

Abstract
Canalization refers to the process by which phenotypes are stabilized within species. Evolution by natural selection can proceed efficiently only when phenotypes are canalized. The existence and identity of canalizing genes have thus been an important, but controversial topic. Recent evidence has increasingly hinted that microRNAs may be involved in canalizing gene expression. Their paradoxical properties (e.g., strongly conserved but functionally dispensable) suggest unconventional regulatory roles. We synthesized published and unpublished results and hypothesize that miRNAs may have dual functions—in gene expression tuning and in expression buffering. In tuning, miRNAs modify the mean expression level of their targets, but in buffering they merely reduce the variance around a preset mean. In light of the constant emergence of new miRNAs, we further discuss the relative importance of these two functions in evolution.
Living organisms process as much information and execute as many instructions as any high-power computers normally do. The difference is that living organisms carry out the tasks under extremely variable environments, whereas no computers are made to withstand even moderate fluctuations in voltage input. Living things must be able to dampen variable inputs (in nutrition, temperature, humidity, genetic background, etc.) to achieve the remarkable stability in the output (development, physiological responses, gene expression, etc.). This phenomenon is the essence of biological homeostasis.
Darwin's The Origin of Species (Darwin 1859), on the other hand, is about evolutionary changes. On the surface, evolutionary changes and phenotypic stability may seem like antithetical concepts, but in the language of genetics they are really two sides of the same issue. Evolution by natural selection can proceed only when biological systems are reasonably stable. Imagine a system in which phenotypic manifestation is highly variable. In it natural selection cannot easily distinguish one genotype from another and would have low efficacy. The original nongenetic version of the Darwinian theory encountered many difficulties. One of them was the blending of genetic materials, potentially leading to the homogenization of phenotypes. The other resulted from the ignorance of the difference between genotype and phenotype and the puzzle over the existence of phenotypic variation. Fisher (1930) pointed out that, in Darwin's thinking, phenotypic variation should have been purged by natural selection—only the fittest should have remained.
The relationship between evolution and biological homeostasis can best be expressed in quantitative genetic terms. Let Vg and Ve denote phenotypic variance due to genotypic and environmental effect, respectively. Vg × e is the interactive term between gene and environment. The total phenotypic variance, Vt, can be expressed as follows:
For simplicity, we assume no dominance here, but the general principle is the same in more complex systems. Any mechanism of biological homeostasis should reduce these variances. While the reduction of Vg might reduce the efficacy of natural selection, as noted above, the more relevant quantity is the heritability, broadly defined as the ratio Vg/Vt. The rate of evolutionary change in response to selection is proportional to the heritability, not Vg. Hence, mechanisms that reduce phenotypic variance in general could indeed improve the efficacy of selection if they reduce Ve and Vg × e more strongly than they reduce Vg.
Waddington (1942, 1959) put biological homeostasis in the evolutionary context and formulated the concept of phenotypic canalization. Developmental program, in the metaphor that water in a valley always flows onto a stable path, is canalized. One may imagine a landscape with multiple valleys. Phenotypes of different species may follow the contours of different valleys, but within each species the phenotypes are robust, stereotypic, and canalized.
This vision was beautifully illustrated by the classical experiments of Rendel (1967) on the genetic control of scutellar bristles in Drosophila. This bristle number does not vary within species (for example, always four in Drosophila melanogaster) but has diverged among Drosophila species, making it a diagnostic trait in species identification. A mutation at the scute locus in D. melanogaster allowed flies to escape canalization and develop fewer (one to three) bristles. With relative ease, artificial selection could move the bristle number up again. (Hence, Rendel provided one of the earliest evidence of hidden genetic variation for morphological characters.) What seems most fascinating is that when the bristle number reached four, it was much more difficult to move out of this number, as if indeed there was a valley of attraction. Furthermore, once this barrier was crossed, it became easy again to select for increased number of bristles. The extra bristles also resemble those of other species in their orders and locations on the scutellum.
Rendel's experiments nicely painted a picture of multiple phenotypic states, each well canalized within a species. In the absence of a molecular mechanism, Rendel's experiments, albeit often discussed, were not followed up in the subsequent decades.
Gene expression as the trait under canalization
In this work, we shall focus on whole-genome gene expression as the phenotype of interest. We assume that whole-genome expression (WGE), like other phenotypes, needs to be “canalized,” as there is substantial stochasticity, both internal and external of the cells, in gene expression (Kaern et al. 2005; Raser and O'Shea 2005). Unlike morphological or other complex phenotypes, gene expression can be both phenotype and genotype (the latter being true in the case of cis-regulation). Hence, the molecular genetic basis of WGE is better understood than morphological phenotypes, for example. Furthermore, with WGE, one can assay tens of thousands of phenotypes in one single experiment.
Do canalizing genes exist?
Whether the phenotype of interest is morphology, behavior, or WGE, the issue of greatest interest is the molecular mechanism(s) of canalization. Do cells need genes that function mainly as canalizing agents? Many studies (von Dassow et al. 2000; Meir et al. 2002; Siegal and Bergman 2002) suggested that complex networks, such as cellular transcription networks, are inherently stable. von Dassow et al. (2000) analyzed the segment polarity phenotype, attempting to simulate the 14-stripe pattern in Drosophila development. Modeling the known segment polarity genes, these authors could reproduce the patterning by simulation. The implication is that canalizing genes are not needed. Nevertheless, analysis of this kind cannot conclude the absence of canalizing genes, since genomes are highly redundant, often with overlapping systems serving similar functions. Lack of evidence in a limited setting does not imply absence.
The search for the elusive canalizing genes has indeed led to some interesting findings. In a series of studies, Lindquist and colleagues (Rutherford and Lindquist 1998) have uncovered a substantial amount of cryptic genetic variation in Drosophila, yeast, and Arabidopsis. These genetic variants do not have detectable phenotypic effects in the wild-type background. However, in the absence of the heat shock protein HSP90, many abnormalities were observed. Given that HSP90 is a chaperon protein that helps the folding of many other proteins, it can conceivably influence the phenotypic consequences of those gene products. The hsp90 gene was suggested to be a canalizing gene (but see Bergman and Siegal [2003] and Hermisson and Wagner [2004] for discussions on the connection between canalization and cryptic genetic variation).
