

Abstract
We compare and contrast the genetic architecture of quantitative phenotypes in two genetically well-characterized model organisms, the laboratory mouse, Mus musculus, and the fruit fly, Drosophila melanogaster, with that found in our own species from recent successes in genome-wide association studies. We show that the current model of large numbers of loci, each of small effect, is true for all species examined, and that discrepancies can be largely explained by differences in the experimental designs used. We argue that the distribution of effect size of common variants is the same for all phenotypes regardless of species, and we discuss the importance of epistasis, pleiotropy, and gene by environment interactions. Despite substantial advances in mapping quantitative trait loci, the identification of the quantitative trait genes and ultimately the sequence variants has proved more difficult, so that our information on the molecular basis of quantitative variation remains limited. Nevertheless, available data indicate that many variants lie outside genes, presumably in regulatory regions of the genome, where they act by altering gene expression. As yet there are very few instances where homologous quantitative trait loci, or quantitative trait genes, have been identified in multiple species, but the availability of high-resolution mapping data will soon make it possible to test the degree of overlap between species.
Recent successes in mapping quantitative trait loci (QTLs) that contribute to phenotypic variation in humans and model organisms make it possible to address important questions about the genetic architecture of quantitative phenotypes, such as the likely number of loci, their mode of genetic action, and interaction with each other and with the environment. An understanding of the genetic architecture of quantitative traits is not only important in its own right, it will also inform studies that seek to proceed to the identification of the quantitative trait genes (QTGs) and the quantitative trait nucleotide (QTN) variants, the functional changes in DNA sequence that contribute to trait variation.
In this work we use examples from two genetically well-characterized model organisms, the laboratory mouse, Mus musculus, and the fruit fly, Drosophila melanogaster, to discuss how an understanding of the genetic architecture of quantitative phenotypes in model organisms informs mapping experiments in human genetics. In our discussion of the latter, we draw upon the recent successful application of genome-wide association studies (GWAS) to both quantitative phenotypes (such as height and weight) and disease phenotypes (assuming that the genetic architecture of common disease is similar, if not identical, to that of quantitative phenotypes).
Genetic architecture has been the subject of a number of recent reviews, most of which deal with information from a single model organism (Mackay 2004) or review a small number of phenotypes (Kendler and Greenspan 2006). Here, our purpose is different. We tackle three related questions. (1) How much does genetic architecture differ between species? (2) How much does genetic architecture vary between phenotypes? (3) Are the same genes involved in the same phenotype in different species? Answers to these questions are important because of the need to identify genes involved in human disease phenotypes and to determine how those genes exert their effect. Currently, model organisms such as the fly and the mouse are the resources of choice for functional genetic studies.
How much does genetic architecture vary between species?
Within a species, the genetic architecture of different phenotypes appears to be similar, but that similarity may not extend to comparisons between species. How much does knowing about one species inform us about another? Below, we review data on the number of loci, their effect sizes, and how they operate on quantitative phenotypes. For quantitative traits, we define the homozygous effect, a, as one-half of the difference in the mean trait phenotype between individuals that are homozygous for alternative QTL alleles (Falconer and Mackay 1996). Homozygous effects are usually reported as either a fraction of the phenotypic standard deviation of the trait in the mapping population (a/σP), or as a fraction of the phenotypic variation (σP2) of the mapping population. In this review, unless otherwise stated, a genetic effect size is meant in this latter sense as a proportion of the phenotypic variance. Heterozygous effects, d, are estimated as the difference between the mean value of the trait for individuals that are heterozygous for the QTL alleles, expressed as a deviation from the average of the two homozygous genotypes. Heterozygous effects are generally expressed as d/a, where d/a = 0 denotes strict additivity (Falconer and Mackay 1996).
Genetic architecture: Many loci of small effect
In the 1990s, with the availability of genome-wide marker sets, it became possible for the first time to map the chromosomal location of genetic effects that contribute to quantitative variation (Lander and Botstein 1989; Hilbert et al. 1991; Jacob et al. 1991; Todd et al. 1991). The methods adopted depended on the organism: Mouse and fly geneticists could use inbred strains, thereby considerably simplifying the genetics. By setting up a cross between two inbred strains, or by using the inbred derivatives of such a cross (recombinant inbred [RI] lines), only two alleles segregated at any locus, and all alleles had the same frequency. In contrast, human geneticists had to make do with what the vagaries of evolution, population history, migration, and mutation had put together.
To map QTLs in rodents, about 100 markers and a few hundred animals were sufficient to provide robust statistical evidence for the location of genetic variants contributing to quantitative phenotypic variation. With falling costs and automated genotyping, plus the development of software packages that made the analyses easily accessible to biologists without statistical expertise, any biologist could, and did, map a phenotype. Consequently, a large literature emerged in the last decade of the 20th century, as biologists turned to genetic mapping to help them identify genes involved in the physiological process or disease that constitutes their special interest.
Mapping QTLs was also relatively easy in flies (and much cheaper than in rodents). Remarkably, the picture that emerged from both organisms was broadly similar. Most of the early mapping studies revealed that a few QTLs with relatively large effects accounted for the bulk of phenotypic variation in most traits. This observation was central to driving attempts to capture common genetic variants segregating in human populations. Critically, the estimates of effect size from model organisms suggested that QTLs explaining as much as 10% of the phenotypic variance might be found, in which case linkage studies using a few hundred families would be sufficient to detect them.
Subsequent studies in mice and flies using larger samples began to reveal a more complex picture. For instance, doubling the number of mice in the mapping population from 800 to 1600 more than doubled the number of QTLs found to influence a measure of fear-related behavior (Turri et al. 2001a, b). A study in Drosophila genotyped over 2000 markers on pools of flies selected from an initial sample of over 10,000 F2 individuals (Lai et al. 2007) and substantially increased the number of QTLs known to influence life span in the fly. Furthermore, high-resolution mapping of QTLs revealed more complex genetic architectures than implicated from the initial genome scans. Single QTLs identified in genome scans typically fractionated into multiple, closely linked QTLs, often with opposite effects. In mouse genetics this finding emerged from studies that chased single effects through the creation of congenics and includes QTLs influencing seizures (Legare et al. 2000), obesity (Stylianou et al. 2004), growth (Christians and Keightley 2004), blood pressure (Frantz et al. 2001; Alemayehu et al. 2002; Garrett and Rapp 2002a,b; Ariyarajah et al. 2004), diabetes (Podolin et al. 1998), antibody production (Puel et al. 1998), and infection (Bihl et al. 1999). In flies, high-resolution mapping efforts using a higher density of recombination or performing quantitative complementation tests to deficiencies revealed similar complexity for QTLs affecting wing shape (Weber et al. 1999, 2001; Mezey et al. 2005), longevity (Pasyukova et al. 2000; De Luca et al. 2003; Wilson et al. 2006), resistance to starvation stress (Harbison et al. 2004), mating behavior (Moehring and Mackay 2004), olfactory behavior (Fanara et al. 2002), locomotor reactivity (a startle response) (Jordan et al. 2006), and numbers of sensory bristles (Dilda and Mackay 2002).
Nevertheless, although more QTLs were discovered, the results were still inconsistent with the “infinitesimal” model of quantitative genetic variation (Falconer and Mackay 1996; Lynch and Walsh 1998), which posits extremely large numbers of loci with very small effects. Rather, the allelic effects followed an exponential distribution, as proposed by Robertson (1967), with a few loci with moderate to large effects, and increasingly larger numbers of loci with increasingly smaller effects.
Despite the success and predictions of genetic architecture from work on model organisms, by the end of the 20th century human genetic studies of complex phenotypes were foundering. A small number of high-profile successes continued to engender optimism (e.g., the identification of the epsilon4 allele of APOE [APOE*4] as a susceptibility gene for Alzheimer's disease [Corder et al. 1993]), but the majority of genetic studies of human quantitative phenotypes became mired in failures to replicate. One simple and unavoidable interpretation of the relative failure was that human studies were underpowered (Risch and Merikangas 1996). Many of the human linkage studies were powered on the basis that the distribution of effect sizes would be approximately exponential, with a few at the top end explaining perhaps 10% of the phenotypic variation, as found in the model organism work. Could it be the case that genetic architecture of quantitative phenotypes in humans was different from mice and flies?
Discrepant findings are largely attributable to differences in the number and frequencies of alleles segregating in outbred populations compared with those in crosses between inbred strains. Estimates of effect size, when expressed as the percentage of phenotypic variance attributable to genetic variation at a locus, vary according to the experimental design. Suppose we identify a QTL that contributes to 1% of the genetic variation in an outbred population. In a model organism we could then set up a breeding program to isolate that chromosomal region so that all other genomic regions are inbred—consequently, the locus now contributes to 100% of the genetic variation. Conversely, QTLs identified in simple systems may have smaller relative effects in outbred populations, because the fraction of the total phenotypic variance explained by a QTL with a given effect depends on both allele and genotype frequencies.
