

SUMMARY
L3mbtl2 has been implicated in transcriptional repression and chromatin compaction but its biological function has not been defined. Here we show that disruption of L3mbtl2 results in embryonic lethality with failure of gastrulation. This correlates with compromised proliferation and abnormal differentiation of L3mbtl2−/− embryonic stem (ES) cells. L3mbtl2 regulates genes by recruiting a Polycomb Repressive Complex1 (PRC1)-related complex, resembling the previously described E2F6-complex, and including G9A, Hdac1, and Ring1b. Presence of L3mbtl2 at target genes is associated with H3K9-dimethylation, low histone-acetylation, and H2AK119-ubiquitination, but the latter is neither dependent on L3mbtl2 nor sufficient for repression. Genome wide studies revealed that the L3mbtl2-dependent complex predominantly regulates genes not bound by canonical PRC1 and PRC2. However, some developmental regulators are repressed by the combined activity of all three complexes. Together, we have uncovered a highly selective, essential role for an atypical PRC1-family complex in ES cells and early development.
INTRODUCTION
In early mammalian development, primitive ectoderm cells forming the inner cell mass of blastocysts are pluripotent and subsequently give rise to all cells of the body. The pluripotent state can be maintained in blastocyst-derived embryonic stem (ES) cells ex vivo, and can be reprogrammed in somatic cells, generating induced pluripotent stem (iPS) cells (Orkin and Hochedlinger, 2011). In pluripotent cells, the expression of differentiation-related genes is suppressed despite a globally open chromatin that allows for access to genes required for rapid self-renewal and for the subsequent initiation of differentiation programs (Orkin and Hochedlinger, 2011). The stem cell program is orchestrated by three core transcription factors, Oct4, Nanog, and Sox2. These factors cooperate with a network of molecules that includes other transcription factors, non-coding RNAs, transcriptional coactivators, corepressors, and chromatin regulators (Young, 2011). However, despite important progress, the molecular mechanisms underlying early development and pluripotency remain incompletely understood.
Gene regulation in pluripotent stem cells is dependent on the activity of several multiprotein complexes that influence transcription by modifying chromatin, including Polycomb Repressive Complexes (PRCs) (Simon and Kingston, 2009). PRCs regulate pattering by repressing Hox genes in Drosophila development, and also control developmental genes in mammalian ES cells (Simon and Kingston, 2009). Two types of mammalian Polycomb complexes are known, PRC1 and PCR2. The composition of these complexes varies in different contexts, and, recently, distinct PRC1-family complexes have been defined (Gao et al., 2012). Germline disruption of components of PRC2 is invariably associated with early embryonic lethality, while Ring1b and Rybp are the only PRC1 components with essential early embryonic roles (Suppl. Table S1). PRC1 and PRC2 act coordinately in ES cells, and disruption of components of either complex leads to derepression of lineage-specific genes. The PRC2 component EZH2 mediates trimethylation of histone 3 lysine 27 (H3K27me3). H3K27me3 serves as a docking site for a PRC1 chromodomain protein (Drosophila: polycomb; mammalian CBX2/4/6/7/8) at a large proportion of PRC2 target genes. Ring1b, the catalytic PRC1 subunit, then mediates mono-ubiquitination of histone 2A lysine 119 (H2AK119ub1). It is unresolved how these modifications mediate gene repression, and Ring1b has the potential to repress genes and compact chromatin independent of histone ubiquitination (Simon and Kingston, 2009). Both mammalian PRCs can also act independently. Repression of some genes is maintained by the remaining complex after selective disruption of either PRC1 or PRC2 (Leeb et al., 2010), and PRC1 can be recruited to PRC2 target genes in PRC2’s absence (Tavares et al., 2012). L3mbtl2 has been implicated as a component of the repressive E2F6-complex that contains Polycomb group proteins also linked to PRC1 in several independent studies (Ogawa et al., 2002; Sanchez et al., 2007; Tahiliani et al., 2007; Trojer et al., 2011).
Malignant Brain Tumor (MBT)-domains participate in the organization of DNA in chromatin by binding histones and compacting chromatin (Trojer et al., 2007). Characteristically, they display a strong preference for binding to mono-and dimethylated lysine residues in histone tails. MBT-domains were first recognized in a family of three genes in Drosophila comprised of l(3)mbt, scm, and sfmbt, all of which are essential for development. Scm and sfmbt strongly repress Hox genes in Drosophila and bind Polycomb responsive elements (Klymenko et al., 2006; Wang et al., 2010). However, neither molecule is a core constituent of Drosophila PRC1 or PRC2 (Klymenko et al., 2006; Wang et al., 2010). In mammals, the family of MBT-domain proteins has expanded to nine members and their functions are less well-defined (Qin et al., 2010). We have previously shown that mice lacking L3mbtl1 are viable (Qin et al., 2010). Similarly, Scmh1−/− mice display only mild phenotypes with variable penetrance (Takada et al., 2007), and disruption of Sfmbt1 in mice is not associated with obvious phenotypes (J.Q., H.H., unpublished data). Mice lacking L3mbtl3 and Mbtd1 die at birth with skeletal defects and compromised hematopoiesis (Arai and Miyazaki, 2005; Honda et al., 2011). However, none of these knockout models have revealed cell types for which MBT-domain proteins are strictly essential, and no requirement in embryonic development has been described. Here we examine the function of L3mbtl2, an orthologue of Drosophila sfmbt also known as h-l(3)mbt-like or m4mbt (Guo et al., 2009).
RESULTS
L3mbtl2 is essential for mouse development
L3mbtl2 is widely expressed (Fig. 1A). We disrupted L3mbtl2 in ES cells and mice by flanking the exons encoding its three C-terminal MBT-domains (residues 315 – 308) with loxP sites and removing them by Cre-mediated recombination (Fig. 1B, Suppl. Fig. S1). This strategy ensured absence the fourth MBT-domain, which binds methylated histones (Guo et al., 2009), and likely resulted in a true “null” allele because we could not detect mutant mRNA (Fig. 1C) or mutant protein (Fig. 2A). Mice heterozygous for the disrupted L3mbtl2 allele appeared normal and were fertile. However after intercrossing heterozygotes, no homozygous pups were born (Fig. 1 D, E). At embryonic day (E) 6.5, L3mbtl2−/− embryos were present at the expected frequency, and their genotype could not be predicted by inspection (Fig. 1D, E). In contrast, at and after E7.5, mutant embryos showed growth retardation (Fig. 1 D, E).
Figure 1. Arrested embryonic development in the absence of L3mbtl2.
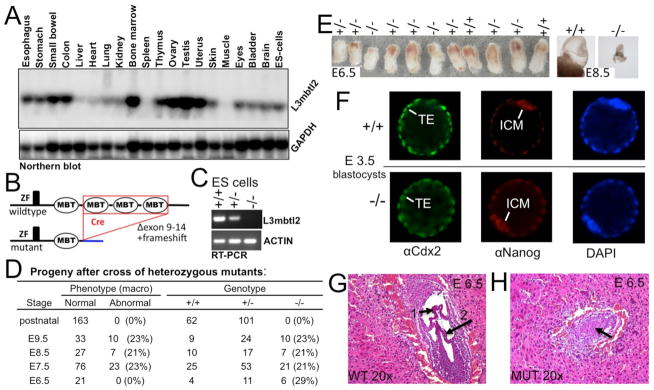
(A) L3mbtl2 is widely expressed. Northern blot analysis of RNA from selected tissues. (B) Illustration of L3mbtl2 conditional allele (for details see Suppl. Fig. S1). (C) RT-PCR analysis of mRNA for L3mbtl2 using primers in the 5-prime region (not deleted in the genome) demonstrates that mutant mRNA is non-detectable and likely unstable (see Fig. 2A for analysis of protein). (D, E) Progeny of heterozygous mutant mice bearing germline-excised L3mbtl2. (D) No homozygous mutants were detected after birth. At macroscopic examination upon dissection at E6.5, L3mbtl2−/− embryos appeared roughly normal in size (E, left). Following E7.5, mutants were growth retarded. At E8.5, mutant embryos were minute (E, right), and at E9.5, only debris was recovered from decidua. (F) Immunohistological analysis of L3mbtl2+/+ and L3mbtl2−/− blastocysts at E3.5 revealed that both form trophectoderm (TE, expressing Cdx2) and inner cell mass (ICM, expressing Nanog). (F, G) Histologic sections from uteri at E6.5 show wildtype embryos with well-defined cavities segmented by chorion (F, arrow 1) and amnion (F, arrow 2) membranes and mutant embryos with unstructured cores (G, arrow) (for further histology see Suppl. Fig. S2).