The hsp90 gene, and, in the example of Rendel (1967), the scute locus, are possible canalizing genes. There is little doubt that individual loci might play a canalizing role. The question is whether the genetic mechanisms are sufficiently general, such that a whole class of genes may underlie canalization.
miRNAs—Their paradoxical function and evolution
One of the major discoveries in the last decade is the existence of microRNAs (miRNA or miR), which constitute a class of post-transcriptional regulators of gene expression (Lee et al. 1993; Wightman et al. 1993; Bartel 2004; Bushati and Cohen 2007). miRNAs are small regulatory molecules 19–24 nucleotides (nt) long (Lee et al. 1993; Wightman et al. 1993; Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001). They normally guide the so-called RNA-induced silencing protein (RISC) complex to down-regulate transcripts that contain miR-matching sequences in their 3′ UTRs.
The rules of matching are different in animals and plants. Nucleotides at positions 2–8 of the miRNA are referred to as the seed. In animals, perfect (or nearly perfect) matching between the seed and the 3′ UTR is required, but the rest of the miRNA follows much less-stringent rules for pairing with targets (Lewis et al. 2003, 2005; Stark et al. 2003; Brennecke et al. 2005; Grun et al. 2005; Lim et al. 2005; Rajewsky 2006; Grimson et al. 2007). (In plants, matching between miRNAs and targets is more extensive [Rhoades et al. 2002].) Thus, in animals, each miRNA may have anywhere between 20 and 200 target genes and many genes are simultaneously the targets of more than one miRNA (Grun et al. 2005; Krek et al. 2005; Lewis et al. 2005; Rajewsky 2006). Not surprisingly, the down-regulation of a target gene by any particular miRNA is often modest and has been shown to be in excess of 50% only infrequently (Nakahara et al. 2005; Baek et al. 2008; Selbach et al. 2008).
Despite extensive studies, miRNAs in animals appear to be paradoxical regulators, as their properties at once suggest extreme functional importance and functional dispensability. Many miRNAs play a role in critical steps of animal development (Carrington and Ambros 2003; Alvarez-Garcia and Miska 2005; Wienholds and Plasterk 2005; Plasterk 2006; Stefani and Slack 2008), and many are highly conserved. For example, about 80 miRNAs are nearly identical within the genus Drosophila (Stark et al. 2007); many of them are conserved even between vertebrates and invertebrates (Sempere et al. 2006; Stark et al. 2007). Developmental regulation and evolutionary conservation should suggest functional importance of miRNAs.
In contrast, another suite of properties may be interpreted to mean that miRNAs are utterly unimportant. First, many miRNAs, even the highly conserved ones, can be individually deleted without causing any obvious defects (Li and Carthew 2005; Miska et al. 2007). It is customary to explain the dispensability in terms of functional redundancy, but it is nevertheless difficult to reconcile the extreme conservation with redundancy. For example, duplicated genes with redundant functions often experience accelerated evolution, likely the result of relaxed selective constraints. miRNAs are exceptional among gene loci in being both highly conserved and dispensable.
Second, the effects of miRNAs on the expression of their targets are usually modest when the miRNAs are mis-expressed (Baek et al. 2008; Selbach et al. 2008). In a most extreme manipulation, the Dicer-1 gene, which is required for the maturation of all miRNAs, was knocked out (Nakahara et al. 2005). Although this is a lethal condition, it is possible to assay gene expression in cell clones homozygous for the Dicer-1 deletion. Since all miRs should be absent, the lethality of Dicer-1 deletion is hardly unexpected. In contrast, it is rather surprising that the number of proteins showing significant mis-expression was modest (at only 9%, 94 out of 1003 proteins) in the same cells (Nakahara et al. 2005). Third, evidence suggests that, even for highly conserved miRNAs, their targets may not be noticeably conserved. We shall return to the subject later, but, again, it is hard to imagine specific functional significance in miRNA–target interactions.
The paradoxes suggest that miRNAs might have a biological role beyond the conventional regulatory functions, which are often well defined and vital (i.e., indispensable). Canalization could be such an unconventional role. Within the limited range of environmental perturbations in most experiments, canalization may not be particularly important, but, in the evolutionary timescale, it may be crucial. Indeed, microRNAs have increasingly attracted attention as candidates for canalizing agents (Cohen et al. 2006; Cui et al. 2006, 2007; Hornstein and Shomron 2006; Li et al. 2006; Choi et al. 2007; Tsang et al. 2007; Varghese and Cohen 2007; Martinez et al. 2008).
The dual functions of miRNAs—Expression tuning and expression buffering
In the literature, the proposed functions of miRNAs may be broadly classified into two categories. The first category is about setting the mean of the expression level of the target genes (referred to as expression tuning) and the second one is about reducing their variance (expression buffering, or homeostasis). The two functions correspond quite well to the circuitry classification of Tsang et al. (2007). As the buffering mechanism may work equally well around various means, these two functions are somewhat independent, and, hence, not mutually exclusive. Given that the tuning function has been the focus of many previous reviews (Plasterk 2006; Bushati and Cohen 2007; Stefani and Slack 2008; Sullivan 2008), it will be addressed only briefly in this section.
miRNAs often tune the expression levels of their target genes (i.e., reset their mean expression levels) to promote cell differentiation. Thanks to the direct action on the transcripts, miRNAs can tune the expression quickly, but the extent of tuning is often modest. An analogy for the role of miRNAs in gene expression may be a rheostat (or a dimmer, as opposed to a switch) (Bartel and Chen 2004). In this sense, the action of miRNAs resembles the genetic effect driving species and racial differentiation (see Wu and Palopoli 1994; Wu and Ting 2004).
Figure 1, A and C, present three simple motifs of the tuning mode in the transcription network. The simple repression motif (Fig. 1A) is illustrated nicely by the study of Xiao et al. (2007) (see also Hobert 2007). In the mouse immune system, miR-150 targets the Myb (also known as c-myb) gene, which then regulates the formation of pre-B and B1 cells. A high level of miR-150 holds Myb in check, preventing the pre-maturation of pro-B cells into pre-B cells. Although miR-150 Myb interaction should be embedded in a more complex network, it appears that this repression dominates other interactions in the particular cellular context.
Figure 1.

Simple network motifs containing miRNA. T denotes the target gene of miRNA; A and B denote other genes. These motifs are classified according to the functions in either tuning or buffering. The expression-tuning motifs include: (A) Simple repression in which miRNA reduces the expression of T; (B) coherent feed-forward loop, where miRNA reinforces the repression of T; (C) double-negative feedback loop, where miRNA and T mutually repress each other. The repression of T and expression of miR, or vice versa, is reinforced by the feedback. This is referred to as a “bistable” motif. The expression-buffering motifs include: (D) incoherent feed-forward loop (FFL), where miRNA buffers the expression of T against the fluctuation in A. (E) Negative feedback loop, where miR and T mutually buffer each other's expression from perturbation. (F) Incoherent FFL with four nodes, in which T1 and T2 buffer gene B against fluctuations in miR.
