

Abstract
Thymic output is a dynamic process, with high activity at birth punctuated by transient periods of involution during infection. Interferon-α (IFN-α) is a critical molecular mediator of pathogen-induced thymic involution, yet despite the importance of thymic involution, relatively little is known about the molecular integrators that establish sensitivity. Here we found that the Dicer-dependent microRNA network, and specifically miR-29a, was critical for reducing the sensitivity of the thymic epithelium to simulated infection signals, protecting the thymus against inappropriate involution. In the absence of Dicer or the miR-29a cluster in the thymic epithelium the amount of IFN-α receptor expressed by the thymic epithelium was increased, allowing suboptimal signals to trigger a rapid loss of thymic cellularity.
Keywords: Thymus, microRNA, interferon, miR-29a
Introduction
Unique among mammalian leukocytes, T cells require a dedicated organ, the thymus, for differentiation. Post-natal thymic output is critical for both effector T cell-mediated immunity 1 and regulatory T cell-mediated tolerance 2. There are, however, circumstances, namely during periodic infections and with aging, that drive a process of severe atrophy, termed thymic involution, which can result in the loss of more than 95% of the cellularity of the thymus. Continued thymic output during these events may be detrimental. The theoretical risk posed by continued thymic function during an infection is that foreign antigens present in the thymus will engender dominant tolerance, protecting the infection from clearance 3. As such, thymic involution during infection is a rapid yet transient phenomenon, with recovery within one to two weeks 4. Thymic involution during aging has also been postulated to have biological functions, from acting as a protective mechanism against autoimmunity 5, to allowing optimization of the peripheral repertoire 6, or simply conserving energy by reducing the inefficient process of T cell differentiation during the declining cost-benefit ratio of old age. The kinetics of thymic involution during aging are distinct from infection-induced involution, with a gradual progressive decline in cellularity, at the rate of roughly 1% per year in humans 7.
Characterization of the direct mediators of thymic involution reveals two distinct pathways. The rapid and transient involution initiated during infection can be reproduced through exposure to pathogen-associated molecular patterns (PAMPs), such as poly(I:C) 4. In this system, thymic involution is dependent upon the viral sensor melanoma differentiation-associated gene 5 (MDA-5) and is mediated through the interferon αreceptor 1 (IFNAR1) expressed on the thymic stroma 4. Age-related thymic involution, by contrast, is mediated through thymic epithelial cell sensitivity to sex hormones 8, and can be reversed through physical or chemical sex hormone ablation 9. Other inducers of thymic involution are known, ranging from psychological stress to inflammation 10, 11, however these phenomena may instead reflect a maladaptive response to non-infectious conditions which mimic signals of infection at the molecular level.
While the main molecular mediators of infection-induced thymic involution have been identified, the mechanism by which appropriate sensitivity is maintained remains unknown. The thymus has conflicting pressures to ensure rapid involution during a high risk infection, without undergoing chronic involution due to the baseline exposure of PAMPs. One potential regulatory system which would be well adapted to tuning involution sensitivity is the microRNA network. MicroRNA (miRNA) are small, non-coding RNA that modulate protein production from messenger RNA (mRNA) through destabilization of the mRNA and inhibition of translation. In monocytes and dendritic cells miRNA can tightly regulate the signaling resulting from exposure to PAMPs. MiR-155 is a positive regulator of Toll-like receptor (TLR) signaling, downregulating SOCS1 and SHIP-1 to enhance TLR signaling 12, 13. MiRNA-146a, miR-147, miR-125b, miR-21 and let-7e, by contrast, are all negative regulators of TLR signaling, reducing sensitivity of the cells to PAMPs 14–17. These miRNA have a complex regulatory system, with both ligand-dependent enhancement and suppression observed 18. As well as modulating the TLR signaling pathway, miRNA-146 is also observed to be involved in the RIG-I antiviral pathway, reducing TRAF6, IRAK1 and IRAK2 levels and hence modulating IFN-α production 19. MiRNA are therefore putative regulators of PAMP signaling in thymic epithelium.