The genetic variance (σG2) of a strictly additive (d = 0) QTL with allele frequencies p and q and homozygous effect a is 2pqa2 in an outbred population (Falconer and Mackay 1996). In progeny derived from a cross of two inbred lines, the frequency of all segregating alleles is p = q = 0.5, whereas in an outbred population, allele frequencies can vary throughout the entire range. Thus, a QTL with homozygous effect a = 0.5σP will explain 12.5% of the phenotypic variance in an F2 population but only 8% of the variance in an outbred population where the minor allele frequency is 0.2%, and 4.5% of the variance in an outbred population where the minor allele frequency is 0.1. Furthermore, only the two homozygous genotypes are present in a population of RI lines, whereas only one homozygous genotype and the heterozygote segregate in a backcross (BC) population. Thus, the genetic variances for a population of RI lines, F2 individuals, and BC individuals are, respectively, a2, a2/2, and a2/4 (Lynch and Walsh 1998); i.e., in the ratio 4:2:1 for RI:F2:BC.
Furthermore, the locus-specific effect on the phenotypic variance depends additionally on environmental variance and the number of other QTLs that contribute. In a cross between two inbred rodent strains a detectable locus contributes to about 5% of the phenotypic variation (Flint et al. 2005). In a complex quasi-outbred stock (known as the heterogeneous stock [HS]) (Valdar et al. 2006a), assuming the QTL allele frequencies remain comparable, the same locus is likely to explain about 2.5% of the variance (Flint et al. 2005). In a fully outbred population, the locus will account for much less.
In the last 2 yr, with the advent of adequately powered genome-wide association studies in humans, we have at last reliable estimates of the locus-specific effect sizes that contribute to variation in complex phenotypes. Genome-wide association analyses of human case-control samples reveal that the genetic architecture of quantitative traits and disease consists of alleles of small effect size. With the exception of the HLA locus on some autoimmune diseases (such as type 1 diabetes) and a few outliers (such as a variant of the HTRA1 gene with an effect on age-related macular degeneration [Dewan et al. 2006]), the per-allele odds ratios are almost all less than 1.5. The genetic architecture of disease and quantitative phenotypes appears to be the same: Small effect sizes characterize quantitative variation in humans. Good examples are height and weight: 54 loci have now been identified that influence height, a great advance on what we knew before the advent of genome-wide association studies (Gudbjartsson et al. 2008). However, each variant contributes less than half a percent to the total phenotype variance. Another way of putting this is that an allele that increases height does so by adding about 0.4 cm. Similarly, for the 10 loci identified that contribute to body mass index (Thorleifsson et al. 2009; Willer et al. 2009), allelic effects range from 0.06 kg/m2 to 0.33 kg/m2.
The small effect sizes reported in human studies do not necessarily mean that the infinitesimal model is a useful description of quantitative genetic variation in human populations. The important parameter is the distribution of effect sizes. Figure 1 shows the distribution of effect sizes at 140 loci discovered for 20 disease phenotypes. In this case, effect size is expressed as a locus-specific odds ratio (so that an allelic odds ratio of two doubles the risk of developing the condition). Note the skewed distribution of the findings, and note also that the distribution is certainly truncated for smaller effects that studies do not have enough power to detect. Despite sample sizes in excess of 10,000, power to detect the QTLs that influence height and weight is about 10% (Willer et al. 2009). In fact, the loci discovered account for remarkably small amounts of the total phenotypic variance. For height, all known loci explain <5% of the variance. For a measure of obesity (body mass index) known loci account for <1% of the variance (Willer et al. 2009). It is quite possible that there is a long tail of much smaller effects remaining to be discovered.
Figure 1.

Effect sizes (expressed as odds ratios) in human genome-wide association studies. Data are from http://www.genome.gov/gwastudies/.
Genetic architecture: Context-dependent effects
While it is generally accepted that the effect of a QTL should be considered in the context of the genetic background on which it occurs, the sex of the animal, and within the environmental context in which the phenotype is measured, systematic measures of the extent of context-dependent effects are still lacking. Results from model organisms unambiguously reveal that these effects exist, largely through the ability to control both genotype and environment, but their detection is not always easy.
Epistatic interactions (defined as a statistical interaction between genotypes at two [or more] loci) are difficult to detect in QTL mapping studies, because the large number of pairwise tests for marker–marker interactions imposes a low experiment-wise significance threshold. Large mapping populations are required to sample individuals in the rarer two-locus genotype classes, and segregation of other QTLs can interfere with detecting epistasis between the pair of loci under consideration. Consequently, most QTL mapping studies report only main QTL effects, or evaluate epistasis only between QTLs with significant main effects.
In flies, epistatic interactions have been documented between QTLs affecting the quintessential additive traits, numbers of sensory bristles (Long et al. 1995; Gurganus et al. 1999; Dilda and Mackay 2002), and wing shape (Weber et al. 1999, 2001), as well as longevity (Leips and Mackay 2000, 2002; Mackay et al. 2006) and locomotor behavior (Jordan et al. 2006). Epistatic effects can be as large as main QTL effects, in opposite directions between different pairs of interacting loci, and between loci without significant main effects on the trait.
Epistatic interactions are more readily detectable in diallel crosses, which produce all possible doubly heterozygous genotypes from a collection of mutations affecting the same trait that have been generated in a common background, or by constructing all nine possible two-locus genotypes for two biallelic loci. The few studies using these designs all reveal extensive epistasis: for metabolic activity (Clark and Wang 1997), olfactory behavior (Fedorowicz et al. 1998; Sambandan et al. 2006), climbing behavior (van Swinderen and Greenspan 2005), and locomotor startle response (Yamamoto et al. 2008). Remarkably, epistatic interactions have also been observed between functional polymorphisms affecting alcohol dehydrogenase protein level within the Adh locus (Stam and Laurie 1996).
Where epistasis has been sought in mice, it has in most cases been found. For example, Cheverud and colleagues, in a series of studies on growth, body weight, and morphometry, have identified numerous interactions in a cross between a large inbred mouse strain (LG/J) and a small inbred strain (SM/J) (Cheverud et al. 2001; Klingenberg et al. 2001, 2004; Workman et al. 2002; Yi et al. 2006). Two locus interaction models indicated the presence of epistasis, as others have independently found for the same phenotypes (Brockmann et al. 2000). Methodological improvements have led to the identification of even more epistatic QTLs in the same cross (between LG/J and SM/J). For instance, a Bayesian method, developed to deal with the problem of testing multiple interactions, estimates epistatic effects by generating posterior samples from the joint posterior distribution of all unknowns, including the main and epistatic effects, given the observed data (Yi et al. 2007). Using this approach, Yi and colleagues detected several epistatic QTL that were not found by frequentist methods (Ankra-Badu et al. 2009). However, this should not lead to the conclusion that epistasis is universal. In some circumstances, despite an extensive search, little evidence for epistasis emerges, as, for example, in the case of fear-related behavior in mice (Flint et al. 2004).
In these examples drawn from model organisms, using experimental resources that reduce genetic heterogeneity, the importance of epistasis has to be placed within the genetic context in which it is measured. The power to detect epistasis between two loci depends on the frequency of the rarest genotype, usually the double homozygous genotypes at both loci. In outbred populations, the least common double homozygote genotype may be rare or even absent if the minor allele frequency at both loci is low. Epistasis between two loci is easier to detect in inbred strain crosses, because the double homozygotes at both loci are present in ∼12.5% of the individuals of an F2 population and 25% of the individuals in a population of RI lines. Methods that further simplify genetic backgrounds, such as the use of chromosome substitution strains (CSS) or genome-tagged mice (GTM), further enhance the ability to detect epistatic interactions in model organisms.
CSS have one chromosome derived from one inbred parental strain and the remainder derived from a second inbred strain; a CSS panel of mice is created by a laborious process of backcrossing so as to replace each chromosome from a host strain with its corresponding partner from a donor strain (Singer et al. 2004). Genetic differences between a panel member and the host arise from differences on the one chromosome they do not share, thereby allowing a relatively quick and simple test or the location of a QTL. Isolating each chromosome also permits a sensitive test of epistasis, and mapping of 90 traits revealed almost half to have significant deviation from an additive model, strong evidence for epistasis (Shao et al. 2008).
GTM are a panel of over 60 strains, each harboring an ∼50 Mb donor segment on a uniform background. Thus, like the CSS, GTM represent a considerable reduction in genetic complexity from a cross between two inbred strains, and the relative contribution of each genetic variant is magnified. Smith and colleagues mapped 97 loci for a variety of behavioral traits, including hyperactivity, anxiety, prepulse inhibition, avoidance, and conditioned freezing (Gale et al. 2008). There was evidence for epistasis at about 60% of the QTLs they identified.
There are two senses in which one can discuss epistatic effects: in reference to the contribution of epistasis to the mean differences between genotypes (“physiological” epistasis) (Cheverud and Routman 1995), or in reference to the contribution of epistatic interactions to the population genetic variance (“statistical” epistasis) (Falconer and Mackay 1996). All of the model organism studies measured the contribution of epistasis in the former sense. Epistatic variance, like other components of genetic variance, depends on both effects and allele frequencies. Therefore, large effects on the mean difference between genotypes may not translate to large effects on the variance, if allele frequencies are low. We cannot compute the expected contributions of the epistasis observed from studies of mutations and QTLs in model organisms to the population genetic variance without knowledge of the allele frequencies in natural populations.