Another example of simple repression that has to be viewed in the context of a larger motif is the miR-9a senseless repression in Drosophila. The gene, senseless, activates proneural gene expression, which plays a key role in the development of sense organ precursors (SOPs) (Li et al. 2006) (see also Cohen et al. 2006). Beside being repressed by miR-9a, the senseless locus is coupled with another gene, E(spl), in a double-negative feedback loop. Such a loop (see Fig. 1C for an example) is a “bistable” motif in which either senseless or E(spl), but not both, would be expressed. The miR-9a senseless interaction would thus determine the eventual state of the bistable motif, which in turn would determine the formation of SOP vs. neighboring cells. In the Cohen et al. (2006) description, miR-9a sets the threshold for SOP formation.
Figure 1B is a coherent feed-forward loop in which the two pathways work coherently to reinforce the silencing of the target gene, T. There are many such examples that are variations on the theme shown in Figure 1B (Hornstein et al. 2005; Stark et al. 2005; Makeyev and Maniatis 2008; Marson et al. 2008). One more motif in the tuning mode is shown in Figure 1C. In the study of Rybak et al. (2008), the human miRNA, let-7, forms a double-negative feedback loop with the mammalian homolog of locus lin-28 in Caenorhabditis elegans. The expression of lin-28 determines the pluripotency of the embryonic stem cells. The locus lin-28 is additionally repressed by miR-125, which, like miR-9a above, tilts the “bistable” circuitry toward one of the two states.
In all of the examples above, miRNAs play a key role in tuning (or resetting) the expression level of the target gene, T, leading to the differentiation of immune cells, sensory organs, or stem cells, respectively.
miRNAs and expression buffering—Evidence from the literature
While tuning is about adjusting the mean expression level of the target gene, buffering is about reducing its variance. When there are no input noises, the buffering function might not be easily noticeable. Such noises could be present at various developmental stages or under environmental conditions that occur infrequently, say, once over many generations. Since most experiments were done in controlled environments with as little noise as possible, it is not surprising that canalizing genes have not been extensively identified. It should also be noted that, at the level of single cells, gene expression is stochastic in nature (Kaern et al. 2005; Raser and O'Shea 2005). Hence, mechanisms for expression buffering may be crucial at the level of single cells.
Figure 1, D and E, are motifs of the buffering mode. Figure 1D is an incoherent feed-forward loop (FFL) in which the two pathways between A and T are “incoherent”—up-regulation in one and down-regulation in the other. In the first pathway, A regulates the level of T, and, hence, the expression of T is not stable if the input value A does not stay constant. The second path, A → miR T, would distribute the variation in the input to miR, reducing the fluctuation in T. In the incoherent FFL, the second path going through miRNA would be dispensable if the input from A is stable. This dispensability may help explain why many miRNAs, even those that are highly conserved, can be deleted without having a strong phenotype. We rarely thought of genes whose primary function is to reduce the variance. The auxiliary path that goes through miRNA in Figure 1D could be considered such an example. Figure 1E is a negative feedback loop, effective in dampening the oscillations in the expression of both genes in the loop.
An example of the incoherent FFL is the MYC (also known as c-Myc) loop reported by O'Donnell et al. (2005). In this example, A of Figure 1D is MYC, T is the E2F1 gene, and miR is miR-17. E2F1 promotes cell cycle progression and may lead to tumorgenesis in humans. The incoherent FFL hence buffers E2F1 expression against variations in the expression of MYC. A good example of negative feedback loop (Fig. 1E) is given by Martinez et al. (2008). In this example, miR is miR-43 and T is lin-26; the latter being expressed in the embryos of C. elegans and involved in epithelial differentiation. Like many other miRNA examples, the deletion of miR-43 does not produce an obvious phenotype.
Buffering motifs of the kinds of Figure 1, D and E were once thought to be relatively uncommon (Alon 2007). As noted by both Tsang et al. (2007) and Martinez et al. (2008), experiments generally are biased against detecting mechanisms of homeostasis, which suppresses phenotypic variation. By bioinformatic analysis, Tsang et al. (2007) have found that incoherent FFLs are no less common than the coherent types. Similarly, Martinez et al. (2008) were able to identify many feedback loops between a transcription factor and a miRNA in the C. elegans genome. In previous reports (Alon 2007), negative feedback loops were rather infrequent, perhaps because only transcription factors were analyzed (TFs).
Figure 1 highlights only the simpler network motifs. In Figure 1F, miR down-regulates a gene B via T1 and, “incoherently” up-regulates B via another target, T2. A recent example of this type of motif is the Nodal signaling pathway (Choi et al. 2007) in which the squint locus is T1 and the lefty locus is T2. These two genes regulate the Nodal pathway antagonistically. The fluctuation in miR-430 is thus dampened and its effect on the Nodal pathway, which controls mesendoderm formation, would be robust.
A complementary approach to the problem of buffering functions is to analyze the features of network wirings involving miRNAs. In a series of studies by Edwin Wang and colleagues (Wang 2008), the authors noted that miRNAs are preferentially wired to positively regulated motifs, highly connected scaffolds, and downstream components of signaling pathways including transcription factors. Wiring to positively regulated motifs could provide buffering as miRNAs down-regulate targets. Wiring to the more heavily trafficked nodes is also an efficient way to buffer the entire system against perturbations. In general, the wiring of miRNAs suggests a role at the systems level, rather than the simple tuning of their immediate targets (see also “paradoxes of miRNAs” discussed earlier). Recently, Mehta et al. (2008) compared the kinetics of gene regulation by TFs vs. by noncoding RNAs in prokaryotes. They found many conditions under which noncoding RNAs can be more effective than TFs in filtering input noises. More general analyses on the relative efficacy of TFs and miRNAs in buffering input noises will be most illuminating on transcription homeostasis.
miRNAs and expression buffering—Evidence from recent experiments
Under the buffering hypothesis, miRNAs stabilize whole-genome expression against input noises. In this sense, they may behave in cells like shock absorbers on an automobile. For example, if a shock absorber works well in an incoherent FFL of Figure 1D, one might expect the expression of miRNAs themselves to vary more than the targets. In fact, miRNAs are expected to fluctuate more in expression than the transcriptome in general. We shall now present some results from our own analyses that may be germane to expression buffering.
Many of the interpretations will depend on the accuracy of target prediction, which remains inexact to this day. Nevertheless, unless the prediction algorithm (or combination of algorithms) is so inaccurate that the proportion of real targets in the predicted set is no better than random assignment, the interpretations should be valid. We will heed the degree to which the inaccuracy might impact the interpretations in relevant sections.