In order to test the function of miRNA regulation in thymic function and involution, we generated a cell-specific deletion of Dicer, the key miRNA biogenesis protein, in thymic epithelial cells. We found that thymic epithelial cells are exquisitely sensitive to loss of the miRNA network, as Dicer-excision drove a progressive degeneration of thymic architecture and function, altering T cell differentiation and peripheral tolerance. In addition to the disruption of thymic architecture, Dicer-deficient thymic epithelium became hyper-sensitive to molecular involution signals, allowing constitutive or suboptimal levels of pathogen-related signals to drive premature and chronic thymic involution. This heightened sensitivity was replicated in mice deficient in the miR-29a cluster (miR-29a/b-1), and was associated with loss of miR-29a-dependent regulation of IFNAR1. Loss of the miR-29a cluster resulted in IFN-α receptor 1-dependent hypersensitivity to pathogen-related signals and hyper-activity of the IFNAR1 signaling pathway. These results implicate the miRNA network, and miR-29a in particular, as the key threshold modulator of the thymic epithelial cell response to peripheral PAMP signals.
Results
miRNAs maintain thymic architecture and T cell development
To determine the function of the miRNA network within thymic epithelial cells we used a Foxn1Cre allele with high specific activity within thymic epithelial cell precursors 20, crossed to a floxed allele of Dicer 21. Histological examination of epithelial architecture in the thymus of Foxn1Cre Dicerfl/fl mice demonstrated a progressive disruption of thymic architecture. Dicer-sufficient siblings (Foxn1wt Dicerfl/fl mice, hereafter referred to as wild-type) typically showed an even distribution of Keratin 8+ epithelial cells in the cortex and Keratin 14+ epithelial cells in the medulla, with relatively few changes in structure during development, aside from the expansion of the medulla during the first weeks after birth (Fig. 1a). Foxn1Cre Dicerfl/fl mice developed normal thymic architecture, with the presence of a discrete cortex and medulla at birth (Fig. 1a). By one week of age, however, disorganization of thymic structure became apparent, with the loss of homogeneity in Keratin 14 expression and the appearance of regions lacking Keratin 14 expression in the medulla (Fig. 1a). These Keratin 14-negative zones were also negative for expression of other epithelial markers (Supplementary Fig. 1), suggesting the presence of ‘epithelial voids’ within the medulla. By three weeks of age, large epithelial voids had also opened up in the cortex and the medullary epithelium was almost absent. These architectural changes were not present in Foxn1Cre Dicerwt/wt mice (Supplementary Fig. 2), and were thus due to thymic epithelial cell-specific excision of Dicer, rather than any cytotoxicity effect of Cre. The heightened sensitivity of medullary epithelium to architectural degradation was reflected in the cellular composition of the epithelial compartment as measured through flow cytometry (Fig. 1b), where the relative fraction of medullary epithelium strongly decreased in Dicer-deficient mice, relative to cortical epithelium, creating a bias of ~25:1 in the ratio of recoverable cortical/medullary epithelium in Foxn1Cre Dicerfl/fl mice compared to the normal ~1:1 ratio in wild-type mice (Fig. 1c).
Figure 1. Loss of miRNA in the thymic epithelium results in progressive loss of thymic architecture.