Figure 2. L3mbtl2 is a critical regulator of ES cell self-renewal.

(A) Cre-mediated disruption of L3mbtl2 results in loss of protein expression. An antiserum against the (non-deleted) N-terminus reacts with a band of the expected size (~ 84kD, 703aa) in protein lysates from L3mbtl2 floxed (+/+) but not excised (−/−) ES cells (first and second lane, upper panel). Note that no mutant protein band is detected (predicted mutant protein: ~45kD from 314 aa encoded by exons 1–7 and 64 aa from abnormal residues after frameshift). Lentiviral expression of FLAG-tagged L3mbtl2 (L3mbtl2-F) results in protein levels comparable to endogenous protein (lanes 1 and 4). (B) L3mbtl2−/− ES cell colony size is strikingly reduced, but fully restored by lentiviral expression of L3mbtl2-F. Shown is colony-size six days after seeding single cell suspensions onto MEF-feeder layers in the presence of LIF. Bar graphs show the mean largest diameter of 20 random ES cell colonies. (C) Growth rates of L3mbtl2−/− ES cell cultures are severely decreased, but are completely rescued by lentiviral expression of L3mbtl2-F (difference between +/+ and −/− on day 6 ⇒ p<0.001). (D, E, F) STAT3-phosphorylation in response to LIF, expression alkaline phosphatase and pluripotency factor proteins. (G) SSEA1 is expressed at reduced levels in L3mbtl2−/− ES cells. Lentiviral expression of L3mbtl2-F restores SSEA1 to normal levels (% SSEA1 expression above threshold, black vertical bar); green histogram shows isotype control. (H) Colony-formation potential of L3mbtl2−/− ES correlates with SSEA1 expression. (I) L3mbtl2−/− ES cells do not show increased apoptosis (representative plots; numbers are from 3 experiments). (J) Loss of L3mbtl2 is associated with diminished G1-S transition. Normal cell cycle is restored by lentiviral L3mbtl2-F. Note that the proportion of cells in the G0/1 phase of the cell cycle is almost doubled (p< 0.001) and the proportion of cells in S is reduced (p<0.001) after L3mbtl2 loss (representative plots, numbers are from three experiments). (K, L) Western blot analysis (K) and real-time quantitative RT-PCR of ES cells (L) show abnormal expression of regulators of the G1-S transition in the absence of L3mbtl2. Cyclin E was reduced and its inhibitors p21 and Lats2, increased. The expression levels of cyclin A, p16, and p53 were not altered. Shown is relative mRNA expression (ratio: measured mRNA/GAPDH mRNA). (M) Chromatin immunoprecipitation with L3mbtl2-F followed by qPCR revealed enrichment for the promoter regions of p21, but not cyclin E, Lats2 or HPRT (shown are means in % of input; p21 binding was also confirmed by ChIP-seq, Suppl. Table S4). (B–M) Significance: two-tailed Student t-tests; means were derived from three biologically independent samples; errors are standard deviations (SD). See Suppl. Fig. S3 for analysis of L3mbtl2−/− MEFs.
Immunohistological analysis of blastocysts at embryonic day (E) 3.5 revealed no differences of trophectoderm (Cdx2+) and inner cell mass (Nanog+) (Fig. 1F). To investigate later effects of L3mbtl2 loss, we analyzed serial histological sections of whole uteri (Fig. 1G, H, Suppl. Fig. S2). At E6.5, mutant embryos were surrounded by mural trophectoderm and a normal outer epithelial layer of primitive endoderm (Suppl. Fig. S2C, D). However, the core of the mutant embryos consisted of an abnormal, unstructured mass of irregular cells (Fig. 1H, Suppl. Fig. S2D). Normal embryos at the egg cylinder stage (E5.5) harbor an inner ectodermal layer surrounding the proamniotic cavity. The latter becomes divided as the chorion and amnion develop (Fig. 1G, Suppl. Fig. S2C). At E6.5, L3mbtl2 mutants did not show a distinct ectodermal epithelial layer, proamniotic cavity, chorion, or amnion (Fig. 1H, Suppl. Fig. S2D). While wildtype E8.5 embryos had progressed in establishing the basic body plan as a result of gastrulation, mutant embryos showed little growth or development (Suppl. Fig. S2E, F, G, H). Thus, L3mbtl2 is not required for implantation or formation of trophectoderm, primitive endoderm, and the inner cell mass. However in its absence, the inner cells mass fails to form a normal primitive ectoderm capable of gastrulation. This knockout phenotype bears similarity with those associated with other Polycomb group proteins (Suppl. Table S1).
L3mbtl2 regulates ES cell proliferation, but is not required for maintenance of ES cell identity
The embryonic phenotype suggested that L3mbtl2 might be required for the function of pluripotent cells of the inner cell mass, which give rise to ES cells ex vivo. Therefore, we disrupted both alleles of L3mbtl2 in ES cells (Suppl. Fig. S1E). This resulted in complete loss of L3mbtl2 protein (Fig. 2A). Upon loss of L3mbtl2, ES cell colony-size was drastically reduced (Fig. 2B), and we observed a severe proliferation defect (Fig. 2C, doubling time increased from ~13 hours to ~33 hours). Remarkably, however, lentiviral expression of FLAG-tagged L3mbtl2 entirely restored normal ES cell growth and protein expression (Fig. 2A, B, C). Importantly, despite expression of L3mbtl2 in murine embryonic fibroblasts (MEFs, E13.5), its disruption did not alter their growth (Suppl. Fig. S3). Thus, L3mbtl2 selectively regulates proliferation in the context of ES cells.
Despite these growth abnormalities, L3mbtl2−/− ES cells retained characteristics of pluripotent cells. Stat3 phosphorylation in response to LIF was not altered (Fig. 2D), and L3mbtl2−/− ES cells expressed Alkaline Phosphatase (Fig. 2E) as well as the pluripotency factors Oct4, Nanog, and Sox2 at normal levels (Fig. 2F). The pluripotency marker SSEA1 was expressed by L3mbtl2−/− ES cells, but its levels were reduced in a proportion of cells (Fig. 2G). L3mbtl2−/− ES cells with diminished SSEA1 expression had a significantly reduced capacity to give rise to ES cell colonies, but L3mbtl2−/− ES cells with high SSEA1 levels gave rise to colonies almost as efficiently as wildtype ES cells (Fig. 2H). Moreover, we were able to stably maintain L3mbtl2−/− ES cells in culture for >36 passages (not shown). Thus, L3mbtl2−/− ES cells are subject to a higher propensity for spontaneous differentiation, but unlimited self-renewal is preserved, albeit at a drastically reduced rate.
The impaired growth of L3mbtl2−/− ES cells was not due to increased apoptosis (Fig. 2I), but correlated with an altered cell cycle profile (Fig. 2J). This was associated with markedly increased proportions of cells in the G0/1-phases of the cell cycle and a reduction of cells in S-phase. Consistently, expression of cyclin E, which promotes transition from the G1- to the S-phase, was reduced (Fig. 2K, L) and its inhibitors p21cip1/waf1 and Lats2 were increased (Fig. 2K, L). Expression levels of p53, a transcriptional activator of p21cip1/waf1, were not altered. Intriguingly, Oct4 loss results in a similar G1/S block with p53-independent up-regulation of p21cip1/waf1 (Lee et al., 2010). Remarkably, ChIP analysis revealed binding of L3mbtl2 to the proximal promoter p21cip1/waf1, but not Lats2 or cyclin E (Fig. 2M, confirmed by ChIP-seq, Suppl. Table S4). Thus, p21cip1/waf1 is a direct transcriptional target of L3mbtl2 in ES cells.