Perturbation of the expression level of target vs. nontarget genes
A straightforward test for the expression-buffering hypothesis is to change the level of miRNAs and observe the consequences on whole-genome expression. Normally, we expect the direct targets to be more affected by the perturbation than the indirect ones, which, in turn, should change more than the transcriptome as a whole. However, if miRNAs are part of the mechanisms of expression buffering, then the trend might be reversed and the direct targets could be least affected by miRNA mis-expression.
There are many studies that perturb whole-genome expression (Farh et al. 2005; Lim et al. 2005; Rehwinkel et al. 2006; Grimson et al. 2007; Karres et al. 2007; Linsley et al. 2007; Baek et al. 2008; Selbach et al. 2008). Most of these studies were designed to study the interactions between miRNAs and their targets. In order to detect the effects of these interactions, the buffering mechanisms were generally circumvented. For example, many studies assayed whole-genome expression immediately following the perturbation, intending to limit the downstream feedbacks. These studies also relied mostly on transfection or knockout of miRNAs, and, hence, were free from feedbacks on these miRNAs.
A direct test of the buffering hypothesis has to assay whole-genome stead-state expression that is perturbed by transgenic miRNAs driven by their own promoters. For control, mutant forms of the transgenic miRNAs would have to be used. Such experiments have recently been carried out by Tang et al. (T. Tang, S. Kumar, S. Yang, J. Lu, S. Shi, and C.-I. Wu, in prep). In that study, a cluster of four miRNAs were used to transform D. melanogaster. These four miRNAs from D. melanogaster (miR-310, miR-311, miR-312, and miR-313, collectively referred to as Dm310s) are duplicates of each other, sharing the same seed but having divergent tails (nonseed segment of miR).
The results, shown in Figure 2, are illuminating of the buffering function of miRNAs. Widely scattered points mean strong responses to the expression of the transgenic miRNAs. Interestingly, the responses of the predicted target genes of Dm310s (Fig. 2B) are milder than those of the transcriptome as a whole (Fig. 2A). The difference is highly significant by various target prediction programs (T. Tang, S. Kumar, S. Yang, J. Lu, S. Shi, and C.-I. Wu, in prep.). Although methods of target prediction are not fully accurate, they should not all be so bad that there are, in fact, fewer real targets in the predicted set than in the control. Furthermore, at least three target genes have been experimentally verified previously and none of them showed significantly reduced expression in Figure 2A. In a control experiment for the experiments of Figure 2, we used nonnative miRNAs as the transgene. In the control, predicted target genes are indeed more strongly misregulated than in the experiment of Figure 2B. The overall results suggest that the predicted targets of Dm310s may indeed be “buffered” in expression (Fig. 2B) and the observed pattern is not likely an experimental artifact or bioinformatic misinference.
Figure 2.

Changes in gene expression with transgenic miR-310s (the “volcano” plots). The x-axis is the log2 value of fold change between the transgenic line and control. The dashed lines at ±0.585 and ±1 indicate relative expression changes of 33% and 50%, respectively. The y-axis is the log10 value of the likelihood of observing the change. (A) Whole-genome expression changes between miR-310s transgenic lines and the wild-type line. (B) Changes in the expression of the predicted targets between miR-310s transgenic lines and the wild-type line. Points of the lighter shade represent significantly misregulated genes.
An additional point concerns the evolution between the two functions of miRNAs (tuning and buffering). The miR-310s cluster is relatively young in Drosophila and each miRNA is still evolving (Lu et al. 2008a). New miRNAs, in the process of becoming integrated into the genome, may have to minimize the disruption to the transcriptome while providing some benefits. Genetic buffering appears to be a possible early function in evolution. We will return to this hypothesis later.
Between-species divergence in target gene expression in relation to miRNA expression divergence
We may also consider natural divergence in the level of miRNA expression between species as some sort of perturbation. We ask whether the expressions of the predicted target genes behave according to the simple repression model of Figure 1A. In such a model, the more highly expressed miRNAs are, then, on average, the less abundant their target transcripts should be.
Cui et al. (2007) previously reported that the divergence in gene expression between species is smaller for miRNA target genes than for nontarget genes. The analysis was not able to incorporate divergence in miRNA expression as such information was generally unavailable. Fortunately, we have previously measured the divergence in miRNA expression in the heads of D. melanogaster and Drosophila simulans males by deep sequencing (Lu et al. 2008b). In the same tissues, we have also measured whole-genome expression by cDNA microarrays (Wang et al. 2008). We then calculated the ratio of expression level in D. melanogaster over that in D. simulans for each miRNA that is significantly up-regulated in either species. These ratios are shown on the x-axis of Figure 3.
Figure 3.

Correlation between the expression ratios in miRNAs and their targets. The ratio is the level of expression in D. melanogaster over that in D. simulans. (A) The scatter plot and (B) the box plot. In A, the x-axis is the miRNA expression ratio, but in B the ratios are given by rank. Note that there is no correlation at all between the expression level of miRNAs and their predicted targets, in contrast to the prediction of the simple repression motif.
The predicted target genes for each miRNA in both species were identified by TargetScan (Lewis et al. 2003, 2005) and their expression level in male heads was evaluated by cDNA microarray in both species, as was done in Wang et al. (2008). The ratio of the value of D. melanogaster over that of D. simulans is shown on the y-axis in Figure 3. As can be seen clearly in Figure 3B, there is no correlation at all between the expression ratio of the target genes and the expression ratio of miRNAs. The result was unexpected as the highest (D. mel/D. sim) ratio is more than 10 times higher than the lowest ratio in relative terms, and yet the average expression level of their target genes remains about the same. The lack of correlation cannot be easily attributed to inaccuracy in target prediction, as we chose putative miRNA targets that are highly conserved among the 12 species of Drosophila. Such stringency usually yields greater than 50% accuracy when the predicted targets were experimentally verified.