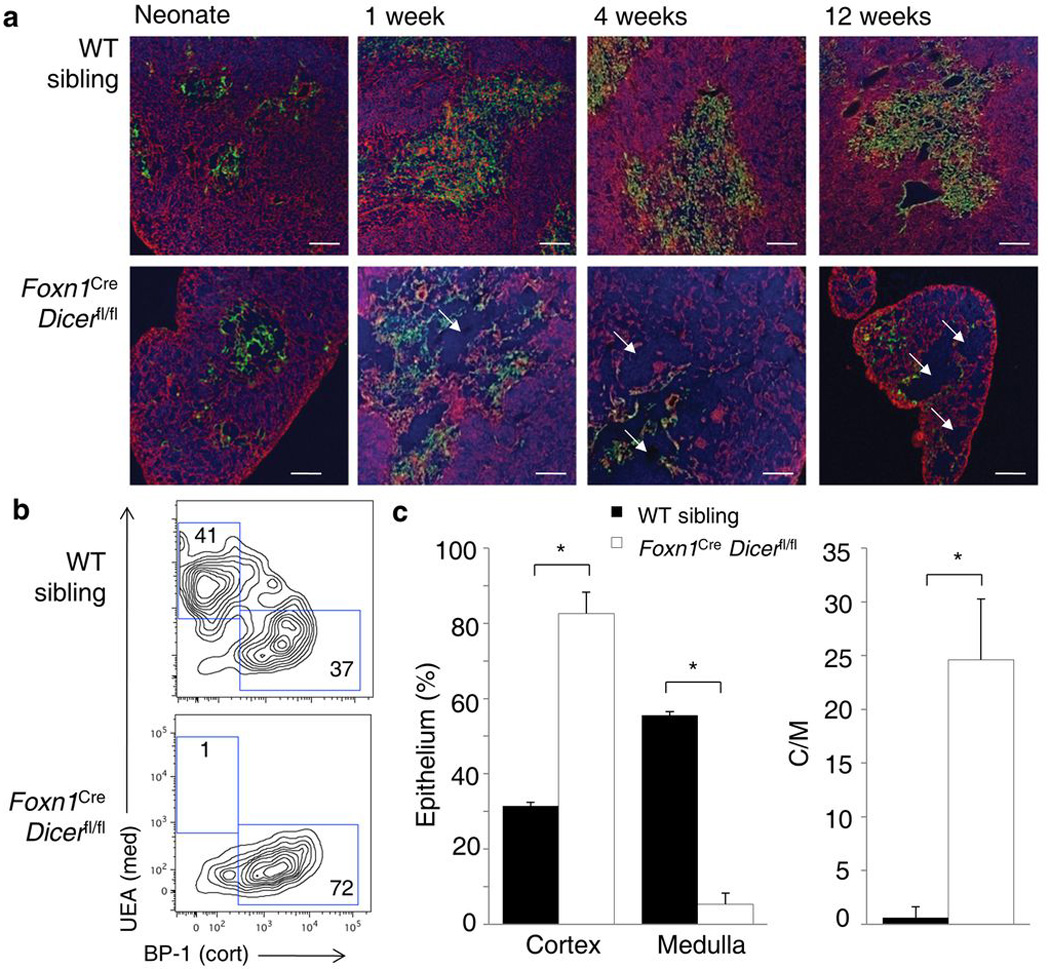
(a) Immunofluorescent staining of the thymus of Foxn1Cre Dicerfl/fl mice and wild-type siblings assessed at birth, 1, 4 and 12 weeks of age. Representative pictures from five experiments are shown. Keratin 8 (cortex; red), Keratin 14 (medulla; green) and DAPI (blue). Arrows indicate epithelial voids. Scale bar represents 100µm. (b) Frequencies of cortical (BP-1+UEA) and medullary (BP-1UEA+) epithelium measured by flow cytometry in wild-type and Foxn1Cre Dicerfl/fl thymi and gated on CD45G8.8+ epithelium at 12 weeks of age. Representative profiles of five experiments are shown. (c) Average proportion of cortical and medullary epithelium and the cortical/medullary ratio in mice analyzed as in b (n=5,6). *p< 0.001.
The progressive loss of epithelium from the thymus in Foxn1Cre Dicerfl/fl mice suggested a role for the miRNA network in preventing apoptosis of epithelial cells, with medullary epithelium showing a heightened sensitivity to miRNA loss. Directly supporting a model of epithelial voids being generated by increased apoptosis, co-staining of Keratin 8 and activated Caspase 3 demonstrated the presence of apoptotic epithelial cells along the boundary of the epithelial voids in Dicer-deficient mice only (Fig. 2a). By activated Caspase-3 staining there was a significant increase in apoptotic epithelium in Foxn1Cre Dicerfl/fl mice, especially in the medullary compartment (Fig. 2b). Despite the increase in apoptosis in the Dicer-deficient epithelium, no evidence was found for selection of Dicer-sufficient clones, with Dicer mRNA absent in purified thymic epithelium (Supplementary Fig. 3). Corresponding with an increase in the apoptosis rate, epithelial markers indicative of specialized or terminal lineages, such as UEA, Aire, CDR1, Claudin 3 and Claudin 4, were reduced in expression from birth (Supplementary Fig. 1, 4, and data not shown). By contrast, immature epithelium appeared to be increased in relative frequency, with increased numbers of Keratin 5+8+ clusters and p63+ epithelium (Supplementary Fig. 4). Increased proliferation may operate in direct response to the elevated apoptosis rates, with a higher percentage of epithelial cells expressing the proliferation marker Ki67, especially within the medullary compartment (Fig. 2c). An increase in epithelial turnover, with enhanced apoptosis and increased compensatory proliferation, would account for the skewing of epithelium towards relatively immature stages, although an additional block in epithelial differentiation cannot be excluded.
