IM, Tucker EE. Detection and quantitation of calcific atherosclerosis by ultrafast computed tomography in children and young adults with homozygous familial hypercholesterolemia. Arteriosclerosis, Thrombosis and Vascular Biology. American Heart Association. 1994 Jul;14(7):1066–1074. doi: 10.1161/01.atv.14.7.1066. [DOI] [PubMed] [Google Scholar]
- 7.Graesdal A, Bogsrud MP, Holven KB, et al. Apheresis in homozygous familial hypercholesterolemia: The results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. J Clin Lipidol. 2012;6(4):331–339. doi: 10.1016/j.jacl.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102(11):1438–1443. doi: 10.1016/j.amjcard.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 9.Richter WO. Die familiäre Hypercholesterinämie. [Familial hypercholesterolemia] Lipidreport. 1995;1:5–14. [Google Scholar]
- 10.Fahed AC, Safa RM, Haddad FF, et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol Gen Met. 2011;102(2):181–188. doi: 10.1016/j.ymgme.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Mabuchi H, Nohara A, Noguchi T, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214(2):404–407. doi: 10.1016/j.atherosclerosis.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Kusters DM, Huijgen R, Defesche JC, et al. Founder mutations in the Netherlands: geographical distribution of the most prevalent mutations in the low-density lipoprotein receptor and apolipoprotein b genes. Neth Heart J. 2011;19(4):175–182. doi: 10.1007/s12471-011-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moorjani S, Roy M, Gagne C, et al. Homozygous familial hypercholesterolemia among French Canadians in Quebec Province. Arteriosclerosis. 1989;9(2):211–216. doi: 10.1161/01.atv.9.2.211. [DOI] [PubMed] [Google Scholar]
- 14.Seftel HC, Baker SG, Sandler MP, et al. A host of hypercholesterolaemic homozygotes in South Africa. Br Med J. 1980;281(6241):633–636. doi: 10.1136/bmj.281.6241.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slimane MN, Lestavel S, Sun X, et al. Fh-Souassi: a founder frameshift mutation in exon 10 of the LDL-receptor gene, associated with a mild phenotype in Tunisian families. Atherosclerosis. 2001;154(3):557–565. doi: 10.1016/s0021-9150(00)00572-4. [DOI] [PubMed] [Google Scholar]