We may broadly refer to the observed pattern (lack of correlation between miRNA and target abundance between closely related species) as “evolutionary buffering.” While expression buffering should lead to evolutionary buffering, the latter is not necessarily caused by the former. In other words, the data of Figure 3 are only broadly compatible with the expression-buffering hypothesis. Direct support will have to come from experiments like those of Figure 2. One also notices in Figure 3 that a sizable fraction of miRNAs vary in expression by more than twofold between the two species of Drosophila. In contrast, almost no target genes in this analysis diverge by more than twofold in expression in the same comparison. If miRNAs serve as shock absorbers, they are expected to fluctuate more than the transcriptome in general. An extensive survey of the relative magnitude of expression fluctuation between miRNAs and their target transcripts might be informative about miRNA functions.
miRNAs and expression buffering—Evidence from evolutionary conservation
The two types of miRNA functions (tuning and buffering) may impose very different constraints on the evolution of miRNA–target interactions. Given that the tuning functions attributed to miRNAs often underlie important phenotypes, one might expect strong conservation in the interaction between miRNAs and their targets. Several examples cited above, including the maturation of immune cells in mouse, the development of sensory organs in Drosophila, and the maintenance of embryonic stem cells in C. elegans, are regulated by miRNAs. Curiously, while these phenotypes are conserved, the underlying miRNA–target interactions do not appear to be so.
The number of cases where the conservation in the miRNA–target coupling has been experimentally verified is surprisingly small (Chen and Rajewsky 2007). Between Drosophila and human, a computational analysis shows that, of the 8136 genes that are putative miRNA targets in either species, only 50 homologous pairs are targeted by the same miRNA in both species (Grun et al. 2005). These authors found that, on average, the number of predicted targets is several times larger in human than in Drosophila; hence, broad-scale nonconservation seems possible.
It is perhaps more informative to compute the relative target richness for each miRNA within each species. For example, miR-125 has a value of 0.17 in Drosophila (17% as target rich as the average among all miRNAs) and 1.44 in human. In contrast, the ratios are 1.02 and 0.32, respectively, for miR-304. Thus, miR-125 is relatively target poor in Drosophila and target rich in humans, but miR-304 is the opposite. The relative target richness between these two miRNAs in these two taxa is more than 25-fold [= 1.44/0.17]/[0.31/1.02].
While the lack of conservation in miRNA–target coupling seems puzzling under the tuning hypothesis, specific miRNA–target interactions are not expected to be highly conserved under the buffering hypothesis. From a network perspective, there may be no strong constraint on individual regulatory links (Wagner 2005; Ciliberti et al. 2007a, b). A configuration of a transcription network with defined links between genes is referred to as the network “genotype.” Two genotypes are connected if changing only one link can convert one genotype to the other. Previous studies have shown that many different network genotypes can have the same transcription output (or phenotype). Importantly, many of these “genotypes” having the same phenotype are interconnected, making it possible for the species to evolve in a meta-genotypic space (Wagner 2005; Ciliberti et al. 2007a).
If miRNAs are not constrained by individual target genes, they may likely function at the higher level of pathway or systems. For example, miR-1 targets genes of muscle development in both fly and human (Stefani and Slack 2008) and miR-9 is involved in sensory organ development (Cohen et al. 2006). Similarly, miR-8 targets genes on the Wnt pathway in both Drosophila and mammals (Kennell et al. 2008). If miRNAs interact with targets mainly to maintain the stability of the system, then each specific link between a miRNA and a target gene might be “evolvable,” as observed.
Evolution of miRNA targets between human and mouse
The analysis on the nonconservation of miRNA targets discussed above was based on comparisons between very distantly related taxa such as human vs. Drosophila. It might then be informative to compare species that are not as different. Hence, we compared human and mouse for miRNA target conservation. We chose nine miRNAs that have been assayed by transfection experiments on human cell lines (Grimson et al. 2007). Human genes that were significantly down-regulated at both 12 and 24 h by the transfected miRNA and have a perfect match with its seed (position 2–8) in their 3′ UTRs are considered direct targets. In total, we assayed 417 direct target sites distributed over 319 genes. A target site is the stretch of 7 bp that match perfectly with the miR seed plus one extra base pair on the 3′ end (or the 5′ end of miR) and 10 bp on the 5′ end, hence, totaling 18 bp.
We analyzed the evolutionary conservation of the 417 target sites between human and mouse. Figure 4, A and B show the conservation of the first 8 bp (referred to as the core) and all 18 bp, respectively. For comparison, fragments of 8 or 18 bp were selected randomly from the same 3′ UTR, where the target site was identified. Figure 4 shows that miRNA target sites are indeed more conserved than the rest of the 3′ UTR, but the degree of conservation is modest. Figure 4A shows that 40% of the 8-bp core in the target genes are fully conserved (first bin). In contrast, only 15% of the random 8-bp sequences are fully conserved between human and mouse. The second bin with one change in the core is roughly the same between the two panels of Figure 4A. Thus, the excess in the first bin is at the expense of the third and fourth bin (with two and three changes in the 8-bp core, respectively). In short, ∼25% (= 40%–15%) of the miRNA target sites are more conserved than expected between human and mouse. If we compare the whole 18 bp, the level of conservation is much smaller, as can be seen in Figure 4B.
Figure 4.

Nucleotide divergence in miRNA target sequences between human and mouse. The distributions of the divergence (proportion of base-pair changes) are given for the core (8 mer) and the minimal miRNA sequence (18 mer) separately. Each sequence is counted as a site. For both types of sequences, random sites of the same length are given for comparison. In total, 447 sites from 349 genes were analyzed.
Evolution of targets of evolving miRNAs between Drosophila species
Another approach to the problem of target-site conservation is to compare the rate of evolutionary turnover of targets between conserved and unconserved miRNAs. If miRNA–target interactions are under strong selective constraints, we should expect the turnover rate to be lower for conserved miRNAs than for unconserved ones. To carry out this type of analysis, it is best to compare miRNAs of the same family, some of which are conserved, while others are evolving. We use the miR-92a/b–miR-310s family (Lu et al. 2008a) for this purpose. miR-92a/b are two highly conserved miRNAs in Drosophila. The duplication of either miR-92a or miR-92b led to a cluster of new miRNAs—miR-310/311/312/313 (or simply miR-310s). The miR-310s cluster have been evolving since their formation 40–50 Mya and are still evolving adaptively in the recent past (Lu et al. 2008a). Despite the differences, all six miRNAs share the same seed.
In Table 1, we used the procedure as implemented in PITA (Kertesz et al. 2007) to predict targets. The procedure does not rely on sequence conservation, and hence, would allow for the inference on evolutionary conservation vis-à-vis turnover. The first observation is the low percentage of shared targets (< 20%) between D. melanogaster and Drosophila pseudoobscura, estimated to have diverged for about 50 million years. We are interested in the relative evolutionary rate of the targets of conserved and evolving miRNAs, thus permitting some inaccuracies in target prediction in both sets.
Table 1.