Figure 2. Medullary thymic epithelium exhibits enhanced apoptosis in the absence of miRNA.
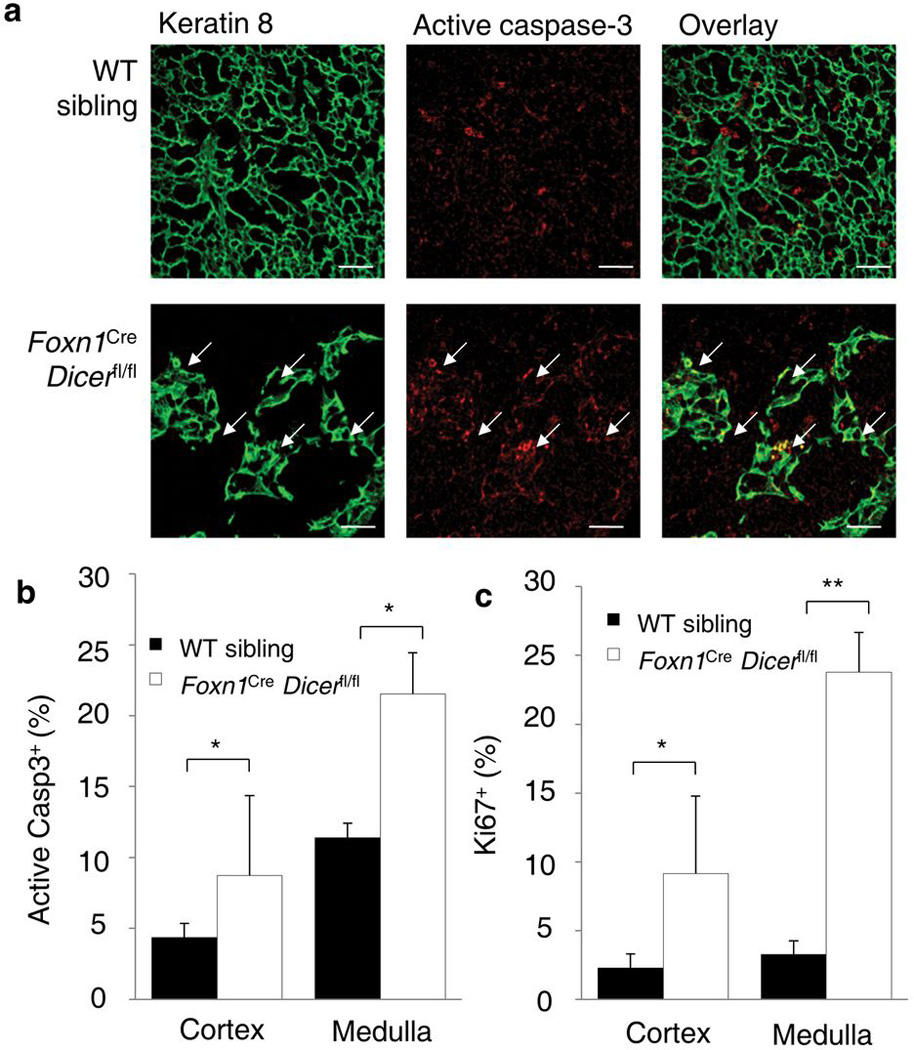
(a) Immunofluorescent staining of the thymus of Foxn1Cre Dicerfl/fl mice and wild-type siblings assessed at 12 weeks of age. Representative pictures of from five experiments are shown. Keratin 8 (green, left), activated Caspase-3 (red, center) and overlay (yellow, right). Arrows indicate apoptotic epithelial cells. Scale bar represents 50µm. (b) Average proportion of apoptotic cells (activated Caspase-3+ cells) within cortical (BP-1+UEA) and medullary (BP-1UEA+) epithelium, as measured by flow cytometry at 12 weeks of age in wild-type and Foxn1Cre Dicerfl/fl siblings (n=4,5). (c) Average frequency of proliferating cells (Ki67+ cells) among cortical (BP-1+UEA) and medullary (BP-1UEA+) epithelium, as measured by flow cytometry at 12 weeks of age in wildtype and Foxn1Cre Dicerfl/fl siblings (n=4,5). *p< 0.05, **p< 0.001.