L3mbtl2 controls differentiation programs in ES cells and early development
We utilized EBs as a model system of early development (Fig. 3A). L3mbtl2−/− ES cells did form EBs, but these were smaller and failed to develop cystic structures (Fig. 3B, day 12). L3mbtl2−/− EBs expressed markers for definitive ectoderm (Fig. 3C, left, top) mesoderm (Fig. 3C, left, middle) and endoderm (Fig. 3C, bottom)(see Suppl. Experimental Procedures for references), However, the expression of markers for all germ layers was abnormal (Fig. 3C, left). Strikingly, we detected increased expression of Sox17, Gata6, Gata4, and Foxa2 in undifferentiated L3mbtl2−/− ES cells and throughout EB culture, indicating that L3mbtl2 suppresses endoderm (Fig. 3C, bottom). In normal EB maturation, early populations (mesoendoderm and primitive ectoderm) fade as more mature tissues are formed (Shen et al., 2009). Accordingly, brachyury (mesoendoderm marker, left middle), Fgf5 (primitive ectoderm marker, Fig. 3C, right, top) as well as Oct4 and Nanog (pluripotent cell markers, Fig. 3C, right, top), were highly expressed in wildtype day 7 EBs, but strikingly reduced at day 12. In contrast, these markers persisted abnormally in L3mbtl2−/− EBs, suggesting that primitive ectoderm and mesoendoderm cells did not efficiently progress in maturation. Finally, we observed increased expression of Eomes, Cdx2, and Hand1 (Fig. 3C, right, bottom, particularly at day 12), suggesting that L3mbtl2 also suppresses the trophectoderm.
Figure 3. L3mbtl2 is a critical regulator of ES cell differentiation.

(A–C) Embryoid body (EB) formation and differentiation. (A) Scheme of experiment; EBs were formed in hanging drops and subsequently maintained in rotating cultures. (B) ES cells lacking L3mbtl2 retained the potential to form EBs after LIF-withdrawal (B, day 3, left column), but EBs were growth-retarded (b, day 7, middle column) and not did develop cystic structures like wildtype EBs (B, day 12, right columns). Bar graphs on the right show mean diameters of 20 EBs from cultures shown on the left. (C) Differentiation and pluripotency gene expression analysis by real-time RT-PCR of mRNA derived from EBs. Marker genes for all germ layers were expressed in mutant and wildtype EBs, but gene expression was highly abnormal in the absence of L3mbtl2. Note that endodermal marker genes were significantly overexpressed at baseline and at later time points in L3mbtl2−/− EBs. Trophectodermal markers were highly overexpressed in day 7 and day 12 in L3mbtl2−/− EBs. On day 12, the expected decrease in Oct4, Nanog, Fgf5 and Brachyury expression was delayed in L3mbtl2−/− EBs. Shown is mean relative mRNA expression (ratio of measured mRNA/ to GAPDH mRNA). (D–K) Analysis of teratomas after subcutaneous injection of ES cells. (D) Scheme of experiment. (E) ES cells lacking L3mbtl2 formed teratomas after an increased latency period, but grew normally after lentiviral rescue with L3mbtl2-F. (F–K) L3mbtl2−/− teratomas displayed poor differentiation, but contained tissue elements derived from all germlayers (histology after Hematoxylin/Eosin stain). (F, G) At low power, wildtype teratomas appeared more pleomorphous than mutant teratomas. The mutant tissue predominantly featured monomorphous “blue” areas (G, left; see also Suppl. Fig. S4B, C, note cells with bizarre nuclei, frequent mitosis and little cytoplasm) and monomorphous areas resembling mature nervous system tissue (G, right, see also Suppl. Fig. S4B, D). (K) Areas resembling choriocarcinoma and containing trophoectodermal giant cells were also frequent in L3mbtl2−/− tumors. Such areas were found in 5/5 L3mbtl2−/− but in 0/5 wildtype teratomas (K, Suppl. Fig. S4M–O). In addition, areas of differentiation into skin (H, ectoderm), cartilage (I, mesoderm), and gut (J, endoderm) could be demonstrated but were very rare compared to wildtype teratomas. (B–E) Significance was determined with two-tailed Student t-tests; means are from three independent samples; error bars represent SD. More histological analysis is shown in Suppl. Fig. S4; for synopsis of findings after knockout of other Polycomb group proteins see Suppl. Table S1.
To assess whether L3mbtl2−/− ES cells give rise to more differentiated cells, we assayed teratoma formation (Fig. 3D–K). Remarkably, growth of L3mbtl2−/− teratomas was delayed for several weeks (Fig. 3E). By comparison, teratomas lacking Eed or Ring1b form after a normal interval, even though they 50% smaller (Leeb et al., 2010). In histologic analysis, L3mbtl2−/− teratomas featured a striking paucity of mature elements (Fig. 3F, G, Suppl. Fig. S4). Yet, rare examples of differentiation into tissues from all three germlayers could be found (mesoderm: cartilage, smooth muscle (Fig. 3I, Suppl. Fig. S4G, H); ectoderm: skin, nervous system tissue (Fig. 3H, Suppl. Fig. S4E, F); endoderm: gut (Fig. 3J, Suppl. Fig. S4I, J). Thus, L3mbtl2 is not required for the later development of many diverse cell types. However, the most abundant components of L3mbtl2−/− teratomas were undifferentiated areas, containing cells of (likely) epithelial origin (Fig. 3G-left, Suppl. Fig. S4B, C, I–L), and areas resembling mature neural tissue (Fig. 3G-right, Suppl. Fig. S4B, C). L3mbtl2−/− teratomas also exhibited trophectoderm differentiation (Fig. 3K, Suppl. Fig. S4M–O), which is never seen in control teratomas. Together, these data reveal a complex role for L3mbtl2 in early development. It is not essential for the potential to give rise to derivatives of all three germlayers (pluripotency)(Fig. 3C). However, it is required for suppressing endodermal and trophectodermal genes in ES cells and EBs. Moreover, L3mbtl2 promotes differentiation programs: early cell populations, expressing Oct4, Fgf5, and Brachyury, persist in late stage L3mbtl2−/− EBs (Fig. 3C) and both L3mbtl2−/− teratomas and EBs lack abundant differentiated elements (Fig. 3B, F, G).
L3mbtl2 physically interacts with factors mediating gene repression and pluripotency
To investigate the molecular basis of L3mbtl2’s role in ES cells, we searched for proteins interacting with lentiviral vector-expressed, FLAG-tagged L3mbtl2 in rescued knockout ES cells by mass spectrometry (Fig. 4A, B). We identified peptides representative of 39 proteins involved in transcriptional regulation and chromatin organization (listed in Fig. 4C). Surprisingly, 23 of these (Fig. 4C, last column, Suppl. Table S2) have previously been shown to interact with pluripotency factors or have been implicated in self-renewal and/or differentiation of pluripotent stem cells. The list included Oct4 and we could verify this interaction by co-immunoprecipitation (CoIP)(Fig. 4D), reverse CoIP in rescued ES cells (Fig. 4G), and by CoIP of the endogenous proteins in wildtype ES cells (Fig. 4H). The interactions of L3mbtl2 with Oct4 and other pluripotency factors may explain its selective role in ES cells.
Figure 4. L3mbtl2 physically interacts with components of repressive transcriptional complexes and molecular regulators of ES cell self-renewal and differentiation.