Number of predicted targets of miR-92 and miR-310s clusters in D. melanogaster and D. pseudoobscura
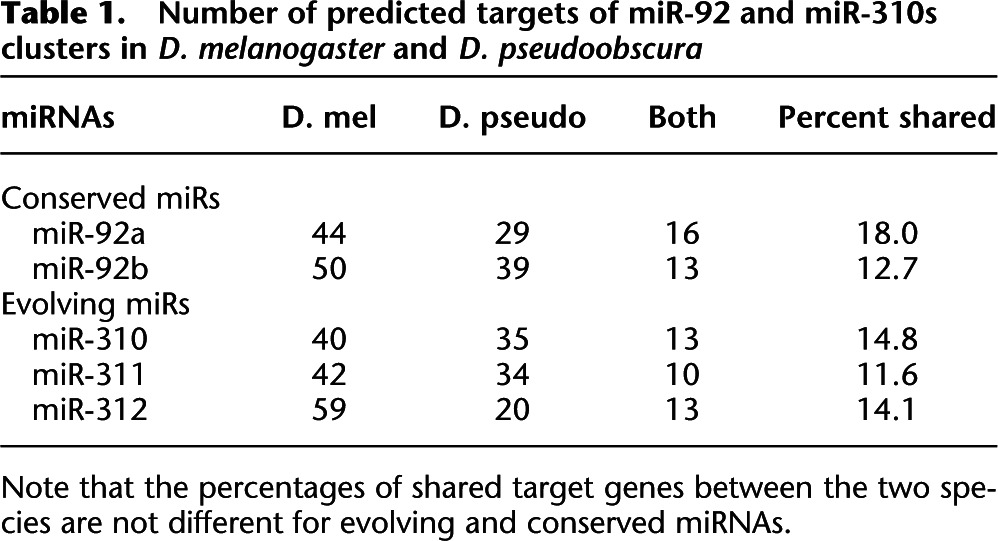
Note that the percentages of shared target genes between the two species are not different for evolving and conserved miRNAs.
The surprising observation of Table 1 is that the rates of evolutionary turnover in targets are rather comparable for conserved vs. evolving miRNAs. Although the results in Table 1 do not lend themselves to a quantitative estimate of target-site conservation, they do suggest, qualitatively, that the miR–target coupling is labile during evolution.
miRNAs and expression buffering—The emergence of new miRNAs
We now address the issue of the emergence of new miRNAs. How does a new miRNA become integrated into the transcriptome? If tuning is to provide the selective advantage, the likelihood of a brand new miRNA repressing the right targets while avoiding the wrong ones seems quite small. If the tuning model is to work, it may have to follow the nearly neutral evolution model (Ohta 1973). In this model, evolution can proceed if the deleterious effects of the new mutations (including new genes) are sufficiently small. In addition, mutation rate has to be correspondingly large to drive evolution. Previous studies (Fahlgren et al. 2007; Lu et al. 2008b) suggested that these two conditions might be met in the evolution of miRNAs. Emergence of new miRNAs is rather frequent and new miRNAs seem to be too lowly expressed to exert a strong fitness effect.
Ohta's (1973) nearly neutral evolution model, when applied to the emergence of new miRNAs, has its limitations. First, the rate of evolutionary decay of neutral miRNAs is very high (Lu et al. 2008b). For Drosophila, 95% of them would degenerate in 10 million years or so. Even if these miRNAs emerged without any deleterious effects, the race against time to acquire a positive fitness effect is tight. In other words, the nearly neutral model does allow more new miRNAs to spread in the population. Whether enough new miRNAs would win the race against degeneracy remains questionable.
Second, the nearly neutral model makes other predictions. For example, the emergence rate should be higher in species that have a smaller effective population size, Ne. Analyses of genomic evolutionary pattern have confirmed that species with smaller Ne, such as humans, tend to have smaller selective constraints (as measured by the Ka/Ks ratio) than species like mouse or Drosophila (for review, see Fay and Wu 2003). Currently, there is currently no comparative analysis on miRNA emergence between closely related species that have disparate Ne's (which should be reflected in their genome-wide Ka/Ks ratio). Nevertheless, the emergence rate of miRNAs in Drosophila does not appear to be lower than that in human or chimpanzee (Berezikov et al. 2006; Lu et al. 2008b). Given the much larger Ne, as well as the smaller genome, the high rate of miRNA emergence in Drosophila is somewhat a surprise. The observation raises questions about the near-neutral model that assumes no selective advantage in the emergence of new miRNAs.
Under the expression-buffering hypothesis, some new miRNAs may have a selective advantage, albeit very small initially. As illustrated in Figure 1D, the addition of the A → miR T path to form a feed-forward loop may not change the mean expression level of T, but may reduce its variance. At the systems level, the addition of a new miR may thus increase the stability of the transcriptome. In this view, newly emerged miRNAs would contribute to expression buffering initially without exerting a significant effect on the mean level of gene expression.
Conclusion
miRNAs are paradoxical regulators of the transcription network. On one hand, they are often highly conserved, controlling key steps in development. On the other hand, they can often be deleted with little phenotypic consequences. Furthermore, the sequences of their targets appear to be weakly constrained. Any model on the function and evolution of miRNAs has to account for these paradoxical properties. In this perspective, we suggest that miRNAs may have dual functions, expression tuning and expression buffering, which together might account for the paradox.
Expression tuning means that miRNAs adjust the mean expression levels of their target genes. This is the prevailing view on the function of miRNAs (Bushati and Cohen 2007). Expression buffering refers to the reduction in the variance of the expression level of the target genes. Many studies of the molecular mechanisms of miRNA regulation indeed suggest such functions (Hornstein and Shomron 2006; Bushati and Cohen 2007). The low level of target conservation and the dispensability of many miRNAs can be explained if miRNAs function as the “canalizing agents,” because network stability does not always require specific links to be conserved (Ciliberti et al. 2007a,b).
We further hypothesize that the dual functions of miRNAs may represent two stages in the evolution of miRNAs. As discussed above, new miRNAs are not likely to improve fitness by resetting the mean expression levels of many target genes when they emerge. Instead, they might gain an advantage in homeostasis by reducing the variances in gene expression. The strong buffering effect observed in Tang et al. (T. Tang, S. Kumar, S. Yang, J. Lu, S. Shi, and C.-I. Wu, in prep.) could be attributed to the fact that they analyzed new miRNAs. The tuning functions would evolve subsequently and gradually after the new miRNAs become integrated into the genome.