Considering the progressive disintegration of the thymic architecture in the absence of Dicer we tested if miRNA may contribute to the capacity of the epithelium to support thymocyte differentiation. Despite the large changes in architecture, T cell differentiation remained remarkably robust. Early T cell differentiation through TCR rearrangement and β-selection was intact, with no changes in CD4 and CD8 double negative (DN) and double positive (DP) thymocyte populations (Supplementary Table 1, Supplementary Fig. 5). At the neonate stage, positive selection was also unmodified, with relatively normal DP to CD4 or CD8 single positive (SP) ratios observed (Fig. 3a,b). Only following the development of large epithelial voids in the thymic cortex at 3 weeks of age, notable defects in positive selection were observed, with reductions in post-positive selection CD3+CD69+ DP thymocytes (Supplementary Table 1, Supplementary Fig. 5) and CD4+ and CD8+ SP thymocytes (Fig. 3a,b, Supplementary Fig. 5). Functionally, the reduced thymic output resulted in a net decrease of CD4+ and CD8+ T cells in the periphery (Supplementary Fig. 6a,b). The combined effects of reduced T cell production and almost a complete loss of medullary thymic epithelial cells (Fig. 1c), including Aire-expressing cells (Supplementary Fig. 4), produced complex effects on autoimmune susceptibility, with Foxn1Cre Dicerfl/fl mice simultaneously demonstrating an increased susceptibility to collagen-induced arthritis and a reduced severity of disease (Supplementary Fig. 6c–e, Supplementary Table 2).
Figure 3. Progressive decay in thymocyte differentiation associated with the presence of epithelial voids in Dicer-deficient mice.
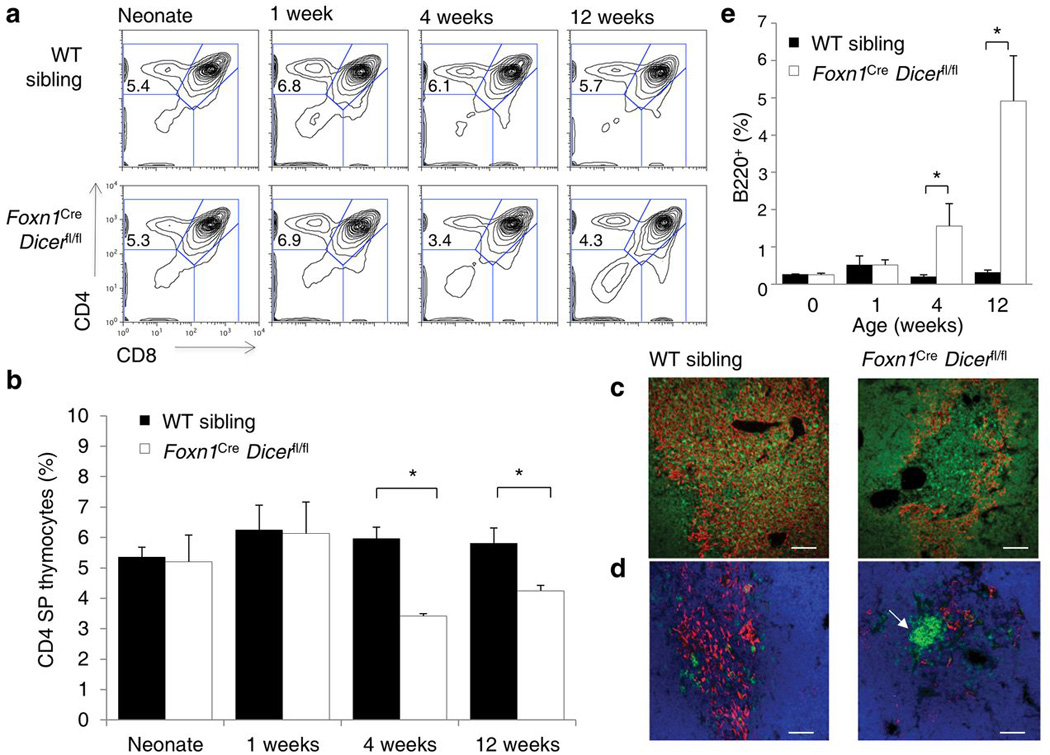
(a) Frequencies of CD4 SP thymocytes measured by flow cytometry in wild-type and Foxn1Cre Dicerfl/fl thymi. Representative CD4-CD8 thymocyte profiles from six experiments are shown. (b) Average CD4 single positive thymocyte frequency in mice analyzed as in a (n=2–5/group). (c) Immunofluorescent staining of the thymus of Foxn1Cre Dicerfl/fl mice and wild-type siblings assessed at 4 weeks of age. Representative pictures of from three experiments are shown. Keratin 14 (red) and CD4 (green). Scale bar represents 100µm. (d) Immunofluorescent staining of the thymus of Foxn1Cre Dicerfl/fl mice and wild-type siblings assessed at 4 weeks of age. Representative pictures of from three experiments are shown. Keratin 14 (red), CD19 (green) and DAPI (blue). 62% of epithelial voids showed CD19 foci (n=53). Scale bar represents 100µm. (e) Average proportion of B cells found within the thymus as measured by flow cytometry of wild-type and Foxn1Cre Dicerfl/fl siblings (n=2–5/group). *p< 0.05.