(A–C) Identification of L3mbtl2-associated proteins by mass spectrometry. (A) Scheme of experiment. (B) Proteins isolated by affinity purification revealed by Coomassie Blue stain. Four gel slices per lane (as indicated on by lines, right border) were processed for analysis in each experiment. (C) List of L3mbtl2-F interacting proteins with known roles in transcriptional regulation. Columns indicate name, International Protein Index identifier, number of peptides obtained in two independent experiments, known roles in NuRD complex (red), E2F6-complex (blue), Polycomb-complexes (green), identification as transcription factor, transcription coregulator, chromatin regulator (grey), and known role in ES cell biology (see Suppl. Table S2). (D–H). (D–F) Analysis of selected protein interactions by co-immunoprecipitation (IP) from nuclear extracts of L3mbtl2-F–rescued knockout ES cells followed by Western blot (WB) analysis using antibodies against the indicated proteins. (D) L3mbtl2-F is associated with Oct4, but not Sox2 or Polycomb group proteins Bmi1, Suz12, or YY1. (E, F) Confirmation of L3mbtl2-F’s association with components of the NuRD (E) and E2F6 complexes (F). (G) Reverse Co-IP with antiserum against Ring1B, Mbd3, Hdac1, and Oct4 followed by Western blot analysis with αFLAG (L3mbtl2-F); (G, right) interaction is not diminished in the presence of ethidium bromide (50μg/ml). (H). Immunoprecipitation of candidate interactors followed by Western blot with an antiserum against L3mbtl2 (top), and antisera directed at the immunoprecipitated proteins (bottom 8 panels, IP controls). Note that endogenous L3mbtl2 interacts with Ring1B, E2F6, Oct4 and Hdac1. See Suppl. Fig. S5 for Western blot analysis of expression levels of selected interactors.
Our analysis also identified proteins that participate in several multi-protein complexes. These included 10 constituents of the E2F6-complex (blue in Fig. 4C), known to interact with L3mbtl2 (Ogawa et al., 2002). Three E2F6-complex components (Ring1a, Yaf2, and GLP) were not detected. We also detected all components of the NuRD-complex (red in Fig. 4C) and multiple Swi/Snf-complex components (Fig. 4C). We confirmed the interaction of L3mbtl2-F with 7 members of the NuRD-complex and 5 members of the E2F6-complex by CoIP (Fig. 4D, E). However, the CoIP with Mi2β, a central NuRD component, was weak (Fig. 4E). The interactions with members of complexes do not necessarily implicate L3mbtl2 as part of these complexes as they may be indirect. Nevertheless, we showed that key interactions (Ring1b, Mbd3, Hdac1, and Oct4) are not the result of proximity at promoters, as they were stable in the presence of ethidium bromide, which disrupts DNA bridges (Fig. 4G).
Candidate L3mbtl2 interactors also included Polycomb group proteins, specifically Ring1b, Rybp, Pcgf6, Rbbp4, and Rbbp7 (Fig. 4C) (Simon and Kingston, 2009). Rbbp4 and Rbbp7, confirmed by CoIP (Fig. 4E), were the only subunits of PRC2 detected by mass spectrometry (Fig. 4C). Consistent with this, we failed to CoIP L3mbtl2 with Suz12 or Ezh2 (Fig. 4H). Moreover, analysis of L3mbtl2−/− ES cells showed no differences in Ezh2 protein (Suppl. Fig. S5A) or global changes in H3K27me3 levels (Suppl. Fig. 5B). Thus, L3mbtl2 does not interact with PRC2.
L3mbtl2 interacts with the E2F6-complex that harbors three proteins also linked to PRC1 (Ring1b, Pcgf6/Mblr, and Rybp, Fig. 4C). We confirmed the interaction with Rybp (Fig. 4F) and Ring1b (Fig. 4F, G, H) by CoIP. However, we did not detect peptides derived from Bmi1, known to associate with Ring1b at most PRC1 target genes in ES cells (Ku et al., 2008). We also failed to detect an interaction of L3mbtl2 with Bmi1 by CoIP. Disruption of Ring1b in ES cells is associated with loss of protein expression of other PRC1 components, including Rybp and Bmi1 (see Suppl. Table S1). However, the protein expression levels of Ring1b, Rybp, and Bmi1 were unchanged (Suppl. Fig. S5A) and so were the global levels of H2AK119ub1 (Suppl. Fig. S5B). Importantly, we detected Ring1b after IP with either E2F6 or Bmi1 (Fig. 4H, 6th row). However, Bmi1 was only detectable after IP with Ring1b, but not E2F6 (Fig. 4H, bottom). These data suggest that two separate Ring1b-containing complexes co-exist in ES cells, the canonical PRC1-complex that contains Bmi1 and the E2F6-complex that does not contain Bmi1. Only the latter interacts with L3mblt2 in ES cells.
Both zinc finger and MBT-domain are essential for the function of L3mbtl2
To explore the molecular function of L3mbtl2’s domains, we generated mutants and tested their potential to rescue self-renewal and differentiation (Fig. 5). Deletion of each of L3mbtl2’s four MBT domains ablated its ability to rescue the colony-growth defect (Fig. 5A). In contrast, combined mutations of four highly conserved amino acid residues, critical for the binding of methylated histones (Guo et al., 2009), had no effect on L3mbtl2’s potential to rescue proliferation (Fig. 5B, bottom) or expression of SSEA1 (Fig. 5C, bottom). Remarkably however, deletions of the atypical C2C2 zinc finger domain (Fig. 5A) and even point mutations of the zinc-coordinating residues abolished L3mbtl2’s potential to rescue proliferation and differentiation (Fig. 5 B, C). This type of zinc finger, which is also found in Drosophila scm and polyhomeotic is not thought to confer sequence-specific DNA binding (Wang et al., 2010) and may be involved in binding RNA. The proliferation defects in L3mbtl2−/− ES cells were not rescued by any of the eight other MBT-protein family members (Suppl. Fig. S6A). Moreover, hybrid proteins with exchanged MBT-domains and N-terminal portions of L3mbtl2 and Mbtd1 were inactive (Suppl. Fig. S6B). We also determined the subcellular localization of mutant proteins lacking either the zinc finger or the MBT-domains. Both types of mutants retained a predominantly nuclear localization similar to wildtype L3mbtl2, but were detected at markedly reduced levels (zinc finger) or were absent (MBT) in the chromatin-bound fraction (Fig. 5E). Similarly, both the zinc finger domain and the MBT domains were required for efficient binding Hdac1, G9A, and Ring1b (Fig. 5F).
Figure 5. Structure/function analysis of L3mbtl2 in ES cells. (A, B).

Shown are representative colonies six days after sorting of lentiviral vector infected L3mbtl2−/− cells for GFP. (A) Lentiviral expression of mutants with deleted individual or combined MBT domains failed to rescue colony growth. Likewise, deletion of the zinc-finger domain of L3mbtl2 disrupted function. (B) Point mutations in the zinc-finger domain abrogated the potential to rescue colony-growth (top two panels), but a mutant with four altered amino acid residues critical for the methyl-binding pocket of the fourth MBT-domain did not impair the potential to rescue colony growth. (C) Mutants with altered amino acids in the zinc finger domain did not rescue SSEA1 expression of L3mbtl2−/− ES cells, but mutants with four point mutations in the methyl-binding pocket rescued similar to wildtype L3mbtl2-F. (D) Western blot analysis confirmed protein expression of all mutants. (E) Western blot analysis of L3mbtl2-F and deletion mutants of zinc finger, and MBT domains showed highly selective enrichment of L3mbtl2-F in nuclear and chromatin-bound extracts. Deletion of the zinc finger domain markedly reduced, and deletion of the MBT domains abolished signal in chromatin-fraction but not the nuclear fraction. Lower panels show controls for the purity of the subcellular fractions. (F) Deletion of the zinc finger or MBT-domains of L3mbtl2 prevent efficient co-immunoprecipitation with Hdac1 (top), G9A (second), and Ring1b (third panel) in nuclear extracts. Note that wildtype and mutant L3mbtl2 are expressed at similar levels (bottom). See Suppl. Fig. S6 for analysis of rescue-potential of other MBT protein family members.
The majority of genes bound and repressed by L3mbtl2 in ES cells are not bound by canonical PRC1 and PRC2
We determined the impact of L3mbtl2 on genome wide mRNA expression. Microarray analysis revealed 1280 genes with >2-fold altered expression levels in L3mbtl2−/− compared to wildtype ES cells (Fig. 6A, column 1 and 4). Conversely expression was changed > 1.5-fold in the opposite direction upon (re)-expression of L3mbtl2-F in 69.5% of these genes (L3mbtl2-F vector infected cells compared with control vector infected cells; Fig. 6A, column 2 and 3). Together, these criteria defined a set of 890 L3mbtl2-regulated genes (Suppl. Table S3). 645 (72.5%) genes were up-regulated in the absence of L3mbtl2 while only 245 genes (27.6%) were down-regulated (Suppl. Table S3). Of note, we confirmed the reversible pattern of gene expression in 10/10 regulated targets by quantitative PCR (not shown).