The remarkably large contribution of epistasis in some experimental designs (such as CSS and GTM) stands in stark contrast to the findings from human genome-wide association studies in which, to date, despite the success of finding main effects, there have been no replicated findings of interactions. We should be clear that this does not mean epistasis does not exist (see, e.g., the triallelic inheritance that gives rise to Bardet-Biedel syndrome [Katsanis et al. 2001] and the requirement for mutations at both RET and additional loci to bring about Hirschsprung disease [Emison et al. 2005]). It means, rather, that given the small main effects, the detection of epistasis in complex phenotypes in our own species will require astronomically large numbers of individuals.
Identical arguments apply to the detection of genotype by environment interactions. Again, it is abundantly clear from the model organism work that these context-dependent genetic effects are pervasive. In mice, quantitative analyses indicate that genotype by environment interaction (GEI) effects occur for all phenotypes, and often makes as large, or larger, contribution to the phenotypic variation than the environmental effects (Valdar et al. 2006b). In flies, whenever QTLs have been mapped in different environments, GEI has been observed: for numbers of sensory bristles (Gurganus et al. 1998, 1999; Dilda and Mackay 2002; Geiger-Thornsberry and Mackay 2002), longevity (Leips and Mackay 2000, 2002; Vieira et al. 2000), competitive fitness (Fry et al. 1998), and immune response to different bacteria (Lazzaro et al. 2006). GEI is mostly attributable to environment-specific expression of QTL alleles, with few cases of opposite effects in different environments (Vieira et al. 2000).
On the face of it, human studies would appear to be one step ahead in having identified a number of variants involved in genes by environment interaction on behavioral phenotypes. One highly cited example is the effect of a functional variant in the promoter of the serotonin transporter gene (SLC6A4 [also known as 5-HTT]) that is only, or primarily, manifest in people who had suffered stressful life events (such as the break-up of a long term relationship) (Caspi et al. 2003). However, meta-analysis of attempts at replication combined with a simulation study of the main effects of genotype and environmental effect indicate that, if the effect is real, it is much smaller than often claimed, and will again need very large sample sizes to detect robustly (Munafo et al. 2009).
Genetic architecture: Pleiotropy
Studies of phenotypic covariance between quantitative traits have long suggested the presence of pleiotropy, a finding supported by artificial selection experiments in which a direct response is frequently accompanied by changes in many genetically correlated traits (Wright 1977; Falconer and Mackay 1996). While strong genetic correlations are to be expected from directional pleiotropic effects among functionally related traits such as body weight and length, pleiotropic effects are also found for loci affecting traits that are less obviously functionally related. The importance of genes having pleiotropic effects on multiple quantitative traits is largely unappreciated and has emerged from the study of model organisms.
In flies, evidence for pleiotropic effects of subtle mutations on multiple quantitative traits comes from evaluating the effects of new P-transposable element mutations in a common inbred genetic background (single genetically engineered P-elements can be induced to hop into new genomic locations by simple crosses to a stock containing a chromosomally stable source of transposase) (Venken and Bellen 2005). In mice, similar observations come from studies of effects of alleles created more onerously by targeted deletion of genes (Austin et al. 2004) or by chemical mutagenesis (Hrabe de Angelis et al. 2000; Nolan et al. 2000).
Analyses of effects of P-element mutations on quantitative traits have revealed that many genes that play a major role in early development also affect adult quantitative traits. Mutations in the developmental loci neuralized, Semaphorin-5c, and Calreticulin affect numbers of sensory bristles as well as olfactory behavior (Sambandan et al. 2006; Rollmann et al. 2007). Mutations in the intergenic region between Tre1 (which affects transepithelial migration of germ cells) and Tre (a trehalose taste receptor also known as Gr5a) affect not only trehalose sensitivity, but also life span and resistance to starvation and heat stress (Rollmann et al. 2006). A mutation in muscleblind is associated with increased aggression (Edwards et al. 2006) and increased resistance to the inebriating effects of ethanol (Morozova et al. 2007), but reduced locomotor startle response (Jordan et al. 2007). Independent evidence for pervasive pleiotropy comes from the substantial overlap of transcripts for which there are correlated responses in expression to selection from the same base population for copulation latency, aggressive behavior, locomotor startle response, and ethanol resistance (Mackay et al. 2005; Edwards et al. 2006; Jordan et al. 2007; Morozova et al. 2007).
In mice, projects to deliver engineered knockouts for every gene in the genome (Collins et al. 2007) are beginning to provide new insights regarding the extent of pleiotropy. Phenotypic data from 250 knockout strains acquired by the knockout project from two companies (Deltagen and Lexicon) are publicly available from the MGI database (http://www.informatics.jax.org/external/ko/). Although not an unbiased selection of mutants, the data are the only instance where a large number of knockouts have been subjected to a broad range of measurements. Table 1, summarizing results for 12 phenotypes, shows that about 7% of mutants have an abnormal phenotype. There seems no reason to expect this figure to be any less for any of the hundreds of other traits that could, and eventually will be examined. If so, pleiotropy will be the rule, not the exception.
Table 1.
Percentage of abnormal phenotypes (at P < 0.05) for 250 mouse knockouts
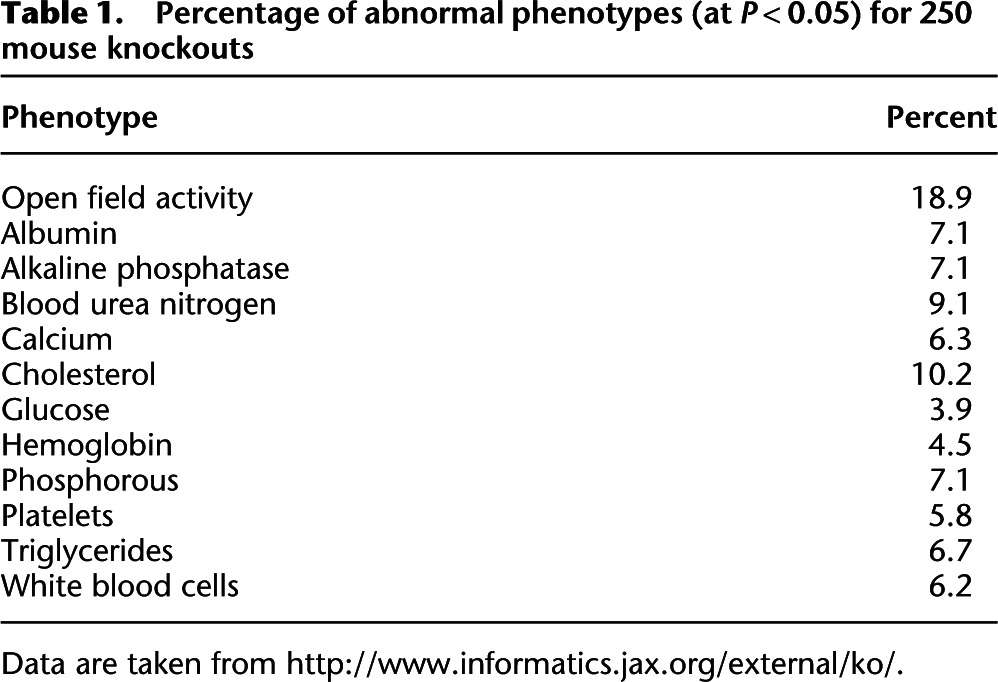
Data are taken from http://www.informatics.jax.org/external/ko/.
Demonstrating pleiotropy in QTL studies in mice and flies is more difficult since QTLs typically contain multiple genes: Disentangling coincidence of location from pleiotropic action is hard. In flies, this problem can be overcome by using genetic association. The structure of linkage disequilibrium in Drosophila is exceptionally fine grained and decays rapidly in regions of normal recombination; so rapidly, in fact, that polymorphic sites adjacent to each other can be in linkage equilibrium (Carbone et al. 2006). This makes it possible to differentiate the effects of single-nucleotide polymorphisms within the same gene. Several studies have evaluated associations of polymorphisms in candidate genes with more than one quantitative trait: the achaete-scute complex (Mackay and Langley 1990; Long et al. 2000), scabrous (Lai et al. 1994; Lyman et al. 1999), Delta (Long et al. 1998), and hairy (Robin et al. 2002) for abdominal and sternopleural bristle number; the epidermal growth factor receptor for wing shape (Palsson and Gibson 2004) and cryptic variation for photoreceptor determination (Dworkin et al. 2003); Dopa decarboxylase for longevity (De Luca et al. 2003) and locomotor startle response (Jordan et al. 2006); and Catecholamines up for locomotor behavior, longevity, starvation resistance, and abdominal and sternopleural bristle number (Carbone et al. 2006). In each case different polymorphic sites were independently associated with the different traits. These observations indicate that pervasive pleiotropy does not necessarily impose evolutionary constraints in the form of strong genetic correlations between traits.
Equivalent high-resolution association methodologies are not available in mice. Instead, statistical approaches have been applied to inbred cross data and typically find evidence for pleiotropy, for example, as in studies of weight and morphometry (Brockmann et al. 2000; Leamy et al. 2002; Wolf et al. 2005, 2006; Pavlicev et al. 2008). However, these methods leave open the possibility that pleiotropy at a locus is attributable to multiple genes of different function that happen to reside next to each on the genome.