In the transcription network, TFs and miRNAs may be complementary in action. In terms of expression tuning, TFs may play a dominant role, setting the mean level of expression. The task of keeping the system close to that mean may require the participation of miRNAs. Martinez et al. (2008) have suggested that miRNAs complement TFs in forming feedback loops. Since miRNAs are negative regulators, the inclusion of both TFs and miRNAs may increase the tunability of the network (Tsai et al. 2008). There is also an emerging interest in comparing the kinetic properties of TFs and noncoding RNAs in the context of dampening input noises (Mehta et al. 2008). In the canalization metaphor, some gene actions are responsible for moving the species to the right valley, while others play the role of making its development canalized. TFs and miRNAs likely play both roles, but with somewhat different emphasis (Sylvestre et al. 2007; Woods et al. 2007; for review, see Hobert 2004, 2008).
Acknowledgments
This work was supported by NIH grants to C.I.W., NSFC (30730008), 973 Program (2007CB815701), SKLBC (08A06) to Suhua Shi and T.T., and China Scholarship Council (CSC, No. 2007102821) to Y.S. The analysis was greatly helped by Jian Lu and Yonggui Fu. Discussions with Ilya Ruvinsky and an earlier visit to Edwin Wang have been most helpful.
Footnotes
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.084640.108.
References
- Alon U. An introduction to systems biology: Design principles of biological circuits. Chapman & Hall/CRC; Boca Raton, FL: 2007. [Google Scholar]
- Alvarez-Garcia I., Miska E.A. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- Baek D., Villen J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel D.P., Chen C.Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Berezikov E., Thuemmler F., van Laake L.W., Kondova I., Bontrop R., Cuppen E., Plasterk R.H.A. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- Bergman A., Siegal M.L. Evolutionary capacitance as a general feature of complex gene networks. Nature. 2003;424:549–552. doi: 10.1038/nature01765. [DOI] [PubMed] [Google Scholar]
- Brennecke J., Stark A., Russell R.B., Cohen S.M. Principles of microRNA-target recognition. PLoS Biol. 2005;3:404–418. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N., Cohen S.M. MicroRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Carrington J.C., Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336–338. doi: 10.1126/science.1085242. [DOI] [PubMed] [Google Scholar]
- Chen K., Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Choi W.Y., Giraldez A.J., Schier A.F. Target protectors reveal dampening and balancing of nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Ciliberti S., Martin O.C., Wagner A. Innovation and robustness in complex regulatory gene networks. Proc. Natl. Acad. Sci. 2007a;104:13591–13596. doi: 10.1073/pnas.0705396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberti S., Martin O.C., Wagner A. Robustness can evolve gradually in complex regulatory gene networks with varying topology. PLoS Comput. Biol. 2007b;3:164–173. doi: 10.1371/journal.pcbi.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.M., Brennecke J., Stark A. Denoising feedback loops by thresholding—a new role for microRNAs. Genes & Dev. 2006;20:2769–2772. doi: 10.1101/gad.1484606. [DOI] [PubMed] [Google Scholar]
- Cui Q., Yu Z., Purisima E.O., Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q., Yu Z., Purisima E.O., Wang E. MicroRNA regulation and interspecific variation of gene expression. Trends Genet. 2007;23:372–375. doi: 10.1016/j.tig.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Darwin C. On the origin of species by means of natural selection. Murray; London, UK: 1859. [Google Scholar]
- Fahlgren N., Howell M.D., Kasschau K.D., Chapman E.J., Sullivan C.M., Cumbie J.S., Givan S.A., Law T.F., Grant S.R., Dangl J.L., et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh K.K.H., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Fay J.C., Wu C.-I. Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 2003;4:213–235. doi: 10.1146/annurev.genom.4.020303.162528. [DOI] [PubMed] [Google Scholar]
- Fisher R.A. The genetical theory of natural selection. Clarendon Press; Oxford, UK: 1930. [Google Scholar]
- Grimson A., Farh K.K.H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D., Wang Y.L., Langenberger D., Gunsalus K.C., Rajewsky N. MicroRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput. Biol. 2005;1:51–66. doi: 10.1371/journal.pcbi.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermisson J., Wagner G.P. The population genetic theory of hidden variation and genetic robustness. Genetics. 2004;168:2271–2284. doi: 10.1534/genetics.104.029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. Common logic of transcription factor and microRNA action. Trends Biochem. Sci. 2004;29:462–468. doi: 10.1016/j.tibs.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Hobert O. miRNAs play a tune. Cell. 2007;131:22–24. doi: 10.1016/j.cell.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–1786. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- Hornstein E., Shomron N. Canalization of development by microRNAs. Nat. Genet. 2006;38:S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Hornstein E., Mansfield J.H., Yekta S., Hu J.K.H., Harfe B.D., McManus M.T., Baskerville S., Bartel D.P., Tabin C.J. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature. 2005;438:671–674. doi: 10.1038/nature04138. [DOI] [PubMed] [Google Scholar]
- Kaern M., Elston T.C., Blake W.J., Collins J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Karres J.S., Hilgers V., Carrera I., Treisman J., Cohen S.M. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell. 2007;131:136–145. doi: 10.1016/j.cell.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Kennell J.A., Gerin I., MacDougald O.A., Cadigan K.M. The microRNA miR-8 is a conserved negative regulator of Wnt signaling. Proc. Natl. Acad. Sci. 2008;105:15417–15422. doi: 10.1073/pnas.0807763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Krek A., Grun D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut R., Lendeckel W., Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lau N.C., Lim L.P., Weinstein E.G., Bartel D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee R.C., Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Shih I.H., Jones-Rhoades M.W., Bartel D.P., Burge C.B. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li X., Carthew R.W. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell. 2005;123:1267–1277. doi: 10.1016/j.cell.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Li Y., Wang F., Lee J.A., Gao F.B. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes & Dev. 2006;20:2793–2805. doi: 10.1101/gad.1466306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M. Microarray analysis shows that some microRNAs down-regulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Linsley P.S., Schelter J., Burchard J., Kibukawa M., Martin M.M., Bartz S.R., Johnson J.M., Cummins J.M., Raymond C.K., Dai H.Y., et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 2007;27:2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Fu Y.G., Kumar S., Shen Y., Zeng K., Xu A.L., Carthew R., Wu C.-I. Adaptive evolution of newly emerged micro-RNA genes in Drosophila. Mol. Biol. Evol. 2008a;25:929–938. doi: 10.1093/molbev/msn040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Shen Y., Wu Q., Kumar S., He B., Shi S., Carthew R.W., Wang S.M., Wu C.-I. The birth and death of microRNA genes in Drosophila. Nat. Genet. 2008b;40:351–355. doi: 10.1038/ng.73. [DOI] [PubMed] [Google Scholar]