Figure 6. Genome wide analysis of L3mbtl2-dependent gene expression and L3mbtl2 gene occupancy.

(A) Expression of L3mbtl2-F in L3mbtl2−/− ES cells reverses gene expression changes associated with L3mbtl2 disruption. Microarray analysis of mRNA expression. Color code for the degree of expression changes is shown at the bottom. Columns show 1280 bars that represent genes with > 2-fold expression differences between L3mbtl2+/+ (first) and L3mbtl2−/− ES cells (fourth). Second and third columns represent analysis of L3mbtl2−/− ES cells infected with L3mbtl2-F or control lentiviral vector. Note that the pattern of L3mbtl2-F infected cells resembles L3mbtl2+/+ ES cells while mock-infected cells resemble L3mbtl2−/− ES cells. See Suppl. Table S3 for 890 genes that were differentially expressed and rescued. (B) A large proportion of genomic L3mbtl2-binding sites are localized at known core promoters. Genome wide distribution of binding sites relative to annotated genes was determined by ChIP-seq. See Suppl. Table S4 for details on 4009 genes that bind close to genes (+/− 10,000 bp from transcriptional start site, TSS). (C) L3mbtl2 occupies active and bivalent genes. Top: shown are L3mbtl2 binding sites (black lines) at active genes (green), bivalent genes (yellow), and silent genes (orange) (defined in Suppl. Experimental Methods, online). Bottom: Distribution of Suz12 is shown for comparison. Note that Suz12 almost exclusively binds to bivalent genes whereas L3mbtl2 also occupies active genes. (D) L3mbtl2 is essential for repression of ~5% of the genes it binds in ES cells. Venn diagram illustrating the overlap of genes bound by L3mbtl2 and genes up-regulated in the absence of L3mbtl2 (L3mbtl2-repressed); see Suppl. Tables S3, S4 for detailed information. (E) L3mbtl2 co-occupies a considerable minority of high confidence combined PRC1 and PRC2 target genes. Venn diagram illustrating the overlap of L3mbtl2-bound genes with a gene set of PRC1/2 target genes derived from published experiments; see Suppl. Tables S5, S4 for details. (F) Most genes that are both L3mbtl2-bound and repressed are not co-occupied by PRC1 and PRC2. Venn diagram; gene sets from Suppl. Table S5, S4. (G) Gene ontology categories associated with the 167 genes that were both bound and repressed by L3mbtl2. See Suppl. Table S6 for information on L3mbtl2-regulated germ cell genes.
Next, we investigated the genome wide localization of L3mbtl2-F in L3mbtl2−/− ES cells by ChIP-seq. L3mbtl2 was found to bind to 5188 genomic sites that showed no signal in the control. ~84% of these were located close to known genes and 36% in core promoters (Fig. 6B, Suppl. Table 4). Remarkably, > 60% of the L3mbtl2-bound genes are transcriptionally active. These findings contrast the binding pattern of the Polycomb protein Suz12 (Fig. 6C, (Marson et al., 2008)), which is almost exclusively bound to bivalent genes. In agreement with our biochemical results (Fig. 4), a genome wide comparison showed that significant proportions of L3mbtl2’s binding sites are shared with Ring1b (32%, p<10−300, GEO number: GSE#22680) and there is also overlap with Oct4 (10%, (Marson et al., 2008)), Mi2β (4%, (Whyte et al., 2012)) (data not shown).
Only ~5% of the L3mbtl2-bound targets were up-regulated after L3mbtl2 loss (Fig. 6D). This suggests redundancy in repression of L3mbtl2-bound genes and is roughly comparable with findings after loss of Ring1b and Eed, which results in up-regulation of only 10–13% or 18% of Polycomb-bound genes, respectively (Leeb et al., 2010; van der Stoop et al., 2008).
To assess the overlap of gene occupancy by L3mbtl2, we defined a gene set of high-confidence, combined PRC1- and PRC2-bound genes (referred to as PRC1/2-bound, below). PRC1/2-bound genes were defined as binding PRC1 or PRC2 components in ≥4 of 6 genome wide analyses reported in two studies (Suppl. Table S5, (Boyer et al., 2006; Ku et al., 2008)); genes that bind Ring1b in the context of the E2F6-complex should not meet these criteria. Remarkably, ~29% (266) of the PRC1/2-bound genes were also bound by L3mbtl2 (Fig. 6E, Suppl. Table S3). However, only 3% (28) of the PRC1/2-bound genes were L3mbtl2-bound and were up-regulated after L3mbtl2 loss (Fig. 6F, Suppl. Table S3). Thus, L3mbtl2 binds a high proportion of PRC1/2 targets, but is not required for repression of most of these genes. Conversely, most of the L3mbtl2-bound genes that were up-regulated after L3mbtl2 loss were not PRC1/2-bound (139/83%) (Fig 6F, Suppl. Table S3). Moreover, none of the ten genes most highly up-regulated (>10-fold) after L3mbtl2 loss was PRC1/2-bound (Suppl. Table S3). Thus, L3mbtl2 predominantly binds and regulates genes in ES cells that are not canonical, combined PRC1 and PRC2 targets.
Finally, we used the David classification tool to identify gene ontology categories associated with the 167 genes both bound and repressed by L3mbtl2. The analysis revealed strong associations with several categories consistent with the phenotype we observed, as well as with “spermatogenesis” (Fig. 6H). Consistent with this, nine of the twelve genes most strikingly regulated genes (7.5 to 74-fold, Suppl. Table S3) had roles in germ cells (Suppl. Table S6).
L3mbtl2 generates and maintains repressive chromatin modifications at its target genes by recruiting a repressive multi-protein complex
L3mbtl2 disruption in ES cells was not associated with global expression changes of its interactors Ring1b, Hdac1, and G9A (Suppl. Fig. 5A). Likewise, the chromatin-modifications altered by these enzymes were not globally changed (i.e. H2AK119ub1, histone 3 and histone 4 acetylation (H3ac and H4ac), and H3K9 mono- and dimethylation (H3K9me1, me2; Suppl. Fig. S5)). To investigate the local impact of L3mbtl2 on chromatin modifications at promoters, we probed a panel of target genes (Fig. 7), selected from genome wide studies (Suppl. Tables S3–5). Their differential expression and L3mbtl2-binding status were confirmed by RT-qPCR and ChIP-qPCR (Fig. 7A, B). The panel included five L3mblt2-bound germ cell genes (Ddx4, Tex19, Piwil2, Stk31, Tcam1, orange dotted boxes, see Suppl. Table S6) and two regulated targets that do not bind L3mbtl2 (Stra8, Cidea) (Fig. 7A, B, left). We also studied four combined PRC1/2 targets (blue dotted boxes), two of which are L3mbtl2-bound (Sox17, Foxa2; note: these genes bind lower, but significant levels of L3mbtl2) and three of which are L3mbtl2-repressed (Sox17, Foxa2, HoxB8) (Fig. 7A, B).
Figure 7. L3mbtl2 represses transcription and recruits chromatin-modifying enzymes that counteract histone-acetylation and enhance H3K9-methylation at promoters.