In human genetic association studies, relatively low linkage disequilibrium makes it possible to argue that pleiotropy is due to the presence of a single gene influencing multiple conditions. In some cases, pleiotropy involves diseases, or physiological processes that have common features. For instance, type 1 diabetes and celiac disease share risk regions (Hunt et al. 2008; Smyth et al. 2008) and polymorphisms in IL23R contribute to susceptibility to three autoimmune disorders: psoriasis, Crohn's disease, and ankylosing spondylitis (Duerr et al. 2006; Burton et al. 2007; Cargill et al. 2007), and a SNP within the intron of CDKAL1 has been associated with type 2 diabetes (T2D) and insulin secretion defects (Gudmundsson et al. 2007; Scott et al. 2007; Zeggini et al. 2007, 2008). In other cases, genetic relationships are beginning to emerge between traits that had not previously been associated: a SNP in HNF1B is associated with protection from type 2 diabetes (Winckler et al. 2007) and for prostate cancer (Gudmundsson et al. 2007); SNPs in TCF7L2 are associated with both type 2 diabetes (Grant et al. 2006) and colon cancer (Slattery et al. 2008); SNPs in JAZF1 are associated with both type 2 diabetes (Zeggini et al. 2008) and prostate cancer (Thomas et al. 2008).
How much does genetic architecture vary between phenotypes?
Evidence from model organisms strongly argues that the distribution of effect size of common variants is the same for all phenotypes. Figure 2 shows a summary of the effect sizes for 98 phenotypes mapped in 2000 heterogeneous stock mice (Valdar et al. 2006a). The phenotypes are classified into six categories: behavior, biochemistry, hematology, immunology, metabolism, and respiratory physiology (as measured in a plethysmograph). There is no significant variation among the categories (F = 1.8, df = 5, P = 0.109). Mapping multiple traits in chromosome substitution strains in mice gives a similar picture of the genetic architecture of quantitative phenotypes (Shao et al. 2008). CSS studies of 90 traits in a mouse panel and 54 traits in a rat panel identified multiple QTLs for each phenotype (Shao et al. 2008). The average phenotypic effects for a CSS were similar across all phenotypes in both mouse and rat panels.
Figure 2.

Effect sizes (expressed as percentage of total variation explained) for six categories of phenotype. Data are taken from analyses of 97 phenotypes mapped in heterogeneous stock mice (Valdar et al. 2006a).
One of the surprises of the Wellcome Trust Case Control Consortium analysis of seven common diseases (Wellcome Trust Case Control Consortium 2007) was that although the same number of individuals was used for each disease (2000) and a common set of controls (3000), the findings for each phenotype varied. While for Crohn's disease (an inflammatory bowel disorder), nine loci exceeded an association signal at P < 5 × 10−7, only one locus was found for coronary artery disease and bipolar disease. In part, the difference in detection can be blamed on experimental design. The WTCCC uses a set of common controls that are not screened for disease. Consequently, for common disorders, such as coronary artery disease, controls include a proportion of affected individuals, and this misclassification will reduce power to detect association (Wellcome Trust Case Control Consortium 2007; McCarthy et al. 2008). But this does not rule out the possibility that failure to detect effects might reflect a different genetic architecture. It could be that the effect sizes of common variants are much less in psychological measures than in anthropometric traits.
It is also possible that the model organism gives an unrepresentative picture. Almost all of the work carried out in mice and flies on quantitative traits uses experimental designs that detect common alleles, not rare variants. In populations derived from crosses between inbred strains, there are a small number of alleles (in most designs only two) at each locus, and the frequencies are large and vary little (in an intercross, each allele will have a frequency of about 0.5). In outbred populations used by human geneticists, this is not the case. Genome-wide association studies detect common variants, so the comparison with the model organism results is probably fair. The impact of rare variants on the genetic architecture of quantitative phenotypes remains relatively unexplored in both model organisms and humans. However, there are clear indications from resequencing studies in humans that it will turn out to be important (Cohen et al. 2004, 2005; Kotowski et al. 2006; Romeo et al. 2007; Ji et al. 2008).
Could the contribution of rare variants differ between phenotypes? There is some evidence from human studies to support this view, coming from studies of the role of copy number variants (CNVs). The extent and distribution of CNVs in the human genome is still being investigated: Current estimates indicate large-scale (>50 kb) CNVs are rare, affecting <5% of the genome, but about 1300 smaller CNVs have minor allele frequencies >1% (McCarroll et al. 2008). Genome-wide association studies typically use array-based methods that can also detect CNVs. Rare CNVs are now known to be common in autism (Sebat et al. 2007) and schizophrenia (International Schizophrenia Consortium 2008; Stefansson et al. 2008; Walsh et al. 2008; Xu et al. 2008) than in controls; in contrast, few common variants have been found in schizophrenia and autism. A similar contribution of rare CNVs has not been reported for other diseases, suggesting that there may be important differences between some phenotypes. Possibly the lower reproductive fitness of schizophrenia and autism have contributed to the concentration of rare variants as a predisposing cause of these psychiatric illnesses, a finding that implies that it may be difficult to model these illnesses genetically using engineered mice.
Are the same genes involved in the same phenotype in different species?
Despite substantial advances in mapping QTLs, the identification of the QTGs and ultimately the QTN sequence variants has proved more difficult. Thus, our information on the overlap at the molecular level is relatively limited. But, the information that we have indicates that sequence positions giving rise to phenotypic variation are not homologous between species. The relevant finding here is from the human work, indicating that QTNs are likely to lie in regulatory regions of the genome (Donnelly 2008), which has implications for their conservation between species. QTNs typically lie outside genes, often very far outside genes: The association signal on chromosome 9 between myocardial infarction and T2D lies more than 100 kb from the nearest genes (CDKN2A and CDKN2B) (Helgadottir et al. 2007; McPherson et al. 2007; Samani et al. 2007); a locus associated with breast, colon, and prostate cancer on the long arm of chromosome 8 is more than a quarter of a megabase from the nearest known gene (Amundadottir et al. 2006; Freedman et al. 2006).
The importance of this finding is that, unlike coding regions, many regulatory regions are not conserved between species. One of the surprises of the Encyclopedia of DNA Elements (ENCODE) Project, which set out to catalog functional elements in 1% of the genome, was the excess of functional elements lying outside regions of sequence conservation (The ENCODE Project Consortium 2007).
In flies, the requirement for dense molecular marker genotypes or, ideally, complete DNA sequence data, for alleles of candidate genes sampled from a natural population has limited the number of association studies performed. However, in all studies reported to date, both common and rare QTNs have been associated with quantitative traits and occur in presumed regulatory regions, as well as nonsynonymous polymorphisms in coding regions (Mackay and Langley 1990; Long et al. 2000; Robin et al. 2002; Palsson and Gibson 2004; Jordan et al. 2006). Although the total number of studies is small, there is a tendency for less common polymorphisms associated with quantitative traits to occur in coding regions and have large effects, while intermediate frequency polymorphisms often occur in presumed regulatory regions (or synonymous SNPs in coding regions) and have smaller effects. All types of polymorphisms have been associated with variation in fly quantitative traits: SNPs, small insertions/deletions, complex SNP/copy number variants, and transposable elements. A more comprehensive picture of the nature and extent of polymorphisms associated with quantitative traits in the fly model will emerge in the near future, when many traits are assessed for a panel of 192 inbred strains for which complete sequences have been determined.
If the position of the variant is not conserved, the gene may well be. Claims that variation within the same gene could contribute to phenotypic variation in both rodents and humans have sustained attempts to map phenotypes in model organisms. However, given the assumption that many physiological processes are conserved between these species, and that, as we have shown, a very large number of loci are likely to be contributing to human phenotypic variation, it is surprising that so few examples of common QTLs have been reported.
One of the earliest examples is the detection of a locus that modifies the severity of cystic fibrosis. A locus was identified on mouse chromosome 7 that modified intestinal disease in homozygous mutant mice (Rozmahel et al. 1996) and subsequently a similar modifier was mapped to human chromosome 19, in the region syntenic to the mouse locus (Zielenski et al. 1999). Unfortunately, the gene involved remains unknown. Polymorphisms in the same gene, cytotoxic T-lymphocyte-associated protein 4 gene (CTLA4), have been shown to contribute to autoimmune disorders in humans and mice (Ueda et al. 2003). A noncoding 3′ region of CTLA4 increases risk of the Graves' disease, autoimmune hypothyroidism, and type 1 diabetes; in a mouse model of type 1 diabetes, susceptibility is associated with variation in Ctla4 gene splicing with reduced production of one splice form. Finally, QTL mapping identified the same susceptibility gene (Fcgr3) contributing to immunologically mediated glomerulonephritis in humans and rats (Aitman et al. 2006). Remarkably, in both rats and humans, copy number variation in the homologous gene was associated with the disease, suggesting that copy number variation may be a conserved molecular mechanism underlying complex and quantitative phenotypes.