- Makeyev E.V., Maniatis T. Multilevel regulation of gene expression by microRNAs. Science. 2008;319:1789–1790. doi: 10.1126/science.1152326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J., et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez N.J., Ow M.C., Barrasa M.I., Hammell M., Sequerra R., Doucette-Stamm L., Roth F.P., Ambros V.R., Walhout A.J.M. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes & Dev. 2008;22:2535–2549. doi: 10.1101/gad.1678608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P., Goyal S., Wingreen N.S. A quantitative comparison of sRNA-based and protein-based gene regulation. Mol. Syst. Biol. 2008;4:221. doi: 10.1038/msb.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir E., von Dassow G., Munro E., Odell G.M. Robustness, flexibility, and the role of lateral inhibition in the neurogenic network. Curr. Biol. 2002;12:778–786. doi: 10.1016/s0960-9822(02)00839-4. [DOI] [PubMed] [Google Scholar]
- Miska E.A., Alvarez-Saavedra E., Abbott A.L., Lau N.C., Hellman A.B., McGonagle S.M., Bartel D.P., Ambros V.R., Horvitz H.R. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007;3:2395–2403. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara K., Kim K., Sciulli C., Dowd S.R., Minden J.S., Carthew R.W. Targets of microRNA regulation in the Drosophila oocyte proteorne. Proc. Natl. Acad. Sci. 2005;102:12023–12028. doi: 10.1073/pnas.0500053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell K.A., Wentzel E.A., Zeller K.I., Dang C.V., Mendell J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Plasterk R.H.A. MicroRNAs in animal development. Cell. 2006;124:877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Rajewsky N. MicroRNA target predictions in animals. Nat. Genet. 2006;38:S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Raser J.M., O'Shea E.K. Noise in gene expression: Origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J., Natalin P., Stark A., Brennecke J., Cohen S.M., Izaurralde E. Genome-wide analysis of mRNAs regulated by Drosha and Argonaute proteins in Drosophila melanogaster. Mol. Cell. Biol. 2006;26:2965–2975. doi: 10.1128/MCB.26.8.2965-2975.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendel J.M. Logos Press; London, UK: 1967. Canalization and gene control. [Google Scholar]
- Rhoades M.W., Reinhart B.J., Lim L.P., Burge C.B., Bartel B., Bartel D.P. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/s0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Rutherford S.L., Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Rybak A., Fuchs H., Smirnova L., Brandt C., Pohl E.E., Nitsch R., Wulczyn F.G. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- Selbach M., Schwanhausser B., Thierfelder N., Fang Z., Khanin R., Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Sempere L.F., Cole C.N., McPeek M.A., Peterson K.J. The phylogenetic distribution of metazoan microRNAs: Insights into evolutionary complexity and constraint. J. Exp. Zoolog. B Mol. Dev. Evol. 2006;306:575–588. doi: 10.1002/jez.b.21118. [DOI] [PubMed] [Google Scholar]
- Siegal M.L., Bergman A. Waddington's canalization revisited: Developmental stability and evolution. Proc. Natl. Acad. Sci. 2002;99:10528–10532. doi: 10.1073/pnas.102303999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A., Brennecke J., Russell R.B., Cohen S.M. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1:397–409. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M. Animal microRNAs confer robustness to gene expression and have a significant impact on 3' UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Stark A., Lin M.F., Kheradpour P., Pedersen J.S., Parts L., Carlson J.W., Crosby M.A., Rasmussen M.D., Roy S., Deoras A.N., et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G., Slack F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- Sullivan C.S. New roles for large and small viral RNAs in evading host defences. Nat. Rev. Genet. 2008;9:503–507. doi: 10.1038/nrg2349. [DOI] [PubMed] [Google Scholar]
- Sylvestre Y., De Guire V., Querido E., Mukhopadhyay U.K., Bourdeau V., Major F., Ferbeyre G., Chartrand P. An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- Tsai T.Y., Choi Y.S., Ma W., Pomerening J.R., Tang C., Ferrell J.E., Jr Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science. 2008;321:126–129. doi: 10.1126/science.1156951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J., Zhu J., van Oudenaarden A. MicroRNA-mediated feedback and feed-forward loops are recurrent network motifs in mammals. Mol. Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese J., Cohen S.M. microRNA miR-14 acts to modulate a positive autoregulatory loop controlling steroid hormone signaling in Drosophila. Genes & Dev. 2007;21:2277–2282. doi: 10.1101/gad.439807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Dassow G., Meir E., Munro E.M., Odell G.M. The segment polarity network is a robust development module. Nature. 2000;406:188–192. doi: 10.1038/35018085. [DOI] [PubMed] [Google Scholar]
- Waddington C.H. Analization of development and the inheritance of acquired characters. Nature. 1942;150:563–565. [Google Scholar]
- Waddington C.H. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183:1654–1655. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- Wagner A. Circuit topology and the evolution of robustness in two-gene circadian oscillators. Proc. Natl. Acad. Sci. 2005;102:11775–11780. doi: 10.1073/pnas.0501094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E. MicroRNA systems biology. In: Erdmann V.A., Poller W., Barciszewski J., editors. RNA technologies in cardiovascular medicine and research. Springer, Berlin-Heidelberg; Germany: 2008. pp. 69–86. [Google Scholar]
- Wang H.Y., Fu Y.G., McPeek M.S., Lu X.M., Nuzhdin S., Xu A.L., Lu J., Wu M.L., Wu C.-I. Complex genetic interactions underlying expression differences between Drosophila races: Analysis of chromosome substitutions. Proc. Natl. Acad. Sci. 2008;105:6362–6367. doi: 10.1073/pnas.0711774105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds E., Plasterk R.H.A. MicroRNA function in animal development. FEBS Lett. 2005;579:5911–5922. doi: 10.1016/j.febslet.2005.07.070. [DOI] [PubMed] [Google Scholar]
- Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern-formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Woods K., Thomson J.M., Hammond S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem. 2007;282:2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- Wu C.-I., Palopoli M.F. Genetics of postmating reproductive isolation in animals. Annu. Rev. Genet. 1994;28:283–308. doi: 10.1146/annurev.ge.28.120194.001435. [DOI] [PubMed] [Google Scholar]
- Wu C.-I., Ting C.T. Genes and speciation. Nat. Rev. Genet. 2004;5:114–122. doi: 10.1038/nrg1269. [DOI] [PubMed] [Google Scholar]
- Xiao C.C., Calado D.P., Galler G., Thai T.H., Patterson H.C., Wang J., Rajewsky N., Bender T.P., Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]