(A) Expression analysis by quantitative real-time RT-PCR of RNA from ES cells. Germ cell genes are marked by dotted blue boxes; canonical PRC1/2 targets are marked by dotted orange boxes. Note: all genes except for Eomes and HPRT were expressed at higher levels in L3mbtl2−/− ES cells. Shown is relative mRNA expression (ratio: measured mRNA/GAPDH mRNA). (B) ChIP of L3mbtl2-F followed by quantitative PCR analysis. Direct target genes (marked in bold print in A–D) showed significantly higher signal in L3mbtl2-F-rescued compared with mock-infected knockout ES cells. (C) Loss of L3mbtl2 at target genes correlates with increased H3- and H4-actetylation as well as reduced H3K9-dimethylation, but does not affect H2AK119-ubiquitination. ChIP of histone modifications followed by qPCR analysis of target gene DNA in ES cells. Left, top: ChIP with IgG control antiserum. Left, second and third panel: ChIP analysis of H3ac and H4ac. Right, top three panels: ChIP analysis of H3K9me1, H3K9me2, and H3K9me3. Bottom, left: ChIP of H3K27me3: note marked enrichment at canonical PRC1/2 targets and slight enrichment at germ cell targets (smallest difference: Tex19 and Foxa2 = 4.3-fold, p<0.05). Bottom, right: ChIP for H2AK119ub1: note that canonical PRC1/2 targets show markedly higher signal than germ cell targets (e.g., difference: STK31 and Foxa2 = 10.1-fold, p<0.01). (D) L3mbtl2 is not required for recruitment of PRC1 and PRC2, but recruits Ring1b, G9A, Hdac1, and E2F6. Top: ChIP of Bmi1 and Suz12. Middle, left: ChIP of Ring1b: note strong enrichment at canonical PRC1/2 targets and much weaker enrichment at germ cell targets (smallest difference: Ddx4 and Sox17 = 11.3-fold, p<0.01); also note that enrichment after Ring1b ChIP was significantly reduced after L3mbtl2 loss at a several L3mbtl2-bound germ cell genes, but not at canonical PRC1/2 targets. Middle, right and bottom left: ChIP of G9A and Hdac1. Bottom, right: ChIP of E2F6. (See also Suppl. Fig. S7) (A–D) Bar graphs represent the mean of 3 independent biological samples and SD. Significance: two-tailed Student t-test. E: Scheme depicting likely components of the L3mbtl2-associated protein complex at target genes. Known chromatin-modifying activities associated with individual components are indicated by color-code.
ChIP analysis of H3ac and H4ac revealed that histone acetylation at L3mbtl2-bound germ cell target promoters increased significantly after disruption of L3mbtl2 (Fig. 7C, left, middle two panels), consistent with reduced local activity of a histone deacetylase, such as Hdac1 (Fig. 4E). Moreover, H3K9me2 was detected at all L3mbtl2-bound targets and was strongly diminished after L3mbtl2 loss (Fig. 7C, right, second panel). H3K9me1 was diminished at some targets after L3mbtl2 loss (Fig. 7C, right, top panel). These changes likely reflect decreased activity of G9A (Shinkai and Tachibana, 2011). Notably, H3K9me3, a mark not dependent on G9A, was not significantly changed at any of the target genes (Fig. 7C, right, third panel). As expected, we detected high levels of H3K27me3 (the mark of PRC2/Ezh2) and H2AK119ub1 (the mark of PRC1/Ring1b) at combined PRC1/2 targets (Fig. 7C, bottom, left). In contrast, at germ cell genes, which are bound by L3mbtl2 but not PRC1/2, we detected only very low levels of both marks (Fig. 7C, bottom). Neither mark was appreciably affected by the absence of L3mbtl2 at combined PRC1/2 targets. However, at some germ cell gene promoters, the low level of H3K27me3 was significantly reduced after L3mbtl2 disruption (Fig. 7C, bottom, left). As these genes do not bind PRC2, the low level of H3K27me3 may be generated by G9A (Shinkai and Tachibana, 2011). In contrast, the low levels of H2A119ub1 at germ cell genes were entirely unaffected by L3mbtl2 loss (Fig. 7C, bottom right).
Next, we probed promoters for the presence of key components of chromatin modifying complexes. As expected, Bmi1, a hallmark of canonical PRC1 in ES cells (Ku et al., 2008), and Suz12, an essential component of PRC2 (Boyer et al., 2006), were strongly enriched at high-confidence PRC1/2 targets (Fig. 7D, top two panels). These data correlate with high levels of H3K27me3 and H2AK119ub1 (Fig. 7C, bottom, left). Enrichment was not diminished after disruption of L3mbtl2. Thus, L3mbtl2 is not required for the recruitment or activity of PRC1 or PRC2 at combined PRC1/2-bound targets, including those that bind L3mbtl2 (Sox17 and Foxa2). As expected, neither Bmi1 nor Suz12 were detected at L3mbtl2-bound germ cell genes, confirming the absence of canonical PRC1 and PRC2.
ChIP analysis of Ring1b showed enrichment of both PRC1/2 targets and germ cell genes (Fig. 7D, middle, left). However, enrichment was at least 10-fold lower at germ cell genes (p<0.001). Disruption of L3mbtl2 did not reduce enrichment at high-confidence PRC1/2 targets, but significantly reduced enrichment at germ cell genes. Notably however, Ring1b was not entirely lost at germ cell genes after L3mbtl2 disruption, but was merely reduced and sufficient to maintain H2AK119ub1 at normal levels (Fig. 7C, bottom, right). These data demonstrate two different modes of binding of Ring1b to promoters in ES cells: high-level binding at PRC1/2 targets that is independent of L3mbtl2, and low-level binding at germ cell genes that is partially dependent on L3mbtl2.
G9A and Hdac1 were strongly enriched at all germ cell gene promoters (Fig. 7D, middle right and bottom). For both, enrichment was strikingly reduced after L3mbtl2 loss. This correlated with increased histone acetylation and reduced H3K9me2 (Fig. 7C). We did not detect enrichment for either enzyme at PRC1/2 targets, including Sox17 and FoxA2 (Fig. 7D). As L3mbtl2 binds the latter two genes at comparatively at low levels (Fig. 7B), it is possible that G9A and Hdac1 were present at these genes at low levels, not detected by our assay.
Our data show that L3mbtl2 loss at target genes results in reduction or loss of three enzymes (Ring1b, G9A, Hdac1) that have been linked to a protein complex associated with E2F6 in somatic cells. Consistent with this, ChIP analysis revealed that E2F6 bound to all five L3mbtl2-bound germ cell genes (Fig. 7D) and its binding was reduced after L3mbtl2 disruption (Fig. 7D). Thus, L3mbtl2 is recruits a repressive complex to target genes in ES cells that bears strong resemblance with the previously described E2F6-complex (Ogawa et al., 2002) (Fig. 7E).
DISCUSSION
Our study reveals essential roles for L3mbtl2 in early embryonic development and pluripotent stem cells. The consequences of L3mbtl2 loss include embryonic failure after implantation, compromised ES cell proliferation, failure to suppress differentiation gene expression in ES cells, and reduced competence for normal differentiation within EBs and teratomas. Our genomic studies revealed that L3mbtl2 loss in ES cells results in reversible gene expression changes of ~ 900 genes. Thus, the complex phenotypes observed here may depend on the cooperation of multiple, possibly hundreds, of genes. Nevertheless, several striking aspects of the phenotype correlate with functions of individual, direct L3mbtl2 targets: First, L3mbtl2 directly binds to and represses the p21Cip1/waf1 promoter, and p21Cip1/Waf1 overexpression is known to block G1 to S-phase transition (Lee et al., 2010), precisely as we observed it in L3mbtl2−/− ES cells. Second, L3mbtl2 binds and represses Foxa2 and Sox17, which may contribute to the increased bias towards endoderm differentiation in L3mbtl2−/− EBs. Third, L3mbtl2 binds and represses Lefty2 (Suppl. Fig S7A, Supp. Table S3), which inhibits gastrulation by antagonizing Nodal. In addition, overexpression of Cdx2, an indirect L3mbtl2 target, may explain the trophoectoderm bias we observed in L3mbtl2−/− EBs and teratomas (Niwa et al., 2005).
Aspects of the phenotype after L3mbtl2 loss resemble findings after disruption of other Polycomb group proteins. For example, embryos lacking Ezh2, Eed, Suz12, and Ring1b exhibit gastrulation failure (Simon and Kingston, 2009)(Suppl. Table S1). Likewise, increased expression of endoderm genes (Fig. 3) has been observed in ES cells lacking Ring1b, Eed, and Suz12 (Suppl. Table S1). Moreover, like L3mbtl2−/− ES cells, Ring1b−/− ES cells fail to repress trophectoderm genes (Suppl. Table S1). Finally, like L3mbtl2−/− EBs (Fig. 3), EBs lacking Eed, Ezh2 and Jarid2 fail to extinguish primitive populations expressing Oct4, Fgf5, or Brachyury (Suppl. Table S1). Despite these similarities, none of the phenotypes associated with loss of Polycomb group proteins phenocopy our findings (Suppl. Table S1). Perhaps the most unique feature is L3mbtl2’s strong impact on proliferation. Among the Polycomb group proteins, only Eed is associated with an appreciable (but less severe) ES cell growth defect upon gene disruption (Leeb et al., 2010) (Suppl. Table S1).