A number of other genes have been identified in rodents and then tested by association in humans, but results have often been equivocal. For example, evidence from QTL mapping and analyses of knockouts implicates Tnfsf4 in diet-induced atherosclerosis in mice (Wang et al. 2005b); however, genetic association studies have not provided consistent evidence for its involvement in humans (Koch et al. 2008). Similar difficulties are found for the involvement of type II SH2 domain-containing inositol 5-phosphatase in the metabolic syndrome (Kaisaki et al. 2004; Marcano et al. 2007) and regulators of G protein signaling in anxiety (Fullerton et al. 2008).
The more general question, of the extent of overlap between QTLs identified in different species, remains unclear. Data from mouse QTL and human linkage studies were used by Paigen and colleagues to show that more than half of human atherosclerosis QTLs are located in regions homologous to mouse QTLs (Wang et al. 2005a). Human linkage results are, however, notoriously unreliable and, as Risch and colleagues pointed out, concordance is unlikely given that the genetic effect in humans depends on disease allele frequencies and such allele frequencies are unpredictable (Risch et al. 1993).
The more recent GWAS data present a new opportunity to revisit this question. The relatively large number of genes now identified in humans, and progress in mapping QTLs at high resolution in mice (Valdar et al. 2006a) should allow comparisons of the extent of overlap. These analyses have yet to be reported.
Overlaps between mammals and Drosophila at a QTL level have not been reported. At a mechanistic level there are examples of genes involved in similar processes, including learning and memory (Dubnau and Tully 1998). More intriguing is the possibility that, at a systems level, there may be commonalities. Could networks of coregulated genes, representing physiological processes, be common between species, and networks accessed by their genetic determinants? A recent study of liver and adipose gene expression in human and mouse tissue suggested that this could be the case: A common macrophage-enriched network was found (Chen et al. 2008; Emilsson et al. 2008). While it is not clear to what extent this may simply reflect common histological characteristics of fatty liver infiltrated by macrophages, the idea of searching for commonality at the systems level is intriguing. As transcriptomic data become more widely available, it will become possible to explore this idea.
Conclusions
Our review of the genetic architecture of quantitative traits has highlighted considerable agreement between analyses carried out in flies, rodents, and our own species. A picture emerges of large numbers of loci of small effect, although the distribution of allelic effects is not consistent with the “infinitesimal” model of quantitative genetic variation (Falconer and Mackay 1996; Lynch and Walsh 1998). Discrepant findings between species are largely attributable to differences in experimental design. For example, geneticists working with model organisms frequently exploit the opportunities provided to analyze inbred strains or derived crosses, while human geneticists are restricted to working with outbred populations. Differences in the number and frequencies of alleles segregating in outbred populations, compared with those in crosses between inbred strains, contribute to differences in inferred quantitative genetic architecture.
Systematic measures of the extent of context-dependent effects in human populations are still lacking, despite robust evidence from model organisms of their importance. Again, extrapolating findings from one species to another is problematic: We cannot compute the expected contributions of the epistasis observed from studies of mutations and QTLs in model organisms to the population genetic variance without knowledge of the allele frequencies in natural populations. To restate, epistatic variance depends on both the size of genetic effects and allele frequencies. Large effects on the mean difference between genotypes may not translate to large effects on the variance if allele frequencies are low. The same arguments apply to studies of genes by environment interaction.
We have highlighted what we believe to be the largely unappreciated importance of pleiotropy. We do not refer here to the expected correlation of genetic effects between related phenotypes, such as height, weight, or morphology. Nor do we refer to pleiotropy consequent upon linkage, that is to say, when mapping of two apparently unrelated traits identifies the same genetic locus because of the close proximity on the genome of two genes that have separate and independent functions. We refer, rather, to the observation from studies of mutations in flies and mice, and now, increasingly, from high-resolution mapping studies in humans, that one gene may have a role in two or more phenotypes previously thought to be unrelated. An appreciation of pleiotropy, of its extent, and how it works, will be critical for interpretations of the functional analysis of every gene in the vertebrate genome, accessible through the creation of a complete set of mouse knockouts.
Finally, when we consider the molecular basis of quantitative traits, we find an important role for noncoding DNA. Many quantitative trait nucleotides lie in what are assumed to be regulatory regions of the genome, though whether this is necessarily the regulation of gene expression or of other features of genome function remains unknown. Given the differences in gene function and population structure between the organisms studied, it is not surprising that there is little evidence for conservation of the position of QTLs between species; however, there has been no thorough search for such loci, largely because until recently the necessary resources of high-resolution high-quality QTL maps were not present. Finally, as more data accumulate and, particularly, with access to whole-genome sequences of multiple individuals in any species, the coming years will undoubtedly see remarkable advances in our understanding of the molecular basis of the genetic architecture of quantitative traits.
Acknowledgments
J.F. is supported by the Wellcome Trust, and T.F.C.M. by grants from the National Institutes of Health.
Footnotes
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.086660.108.
References
- Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E., et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Alemayehu A., Breen L., Krenova D., Printz M.P. Reciprocal rat chromosome 2 congenic strains reveal contrasting blood pressure and heart rate QTL. Physiol. Genomics. 2002;10:199–210. doi: 10.1152/physiolgenomics.00065.2002. [DOI] [PubMed] [Google Scholar]
- Amundadottir L.T., Sulem P., Gudmundsson J., Helgason A., Baker A., Agnarsson B.A., Sigurdsson A., Benediktsdottir K.R., Cazier J.B., Sainz J., et al. A common variant associated with prostate cancer in European and African populations. Nat. Genet. 2006;38:652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- Ankra-Badu G.A., Pomp D., Shriner D., Allison D.B., Yi N. Genetic influences on growth and body composition in mice: Multilocus interactions. Int. J. Obes. (Lond). 2009;33:89–95. doi: 10.1038/ijo.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyarajah A., Palijan A., Dutil J., Prithiviraj K., Deng Y., Deng A.Y. Dissecting quantitative trait loci into opposite blood pressure effects on Dahl rat chromosome 8 by congenic strains. J. Hypertens. 2004;22:1495–1502. doi: 10.1097/01.hjh.0000133720.94075.6f. [DOI] [PubMed] [Google Scholar]
- Austin C.P., Battey J.F., Bradley A., Bucan M., Capecchi M., Collins F.S., Dove W.F., Duyk G., Dymecki S., Eppig J.T., et al. The knockout mouse project. Nat. Genet. 2004;36:921–924. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihl F., Brahic M., Bureau J.F. Two loci, Tmevp2 and Tmevp3, located on the telomeric region of chromosome 10, control the persistence of Theiler's virus in the central nervous system of mice. Genetics. 1999;152:385–392. doi: 10.1093/genetics/152.1.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann G.A., Kratzsch J., Haley C.S., Renne U., Schwerin M., Karle S. Single QTL effects, epistasis, and pleiotropy account for two-thirds of the phenotypic F2 variance of growth and obesity in DU6i x DBA/2 mice. Genome Res. 2000;10:1941–1957. doi: 10.1101/gr.gr1499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M.A., Jordan K.W., Lyman R.F., Harbison S.T., Leips J., Morgan T.J., DeLuca M., Awadalla P., Mackay T.F. Phenotypic variation and natural selection at Catsup, a pleiotropic quantitative trait gene in Drosophila. Curr. Biol. 2006;16:912–919. doi: 10.1016/j.cub.2006.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M., Schrodi S.J., Chang M., Garcia V.E., Brandon R., Callis K.P., Matsunami N., Ardlie K.G., Civello D., Catanese J.J., et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A., Sugden K., Moffitt T.E., Taylor A., Craig I.W., Harrington H., McClay J., Mill J., Martin J., Braithwaite A., et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chen Y., Zhu J., Lum P.Y., Yang X., Pinto S., MacNeil D.J., Zhang C., Lamb J., Edwards S., Sieberts S.K., et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud J.M., Routman E.J. Epistasis and its contribution to genetic variance components. Genetics. 1995;139:1455–1461. doi: 10.1093/genetics/139.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud J.M., Vaughn T.T., Pletscher L.S., Peripato A.C., Adams E.S., Erikson C.F., King-Ellison K.J. Genetic architecture of adiposity in the cross of LG/J and SM/J inbred mice. Mamm. Genome. 2001;12:3–12. doi: 10.1007/s003350010218. [DOI] [PubMed] [Google Scholar]