Our proteomic analysis revealed that L3mbtl2 interacts with known constituents of multiprotein complexes, including the NuRD-(Whyte et al., 2012) and E2F6-complexes (Ogawa et al., 2002) as well as with some components of the Polycomb complexes PRC1 (Ring1b, Rybp, Pcgf6) and PRC2 (Rbbp4, Rbbp7) (Simon and Kingston, 2009). We focused our analysis on deciphering the relationship of L3mbtl2 with other Polycomb complexes.
We demonstrate that L3mbtl2 functions independently of PRC2 in ES cells. L3mbtl2 is not necessary for recruitment or maintenance of PRC2. The majority of PRC2 target genes do not bind L3mbtl2, and L3mbtl2 loss does not diminish H3K27me3 or Suz12 binding at genes bound by both L3mbtl2 and PRC2. Moreover, L3mbtl2 binding is insufficient for recruitment of PRC2, as most genes bound and regulated by L3mbtl2 are not bound by PRC2. In agreement with this, L3mbtl2 did not biochemically interact with PRC2 core components. Despite their independence, L3mbtl2 and PRC2 cooperate in the repression of some genes. For example, Sox17 and Foxa2 are not only up-regulated after L3mbtl2 loss, but also after loss of the PRC2 component Eed (Leeb et al., 2010).
Very recently, PRC1 in human somatic cells has been resolved into six distinct groups of complexes (PRC1.1-1.6) with mutually exclusive ring finger components (PCGF1-6), distinct associated proteins, and genomic localization (Gao et al., 2012). PRC1.2 and 1.4 (containing MEL18/PCGF2 and BMI1/PCGF4, respectively) are most similar to the canonical PRC1 that occupies combined PRC1/2 targets (Boyer et al., 2006; Ku et al., 2008), while L3mbtl2 is associated with PRC1.6 (containing MBLR/PCGF6) (Gao et al., 2012). The notion that “PRC1” represents multiple entities dovetails with our findings. First, both Bmi1 (PRC1.4) and L3mbtl2 (PRC1.6) interacted with Ring1b, but not with each other. Second, L3mbtl2 (PRC1.6) binds only a minority of combined PRC1/2 target genes (PRC1.2, 1.4). Third, loss of L3mbtl2 (PRC1.6) at target genes that are also combined PRC1/2 targets (PRC1.2, 1.4) did not decrease binding of Bmi1, Ring1b, or H2AK119ub1. Finally, in contrast to canonical PRC1 (PRC1.2, 1.4), the majority of L3mbtl2-bound and -repressed genes do not bind PRC2. Together, these data show L3mbtl2 is neither a component of “canonical” PRC1 in ES cells nor required for its stability or enzymatic activity.
Instead, L3mbtl2 is a constituent of a PRC1-family complex in ES cells that has been previously designated E2F6-complex, PRC1L4, or PRC1.6 in somatic cells and was detected in mass spectrometry experiments with E2F6, RING1B, SMCX, and L3MBTL2 (Gao et al., 2012; Ogawa et al., 2002; Sanchez et al., 2007; Tahiliani et al., 2007; Trojer et al., 2011). A biological function of this complex has not been demonstrated. Our data strongly suggest that this PRC1-family complex is tightly linked to the essential requirement for L3mbtl2 in ES cells. We demonstrate that L3mbtl2 is required for optimal recruitment of at least four likely components of this complex to selected germ cell genes (Fig. 7E). Moreover, these promoters bear the marks of the three associated enzymes, H3K9me2 (G9a), low H3ac, H4ac (Hdac1), and H2AK119ub1 (Ring1b). Notably, we detected the latter mark and Ring1b at 10-fold lower levels than that at canonical PRC1 targets. Disruption of L3mbtl2 resulted in markedly increased H3ac and H4ac as well as reduced H3K9me2, but, in contrast to data in somatic cells (Trojer et al., 2011), did not affect H2AK119ub1. Since L3mblt2 loss at promoters is associated with increased target gene expression without changing H2AK119ub1 levels, this mark alone is insufficient to maintain repression. However, Ring1b and/or H2AK119ub1 are likely necessary for optimal L3mbtl2-mediated repression because the germ cell genes regulated by L3mbtl2 in ES cells were shown to be at least partially de-repressed in Ring1b−/− ES cells ((Leeb et al., 2010) and Suppl. Table. S6). Notably, the importance of G9A and Hdac1 in ES cells and early development has also been demonstrated by gene targeting (Dovey et al., 2010; Shinkai and Tachibana, 2011). In addition to histone modification, chromatin compaction by Ring1b (Simon and Kingston, 2009) and L3mbtl2 itself (Trojer et al., 2011) likely contribute to a repressive chromatin structure. Together, our data suggest that L3mbtl2 organizes repressive chromatin at critical targets in ES cells by supporting the assembly and maintenance of an evolved PRC1-family complex that utilizes mechanisms not classically linked with Polycomb proteins (Fig. 7E). Like L3mbtl2, Ring1b and Rybp are essential for development (Suppl. Table S1) and both are constituents of all types of PRC1-complexes (Gao et al., 2012). In contrast, disruption of Bmi1/PCGF4 and Mel18/PCGF2 was not embryonic lethal even in combination (Akasaka et al., 2001). Intriguingly, therefore, the L3mbtl2-associated complex may be the only PRC1-family complex that is essential for early development.
L3mbtl2 is a homologue of Drosophila sfmbt (Guo et al., 2009), which binds the DNA-binding transcription factor pho, a recruiter of Drosophila polycomb complexes to specific genomic sites (Klymenko et al., 2006). L3mbtl2 does not bind to YY1, the mammalian homologue of pho (Fig. 4), but it interacts with E2F6, Mga/Max (binding both E-box motifs and T-box motifs (Ogawa et al., 2002)), and with Oct4 (Fig. 4). However, we did not find a dominant role of these factors in recruitment of L3mbtl2. Oct4 co-occupies 10% of L3mbtl2-bound genomic sites, and conversely, L3mbtl2 is present at only 2% of Oct binding sites, so the two factors are primarily recruited independently (see Suppl. Fig. S7A–C). We did detect a weak, significant association between genomic L3mblt2 binding sites and E-box motifs (p < 10−6), which might be bound by Mga/Max, but no significant association L3mbtl2-binding with the E2F motif. This was surprising because of the strong biochemical interaction and co-localization of E2F6 and L3mbtl2 in somatic cells (Trojer et al., 2011) and in ES cells (Fig. 7D, Suppl. Fig. 7F). Providing an explanation, we found that E2F6-binding is dependent on L3mbtl2 at most (but not all, Suppl. Fig. S7) genes, so E2F6 does not always depend on its DNA-binding site for localization. Moreover, E2F6−/− mice are viable (Storre et al., 2005), so E2F6 is dispensable for the essential role L3mbtl2-dependent repressive complexes in early development. Intriguingly, four germ cell genes, that are bound by E2F6 and upregulated after E2F6 loss in MEFs (Pohlers et al., 2005; Storre et al., 2005), were not upregulated in L3mbtl2−/− ES cells (Suppl. Fig. 7G), and the germ cell genes regulated by L3mbtl2 in ES cells were not up-regulated in L3mbtl2−/− MEFs (Suppl. Fig 7H). Therefore, the importance of different constituents of the complex varies with different cellular contexts and target genes.
The function of L3mbtl2 appears highly selective for pluripotent cells and early embryonic cells. Despite expression, we detected no obvious roles in MEFs (Suppl. Fig. S3), in the maintenance of hematopoiesis, or CNS development (after Mx-Cre or Nestin-Cre deletion, respectively). The striking overlap between L3mbtl2’s interaction partners and the ES cell core transcriptional network provides leads for understanding the molecular basis of its selective role in ES cells.