- Christians J.K., Keightley P.D. Fine mapping of a murine growth locus to a 1.4-cM region and resolution of linked QTL. Mamm. Genome. 2004;15:482–491. doi: 10.1007/s00335-004-3046-0. [DOI] [PubMed] [Google Scholar]
- Clark A.G., Wang L. Epistasis in measured genotypes: Drosophila P-element insertions. Genetics. 1997;147:157–163. doi: 10.1093/genetics/147.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- Collins F.S., Rossant J., Wurst W. A mouse for all reasons. Cell. 2007;128:9–13. doi: 10.1016/j.cell.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- De Luca M., Roshina N.V., Geiger-Thornsberry G.L., Lyman R.F., Pasyukova E.G., Mackay T.F. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 2003;34:429–433. doi: 10.1038/ng1218. [DOI] [PubMed] [Google Scholar]
- Dewan A., Liu M., Hartman S., Zhang S.S., Liu D.T., Zhao C., Tam P.O., Chan W.M., Lam D.S., Snyder M., et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- Dilda C.L., Mackay T.F. The genetic architecture of Drosophila sensory bristle number. Genetics. 2002;162:1655–1674. doi: 10.1093/genetics/162.4.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–731. doi: 10.1038/nature07631. [DOI] [PubMed] [Google Scholar]
- Dubnau J., Tully T. Gene discovery in Drosophila: New insights for learning and memory. Annu. Rev. Neurosci. 1998;21:407–444. doi: 10.1146/annurev.neuro.21.1.407. [DOI] [PubMed] [Google Scholar]
- Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A., et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin I., Palsson A., Birdsall K., Gibson G. Evidence that Egfr contributes to cryptic genetic variation for photoreceptor determination in natural populations of Drosophila melanogaster. Curr. Biol. 2003;13:1888–1893. doi: 10.1016/j.cub.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Edwards A.C., Rollmann S.M., Morgan T.J., Mackay T.F. Quantitative genomics of aggressive behavior in Drosophila melanogaster. PLoS Genet. 2006;2:e154. doi: 10.1371/journal.pgen.0020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J., Carlson S., Helgason A., Walters G.B., Gunnarsdottir S., et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- Emison E.S., McCallion A.S., Kashuk C.S., Bush R.T., Grice E., Lin S., Portnoy M.E., Cutler D.J., Green E.D., Chakravarti A. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–863. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer D.S., Mackay T.F.C. Introduction to quantitative genetics. 4th ed. Longman; Harlow, UK: 1996. [Google Scholar]
- Fanara J.J., Robinson K.O., Rollmann S.M., Anholt R.R., Mackay T.F. Vanaso is a candidate quantitative trait gene for Drosophila olfactory behavior. Genetics. 2002;162:1321–1328. doi: 10.1093/genetics/162.3.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorowicz G.M., Fry J.D., Anholt R.R., Mackay T.F. Epistatic interactions between smell-impaired loci in Drosophila melanogaster. Genetics. 1998;148:1885–1891. doi: 10.1093/genetics/148.4.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J., De Fries J.C., Henderson N.D. Little epistasis for anxiety-related measures in the DeFries strains of laboratory mice. Mamm. Genome. 2004;15:77–82. doi: 10.1007/s00335-003-3033-x. [DOI] [PubMed] [Google Scholar]
- Flint J., Valdar W., Shifman S., Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Frantz S., Clemitson J.R., Bihoreau M.T., Gauguier D., Samani N.J. Genetic dissection of region around the Sa gene on rat chromosome 1: Evidence for multiple loci affecting blood pressure. Hypertension. 2001;38:216–221. doi: 10.1161/01.hyp.38.2.216. [DOI] [PubMed] [Google Scholar]
- Freedman M.L., Haiman C.A., Patterson N., McDonald G.J., Tandon A., Waliszewska A., Penney K., Steen R.G., Ardlie K., John E.M., et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. 2006;103:14068–14073. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry J.D., Nuzhdin S.V., Pasyukova E.G., Mackay T.F. QTL mapping of genotype-environment interaction for fitness in Drosophila melanogaster. Genet. Res. 1998;71:133–141. doi: 10.1017/s0016672398003176. [DOI] [PubMed] [Google Scholar]
- Fullerton J.M., Willis-Owen S.A., Yalcin B., Shifman S., Copley R.R., Miller S.R., Bhomra A., Davidson S., Oliver P.L., Mott R., et al. Human-mouse quantitative trait locus concordance and the dissection of a human neuroticism locus. Biol. Psychiatry. 2008;63:874–883. doi: 10.1016/j.biopsych.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Gale G.D., Yazdi R.D., Khan A.H., Lusis A.J., Davis R.C., Smith D.J. A genome-wide panel of congenic mice reveals widespread epistasis of behavior quantitative trait loci. Mol. Psychiatry. 2008 doi: 10.1038/mp.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett M.R., Rapp J.P. Multiple blood pressure QTL on rat chromosome 2 defined by congenic Dahl rats. Mamm. Genome. 2002a;13:41–44. doi: 10.1007/s00335-001-2114-y. [DOI] [PubMed] [Google Scholar]
- Garrett M.R., Rapp J.P. Two closely linked interactive blood pressure QTL on rat chromosome 5 defined using congenic Dahl rats. Physiol. Genomics. 2002b;8:81–86. doi: 10.1152/physiolgenomics.00080.2001. [DOI] [PubMed] [Google Scholar]
- Geiger-Thornsberry G.L., Mackay T.F. Association of single-nucleotide polymorphisms at the Delta locus with genotype by environment interaction for sensory bristle number in Drosophila melanogaster. Genet. Res. 2002;79:211–218. doi: 10.1017/s0016672302005621. [DOI] [PubMed] [Google Scholar]
- Grant S.F., Thorleifsson G., Reynisdottir I., Benediktsson R., Manolescu A., Sainz J., Helgason A., Stefansson H., Emilsson V., Helgadottir A., et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Walters G.B., Thorleifsson G., Stefansson H., Halldorsson B.V., Zusmanovich P., Sulem P., Thorlacius S., Gylfason A., Steinberg S., et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008;40:609–615. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- Gudmundsson J., Sulem P., Steinthorsdottir V., Bergthorsson J.T., Thorleifsson G., Manolescu A., Rafnar T., Gudbjartsson D., Agnarsson B.A., Baker A., et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- Gurganus M.C., Fry J.D., Nuzhdin S.V., Pasyukova E.G., Lyman R.F., Mackay T.F. Genotype-environment interaction at quantitative trait loci affecting sensory bristle number in Drosophila melanogaster. Genetics. 1998;149:1883–1898. doi: 10.1093/genetics/149.4.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurganus M.C., Nuzhdin S.V., Leips J.W., Mackay T.F. High-resolution mapping of quantitative trait loci for sternopleural bristle number in Drosophila melanogaster. Genetics. 1999;152:1585–1604. doi: 10.1093/genetics/152.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison S.T., Yamamoto A.H., Fanara J.J., Norga K.K., Mackay T.F. Quantitative trait loci affecting starvation resistance in Drosophila melanogaster. Genetics. 2004;166:1807–1823. doi: 10.1534/genetics.166.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir A., Thorleifsson G., Manolescu A., Gretarsdottir S., Blondal T., Jonasdottir A., Jonasdottir A., Sigurdsson A., Baker A., Palsson A., et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- Hilbert P., Lindpaintner K., Beckmann J.S., Serikawa T., Soubrier F., Dubay C., Cartwright P., De Gouyon B., Julier C., Takahasi S., et al. Chromosomal mapping of two genetic loci associated with blood-pressure regulation in hereditary hypertensive rats. Nature. 1991;353:521–529. doi: 10.1038/353521a0. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis M.H., Flaswinkel H., Fuchs H., Rathkolb B., Soewarto D., Marschall S., Heffner S., Pargent W., Wuensch K., Jung M., et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- Hunt K.A., Zhernakova A., Turner G., Heap G.A., Franke L., Bruinenberg M., Romanos J., Dinesen L.C., Ryan A.W., Panesar D., et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob H., Lindpaintner K., Lincoln S., Kusumi K., Bunker R., Mao Y., Ganten D., Dzau V., Lander E. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell. 1991;67:213–224. doi: 10.1016/0092-8674(91)90584-l. [DOI] [PubMed] [Google Scholar]
- Ji W., Foo J.N., O'Roak B.J., Zhao H., Larson M.G., Simon D.B., Newton-Cheh C., State M.W., Levy D., Lifton R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K.W., Morgan T.J., Mackay T.F. Quantitative trait loci for locomotor behavior in Drosophila melanogaster. Genetics. 2006;174:271–284. doi: 10.1534/genetics.106.058099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K.W., Carbone M.A., Yamamoto A., Morgan T.J., Mackay T.F. Quantitative genomics of locomotor behavior in Drosophila melanogaster. Genome Biol. 2007;8:R172. doi: 10.1186/gb-2007-8-8-r172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisaki P.J., Delepine M., Woon P.Y., Sebag-Montefiore L., Wilder S.P., Menzel S., Vionnet N., Marion E., Riveline J.P., Charpentier G., et al. Polymorphisms in type II SH2 domain-containing inositol 5-phosphatase (INPPL1, SHIP2) are associated with physiological abnormalities of the metabolic syndrome. Diabetes. 2004;53:1900–1904. doi: 10.2337/diabetes.53.7.1900. [DOI] [PubMed] [Google Scholar]
- Katsanis N., Ansley S.J., Badano J.L., Eichers E.R., Lewis R.A., Hoskins B.E., Scambler P.J., Davidson W.S., Beales P.L., Lupski J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- Kendler K.S., Greenspan R.J. The nature of genetic influences on behavior: Lessons from “simpler” organisms. Am. J. Psychiatry. 2006;163:1683–1694. doi: 10.1176/ajp.2006.163.10.1683. [DOI] [PubMed] [Google Scholar]
- Klingenberg C.P., Leamy L.J., Routman E.J., Cheverud J.M. Genetic architecture of mandible shape in mice: Effects of quantitative trait loci analyzed by geometric morphometrics. Genetics. 2001;157:785–802. doi: 10.1093/genetics/157.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg C.P., Leamy L.J., Cheverud J.M. Integration and modularity of quantitative trait locus effects on geometric shape in the mouse mandible. Genetics. 2004;166:1909–1921. doi: 10.1534/genetics.166.4.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch W., Hoppmann P., Mueller J.C., Schomig A., Kastrati A. Lack of support for association between common variation in TNFSF4 and myocardial infarction in a German population. Nat. Genet. 2008;40:1386–1387. doi: 10.1038/ng1208-1386. [DOI] [PubMed] [Google Scholar]
- Kotowski I.K., Pertsemlidis A., Luke A., Cooper R.S., Vega G.L., Cohen J.C., Hobbs H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006;78:410–422. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C., Lyman R.F., Long A.D., Langley C.H., Mackay T.F. Naturally occurring variation in bristle number and DNA polymorphisms at the scabrous locus of Drosophila melanogaster. Science. 1994;266:1697–1702. doi: 10.1126/science.7992053. [DOI] [PubMed] [Google Scholar]
- Lai C.Q., Leips J., Zou W., Roberts J.F., Wollenberg K.R., Parnell L.D., Zeng Z.B., Ordovas J.M., Mackay T.F. Speed-mapping quantitative trait loci using microarrays. Nat. Methods. 2007;4:839–841. doi: 10.1038/nmeth1084. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro B.P., Sackton T.B., Clark A.G. Genetic variation in Drosophila melanogaster resistance to infection: A comparison across bacteria. Genetics. 2006;174:1539–1554. doi: 10.1534/genetics.105.054593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy L.J., Pomp D., Eisen E.J., Cheverud J.M. Pleiotropy of quantitative trait loci for organ weights and limb bone lengths in mice. Physiol. Genomics. 2002;10:21–29. [Google Scholar]
- Legare M.E., Bartlett F.S., Frankel W.N. A major effect QTL determined by multiple genes in epileptic EL mice. Genome Res. 2000;10:42–48. [PMC free article] [PubMed] [Google Scholar]
- Leips J., Mackay T.F. Quantitative trait loci for life span in Drosophila melanogaster: Interactions with genetic background and larval density. Genetics. 2000;155:1773–1788. doi: 10.1093/genetics/155.4.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leips J., Mackay T.F. The complex genetic architecture of Drosophila life span. Exp. Aging Res. 2002;28:361–390. doi: 10.1080/03610730290080399. [DOI] [PubMed] [Google Scholar]
- Long A.D., Mullaney S.L., Reid L.A., Fry J.D., Langley C.H., Mackay T.F. High-resolution mapping of genetic factors affecting abdominal bristle number in Drosophila melanogaster. Genetics. 1995;139:1273–1291. doi: 10.1093/genetics/139.3.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A.D., Lyman R.F., Langley C.H., Mackay T.F. Two sites in the Delta gene region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics. 1998;149:999–1017. doi: 10.1093/genetics/149.2.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A.D., Lyman R.F., Morgan A.H., Langley C.H., Mackay T.F. Both naturally occurring insertions of transposable elements and intermediate frequency polymorphisms at the achaete-scute complex are associated with variation in bristle number in Drosophila melanogaster. Genetics. 2000;154:1255–1269. doi: 10.1093/genetics/154.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman R.F., Lai C., Mackay T.F. Linkage disequilibrium mapping of molecular polymorphisms at the scabrous locus associated with naturally occurring variation in bristle number in Drosophila melanogaster. Genet. Res. 1999;74:303–311. doi: 10.1017/s001667239900419x. [DOI] [PubMed] [Google Scholar]
- Lynch M., Walsh B. Genetics and analysis of quantitative traits. Sinauer Associates; Sunderland, MA: 1998. [Google Scholar]
- Mackay T.F. The genetic architecture of quantitative traits: Lessons from Drosophila. Curr. Opin. Genet. Dev. 2004;14:253–257. doi: 10.1016/j.gde.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Mackay T.F., Langley C.H. Molecular and phenotypic variation in the achaete-scute region of Drosophila melanogaster. Nature. 1990;348:64–66. doi: 10.1038/348064a0. [DOI] [PubMed] [Google Scholar]
- Mackay T.F., Heinsohn S.L., Lyman R.F., Moehring A.J., Morgan T.J., Rollmann S.M. Genetics and genomics of Drosophila mating behavior. Proc. Natl. Acad. Sci. 2005;102(Suppl. 1):6622–6629. doi: 10.1073/pnas.0501986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay T.F.C., Roshina N.V., Leips J.W., Pasyukova E.G. Complex genetic architecture of Drosophila longevity. In: Masaro E.J., Austad S.N., editors. Handbook of the biology of aging. 6th ed. Elsevier, Academic Press; San Diego, CA: 2006. pp. 181–216. [Google Scholar]
- Marcano A.C., Burke B., Gungadoo J., Wallace C., Kaisaki P.J., Woon P.Y., Farrall M., Clayton D., Brown M., Dominiczak A., et al. Genetic association analysis of inositol polyphosphate phosphatase-like 1 (INPPL1, SHIP2) variants with essential hypertension. J. Med. Genet. 2007;44:603–605. doi: 10.1136/jmg.2007.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I., Maller J.B., Kirby A., et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- McPherson R., Pertsemlidis A., Kavaslar N., Stewart A., Roberts R., Cox D.R., Hinds D.A., Pennacchio L.A., Tybjaerg-Hansen A., Folsom A.R., et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey J.G., Houle D., Nuzhdin S.V. Naturally segregating quantitative trait loci affecting wing shape of Drosophila melanogaster. Genetics. 2005;169:2101–2113. doi: 10.1534/genetics.104.036988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring A.J., Mackay T.F. The quantitative genetic basis of male mating behavior in Drosophila melanogaster. Genetics. 2004;167:1249–1263. doi: 10.1534/genetics.103.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova T.V., Anholt R.R., Mackay T.F. Phenotypic and transcriptional response to selection for alcohol sensitivity in Drosophila melanogaster. Genome Biol. 2007;8:R231. doi: 10.1186/gb-2007-8-10-r231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo M.R., Durrant C., Lewis G., Flint J. Gene X environment interactions at the serotonin transporter locus. Biol. Psychiatry. 2009;65:211–219. doi: 10.1016/j.biopsych.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Nolan P.M., Peters J., Strivens M., Rogers D., Hagan J., Spurr N., Gray I.C., Vizor L., Brooker D., Whitehill E., et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat. Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- Palsson A., Gibson G. Association between nucleotide variation in Egfr and wing shape in Drosophila melanogaster. Genetics. 2004;167:1187–1198. doi: 10.1534/genetics.103.021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyukova E.G., Vieira C., Mackay T.F. Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics. 2000;156:1129–1146. doi: 10.1093/genetics/156.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicev M., Kenney-Hunt J.P., Norgard E.A., Roseman C.C., Wolf J.B., Cheverud J.M. Genetic variation in pleiotropy: Differential epistasis as a source of variation in the allometric relationship between long bone lengths and body weight. Evolution Int. J. Org. Evolution. 2008;62:199–213. doi: 10.1111/j.1558-5646.2007.00255.x. [DOI] [PubMed] [Google Scholar]
- Podolin P.L., Denny P., Armitage N., Lord C.J., Hill N.J., Levy E.R., Peterson L.B., Todd J.A., Wicker L.S., Lyons P.A. Localization of two insulin-dependent diabetes (Idd) genes to the Idd10 region on mouse chromosome 3. Mamm. Genome. 1998;9:283–286. doi: 10.1007/s003359900749. [DOI] [PubMed] [Google Scholar]
- Puel A., Mevel J.C., Bouthillier Y., Decreusefond C., Fridman W.H., Feingold N., Mouton D. Identification of two quantitative trait loci involved in antibody production on mouse chromosome 8. Immunogenetics. 1998;47:326–331. doi: 10.1007/s002510050365. [DOI] [PubMed] [Google Scholar]
- Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Risch N., Ghosh S., Todd J.A. Statistical evaluation of multiple-locus linkage data in experimental species and its relevance to human studies: Application to nonobese diabetic (NOD) mouse and human insulin-dependent diabetes mellitus (IDDM) Am. J. Hum. Genet. 1993;53:702–714. [PMC free article] [PubMed] [Google Scholar]
- Robertson A. The nature of quantitative genetic variation. In: Brink R.A., Styles E.D., editors. Heritage from Mendel. University of Wisconsin; Madison, WI: 1967. pp. 265–280. [Google Scholar]
- Robin C., Lyman R.F., Long A.D., Langley C.H., Mackay T.F. hairy: A quantitative trait locus for Drosophila sensory bristle number. Genetics. 2002;162:155–164. doi: 10.1093/genetics/162.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollmann S.M., Magwire M.M., Morgan T.J., Ozsoy E.D., Yamamoto A., Mackay T.F., Anholt R.R. Pleiotropic fitness effects of the Tre1-Gr5a region in Drosophila melanogaster. Nat. Genet. 2006;38:824–829. doi: 10.1038/ng1823. [DOI] [PubMed] [Google Scholar]