The function of the major domains of L3mbtl2 remains poorly understood. In our structure/function analysis, the well-characterized methylated histone-binding cavity in the fourth MBT-domain was irrelevant for its functions (Fig. 5). A similar conclusion was recently drawn from testing mutants of in a transcriptional assay (Trojer et al., 2011) and, additionally, it was shown that L3mbtl2 is able to bind unmethylated histones. We demonstrated that both, the zinc finger and the MBT-domains, are absolutely required for its function. Future work is needed to identify the precise ligands of L3mbtl2’s domains and decipher the molecular basis for its interactions. As mutations in L3mbtl2 have been found in brain tumors (Northcott et al., 2009), it will be interesting to investigate whether, like PRC2 (Hock, 2012), L3mbtl2-dependent complexes play important roles beyond development in adult tissues and cancer.
EXPERIMENTAL PROCEDURES
Gene expression analysis
Northern blot analysis of tissues and RT-PCR from ES-cells and embryoid bodies and microarray analysis were performed as described in Extended Experimental Procedures.
Gene targeting of L3mbtl2
Generation of the targeting vector, ES cell culture, mutant ES cells and mice as well as analysis of embryos are described in Extended Experimental Procedures. All animal studies were performed in accordance with the guidelines and under the supervision of the Massachusetts General Hospital Subcommittee on Animal Research (SRAC).
Analysis of ES cells
Colony-size assay, growth curve generation, immunocytochemistry, flow cytometry, apoptosis assay and cell cycle analysis, are described in Extended Experimental Procedures.
Embryoid body differentiation and teratoma formation
The differentiation potential of ES cells was assessed by gene expression analysis after embryoid body formation using the hanging drop method and by histology and immunohistology after teratoma formation following subcutaneous injection in vivo. Details are described in Extended Experimental Procedures.
Generation of expression vectors, mutagenesis, and rescue assays
Full-length MBT-domain protein coding sequences were PCR-amplified from commercially available clones or reverse-transcribed ES cell RNA introducing C-terminal FLAG tags in the 3-terminal primers. Mutagenesis was performed in pBluescript, followed by sequencing of the entire coding region and transfer into a lentiviral expression vector to perform rescue assays. Details are described in Extended Experimental Procedures.
Analysis of protein interactions
Protein purification, mass spectrometry analysis, co-immunoprecipitation, and Western blot analysis are described in Extended Experimental Procedures.
Chromatin immunoprecipitation assays
Preparation of whole cell lysate, immunoprecipitaton, amplification of DNA templates, and ChIP-seq are described in Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We are grateful to Laura Prickett and Kat Folz-Donahue from the HSCI Flow-core at MGH. We thank Denille Van Buren for help with editing the manuscript, Kelly L Shea for help with dissecting embryos, Jose M. Polo for advice on ChIP. We thank G. Mostoslavsky for the PHAGE vector, Peter Rahl the Ring1b ChIP-seq data set, and Daniel Grau and Robert Kingston for help with planning the mass spec experiments and antibody for Bmi1. This work was supported by a contribution from the Ellison Foundation to MGH start-up funds for H.H., by the Federal Share of the Program Income earned by Massachusetts General Hospital on C06 CA059267, Proton Therapy Research and Treatment Center, and by the Silvio O Conte Center “Epigenetic Mechanisms of Depression” (P50MH096890 01). J.Q. was a recipient of an MGH ECOR Fund for Medical Discovery Award.
Footnotes
Supplemental Information includes seven figures, six tables, Extended Experimental Procedures, and supplemental references and can be found with this article online at.
References
- Akasaka T, van Lohuizen M, van der Lugt N, Mizutani-Koseki Y, Kanno M, Taniguchi M, Vidal M, Alkema M, Berns A, Koseki H. Mice doubly deficient for the Polycomb Group genes Mel18 and Bmi1 reveal synergy and requirement for maintenance but not initiation of Hox gene expression. Development. 2001;128:1587–1597. doi: 10.1242/dev.128.9.1587. [DOI] [PubMed] [Google Scholar]
- Arai S, Miyazaki T. Impaired maturation of myeloid progenitors in mice lacking novel Polycomb group protein MBT-1. Embo J. 2005;24:1863–1873. doi: 10.1038/sj.emboj.7600654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242–8247. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Nady N, Qi C, Allali-Hassani A, Zhu H, Pan P, Adams-Cioaba MA, Amaya MF, Dong A, Vedadi M, et al. Methylation-state-specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 2009;37:2204–2210. doi: 10.1093/nar/gkp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev. 2012;26:751–755. doi: 10.1101/gad.191163.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda H, Takubo K, Oda H, Kosaki K, Tazaki T, Yamasaki N, Miyazaki K, Moore KA, Honda Z, Suda T, et al. Hemp, an mbt domain-containing protein, plays essential roles in hematopoietic stem cell function and skeletal formation. Proc Natl Acad Sci U S A. 2011;108:2468–2473. doi: 10.1073/pnas.1003403108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymenko T, Papp B, Fischle W, Kocher T, Schelder M, Fritsch C, Wild B, Wilm M, Muller J. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev. 2006;20:1110–1122. doi: 10.1101/gad.377406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Go Y, Kang I, Han YM, Kim J. Oct-4 controls cell-cycle progression of embryonic stem cells. Biochem J. 2010;426:171–181. doi: 10.1042/BJ20091439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlers M, Truss M, Frede U, Scholz A, Strehle M, Kuban RJ, Hoffmann B, Morkel M, Birchmeier C, Hagemeier C. A role for E2F6 in the restriction of male-germ-cell-specific gene expression. Curr Biol. 2005;15:1051–1057. doi: 10.1016/j.cub.2005.04.060. [DOI] [PubMed] [Google Scholar]
- Qin J, Van Buren D, Huang HS, Zhong L, Mostoslavsky R, Akbarian S, Hock H. Chromatin protein L3MBTL1 is dispensable for development and tumor suppression in mice. J Biol Chem. 2010;285:27767–27775. doi: 10.1074/jbc.M110.115410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Sanchez I, Demmers JA, Rodriguez P, Strouboulis J, Vidal M. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Mol Cell Proteomics. 2007;6:820–834. doi: 10.1074/mcp.M600275-MCP200. [DOI] [PubMed] [Google Scholar]
- Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. 2009;139:1303–1314. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Storre J, Schafer A, Reichert N, Barbero JL, Hauser S, Eilers M, Gaubatz S. Silencing of the meiotic genes SMC1beta and STAG3 in somatic cells by E2F6. J Biol Chem. 2005;280:41380–41386. doi: 10.1074/jbc.M506797200. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Takada Y, Isono K, Shinga J, Turner JM, Kitamura H, Ohara O, Watanabe G, Singh PB, Kamijo T, Jenuwein T, et al. Mammalian Polycomb Scmh1 mediates exclusion of Polycomb complexes from the XY body in the pachytene spermatocytes. Development. 2007;134:579–590. doi: 10.1242/dev.02747. [DOI] [PubMed] [Google Scholar]
- Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664–678. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Cao AR, Gao Z, Li Y, Zhang J, Xu X, Li G, Losson R, Erdjument- Bromage H, Tempst P, et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol Cell. 2011;42:438–450. doi: 10.1016/j.molcel.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation- dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- van der Stoop P, Boutsma EA, Hulsman D, Noback S, Heimerikx M, Kerkhoven RM, Voncken JW, Wessels LF, van Lohuizen M. Ubiquitin E3 ligase Ring1b/Rnf2 of polycomb repressive complex 1 contributes to stable maintenance of mouse embryonic stem cells. PLoS One. 2008;3:e2235. doi: 10.1371/journal.pone.0002235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jahren N, Miller EL, Ketel CS, Mallin DR, Simon JA. Comparative analysis of chromatin binding by Sex Comb on Midleg (SCM) and other polycomb group repressors at a Drosophila Hox gene. Mol Cell Biol. 2010;30:2584–2593. doi: 10.1128/MCB.01451-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.