- Rollmann S.M., Yamamoto A., Goossens T., Zwarts L., Callaerts-Vegh Z., Callaerts P., Norga K., Mackay T.F., Anholt R.R. The early developmental gene Semaphorin 5c contributes to olfactory behavior in adult Drosophila. Genetics. 2007;176:947–956. doi: 10.1534/genetics.106.069781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S., Pennacchio L.A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H.H., Cohen J.C. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozmahel R., Wilschanski M., Matin A., Plyte S., Oliver M., Auerbach W., Moore A., Forstner J., Durie P., Nadeau J., et al. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat. Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- Samani N.J., Erdmann J., Hall A.S., Hengstenberg C., Mangino M., Mayer B., Dixon R.J., Meitinger T., Braund P., Wichmann H.E., et al. Genome-wide association analysis of coronary artery disease. N. Engl. J. Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambandan D., Yamamoto A., Fanara J.J., Mackay T.F., Anholt R.R. Dynamic genetic interactions determine odor-guided behavior in Drosophila melanogaster. Genetics. 2006;174:1349–1363. doi: 10.1534/genetics.106.060574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U., et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J., et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Burrage L.C., Sinasac D.S., Hill A.E., Ernest S.R., O'Brien W., Courtland H.W., Jepsen K.J., Kirby A., Kulbokas E.J., et al. Genetic architecture of complex traits: Large phenotypic effects and pervasive epistasis. Proc. Natl. Acad. Sci. 2008;105:19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer J.B., Hill A.E., Burrage L.C., Olszens K.R., Song J., Justice M., O'Brien W.E., Conti D.V., Witte J.S., Lander E.S., et al. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;304:445–448. doi: 10.1126/science.1093139. [DOI] [PubMed] [Google Scholar]
- Slattery M.L., Folsom A.R., Wolff R., Herrick J., Caan B.J., Potter J.D. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer Epidemiol. Biomarkers Prev. 2008;17:978–982. doi: 10.1158/1055-9965.EPI-07-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth D.J., Plagnol V., Walker N.M., Cooper J.D., Downes K., Yang J.H., Howson J.M., Stevens H., McManus R., Wijmenga C., et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Engl. J. Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam L.F., Laurie C.C. Molecular dissection of a major gene effect on a quantitative trait: The level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics. 1996;144:1559–1564. doi: 10.1093/genetics/144.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H., Rujescu D., Cichon S., Pietilainen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianou I.M., Christians J.K., Keightley P.D., Bunger L., Clinton M., Bulfield G., Horvat S. Genetic complexity of an obesity QTL (Fob3) revealed by detailed genetic mapping. Mamm. Genome. 2004;15:472–481. doi: 10.1007/s00335-004-3039-z. [DOI] [PubMed] [Google Scholar]
- Thomas G., Jacobs K.B., Yeager M., Kraft P., Wacholder S., Orr N., Yu K., Chatterjee N., Welch R., Hutchinson A., et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A., Styrkarsdottir U., Gretarsdottir S., Thorlacius S., Jonsdottir I., et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- Todd J.A., Aitman T.J., Cornall R.J., Ghosh S., Hall J.R., Hearne C.M., Knight A.M., Love J.M., McAleer M.A., Prins J.B., et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 1991;351:542–547. doi: 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- Turri M.G., Datta S.R., DeFries J., Henderson N.D., Flint J. QTL analysis identifies multiple behavioral dimensions in ethological tests of anxiety in laboratory mice. Curr. Biol. 2001a;11:725–734. doi: 10.1016/s0960-9822(01)00206-8. [DOI] [PubMed] [Google Scholar]
- Turri M.G., Henderson N.D., DeFries J.C., Flint J. Quantitative trait locus mapping in laboratory mice derived from a replicated selection experiment for open-field activity. Genetics. 2001b;158:1217–1226. doi: 10.1093/genetics/158.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H., Howson J.M., Esposito L., Heward J., Snook H., Chamberlain G., Rainbow D.B., Hunter K.M., Smith A.N., Di Genova G., et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- Valdar W., Solberg L.C., Gauguier D., Burnett S., Klenerman P., Cookson W.O., Taylor M.S., Rawlins J.N., Mott R., Flint J. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 2006a;38:879–887. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- Valdar W., Solberg L.C., Gauguier D., Cookson W.O., Rawlins J.N., Mott R., Flint J. Genetic and environmental effects on complex traits in mice. Genetics. 2006b;174:959–984. doi: 10.1534/genetics.106.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swinderen B., Greenspan R.J. Flexibility in a gene network affecting a simple behavior in Drosophila melanogaster. Genetics. 2005;169:2151–2163. doi: 10.1534/genetics.104.032631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken K.J., Bellen H.J. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat. Rev. Genet. 2005;6:167–178. doi: 10.1038/nrg1553. [DOI] [PubMed] [Google Scholar]
- Vieira C., Pasyukova E.G., Zeng Z.B., Hackett J.B., Lyman R.F., Mackay T.F. Genotype-environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics. 2000;154:213–227. doi: 10.1093/genetics/154.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A., et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Wang X., Ishimori N., Korstanje R., Rollins J., Paigen B. Identifying novel genes for atherosclerosis through mouse-human comparative genetics. Am. J. Hum. Genet. 2005a;77:1–15. doi: 10.1086/431656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ria M., Kelmenson P.M., Eriksson P., Higgins D.C., Samnegard A., Petros C., Rollins J., Bennet A.M., Wiman B., et al. Positional identification of TNFSF4, encoding OX40 ligand, as a gene that influences atherosclerosis susceptibility. Nat. Genet. 2005b;37:365–372. doi: 10.1038/ng1524. [DOI] [PubMed] [Google Scholar]
- Weber K., Eisman R., Morey L., Patty A., Sparks J., Tausek M., Zeng Z.B. An analysis of polygenes affecting wing shape on chromosome 3 in Drosophila melanogaster. Genetics. 1999;153:773–786. doi: 10.1093/genetics/153.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K., Eisman R., Higgins S., Morey L., Patty A., Tausek M., Zeng Z.B. An analysis of polygenes affecting wing shape on chromosome 2 in Drosophila melanogaster. Genetics. 2001;159:1045–1057. doi: 10.1093/genetics/159.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer C.J., Speliotes E.K., Loos R.J., Li S., Lindgren C.M., Heid I.M., Berndt S.I., Elliott A.L., Jackson A.U., Lamina C., et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R.H., Morgan T.J., Mackay T.F. High-resolution mapping of quantitative trait loci affecting increased life span in Drosophila melanogaster. Genetics. 2006;173:1455–1463. doi: 10.1534/genetics.105.055111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler W., Weedon M.N., Graham R.R., McCarroll S.A., Purcell S., Almgren P., Tuomi T., Gaudet D., Bostrom K.B., Walker M., et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56:685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- Wolf J.B., Leamy L.J., Routman E.J., Cheverud J.M. Epistatic pleiotropy and the genetic architecture of covariation within early and late-developing skull trait complexes in mice. Genetics. 2005;171:683–694. doi: 10.1534/genetics.104.038885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J.B., Pomp D., Eisen E.J., Cheverud J.M., Leamy L.J. The contribution of epistatic pleiotropy to the genetic architecture of covariation among polygenic traits in mice. Evol. Dev. 2006;8:468–476. doi: 10.1111/j.1525-142X.2006.00120.x. [DOI] [PubMed] [Google Scholar]
- Workman M.S., Leamy L.J., Routman E.J., Cheverud J.M. Analysis of quantitative trait locus effects on the size and shape of mandibular molars in mice. Genetics. 2002;160:1573–1586. doi: 10.1093/genetics/160.4.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Evolution and the genetics for populations, Vol III: Experimental results and evolutionary deductions. Chicago University Press; Chicago, IL: 1977. [Google Scholar]
- Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Zwarts L., Callaerts P., Norga K., Mackay T.F., Anholt R.R. Neurogenetic networks for startle-induced locomotion in Drosophila melanogaster. Proc. Natl. Acad. Sci. 2008;105:12393–12398. doi: 10.1073/pnas.0804889105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi N., Zinniel D.K., Kim K., Eisen E.J., Bartolucci A., Allison D.B., Pomp D. Bayesian analyses of multiple epistatic QTL models for body weight and body composition in mice. Genet. Res. 2006;87:45–60. doi: 10.1017/S0016672306007944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi N., Shriner D., Banerjee S., Mehta T., Pomp D., Yandell B.S. An efficient Bayesian model selection approach for interacting quantitative trait loci models with many effects. Genetics. 2007;176:1865–1877. doi: 10.1534/genetics.107.071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E., Weedon M.N., Lindgren C.M., Frayling T.M., Elliott K.S., Lango H., Timpson N.J., Perry J.R., Rayner N.W., Freathy R.M., et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E., Scott L.J., Saxena R., Voight B.F., Marchini J.L., Hu T., de Bakker P.I., Abecasis G.R., Almgren P., Andersen G., et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenski J., Corey M., Rozmahel R., Markiewicz D., Aznarez I., Casals T., Larriba S., Mercier B., Cutting G.R., Krebsova A., et al. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat. Genet. 1999;22:128–129. doi: 10.1038/9635. [DOI] [PubMed] [Google Scholar]