As these phenomena could be due to reduced interactions with epithelium, we investigated the cellular composition of the epithelial voids further. While epithelial voids showed an absence of thymic epithelium, they were not acellular, with thymocytes present in the cortical voids (Fig. 3c). Dendritic cell distribution and vascularization of epithelial voids was largely normal, while fibroblasts were absent from cortical voids (Supplementary Fig. 7). The frequent distribution of F4/80+ macrophages along the boundary of the epithelial voids (Supplementary Fig. 7), further supports a model of enhanced epithelial cell death in the region. An unusual feature common to the epithelial voids was the presence of dense foci of CD19+ B cells, resulting in an expansion in the total number of B cells within the thymus (Fig. 3d,e). These B cells may represent aberrant migration into the thymus or autochthonous production from thymocyte precursors in the absence of epithelial signals. Overall, these results suggest that the Dicer-deficient neonatal thymus has preserved function, but that progressive defects in thymocyte differentiation and selection processes develop in line with altered architecture.
The miRNA network prevents pathogen-associated thymic involution
Coincident with the thymic architectural changes, Dicer-deficiency in the thymic epithelium caused premature thymic involution. Although thymic size in Foxn1Cre Dicerfl/fl mice was normal at birth and grew to near normal sizes by four weeks of age, after this age it began to rapidly involute, dropping by 90% at 12 weeks (Fig. 4a). To test if miRNA protect against thymic involution by modulating sensitivity of the thymic epithelium to sex hormones, we surgically castrated Foxn1Cre Dicerfl/fl mice and wild-type siblings at 7 weeks of age. While both castrated wild-type and Dicer-deficient mice demonstrated a gain in thymic cellularity at 12 weeks (Fig. 4b), the difference between the two conditions was not eliminated, indicating that the loss of thymic epithelial miRNA did not affect sensitivity to a sex hormone signal.
Figure 4. Thymic epithelial defects in the miRNA network result in hyper-sensitivity to IFN-α and premature thymic involution.
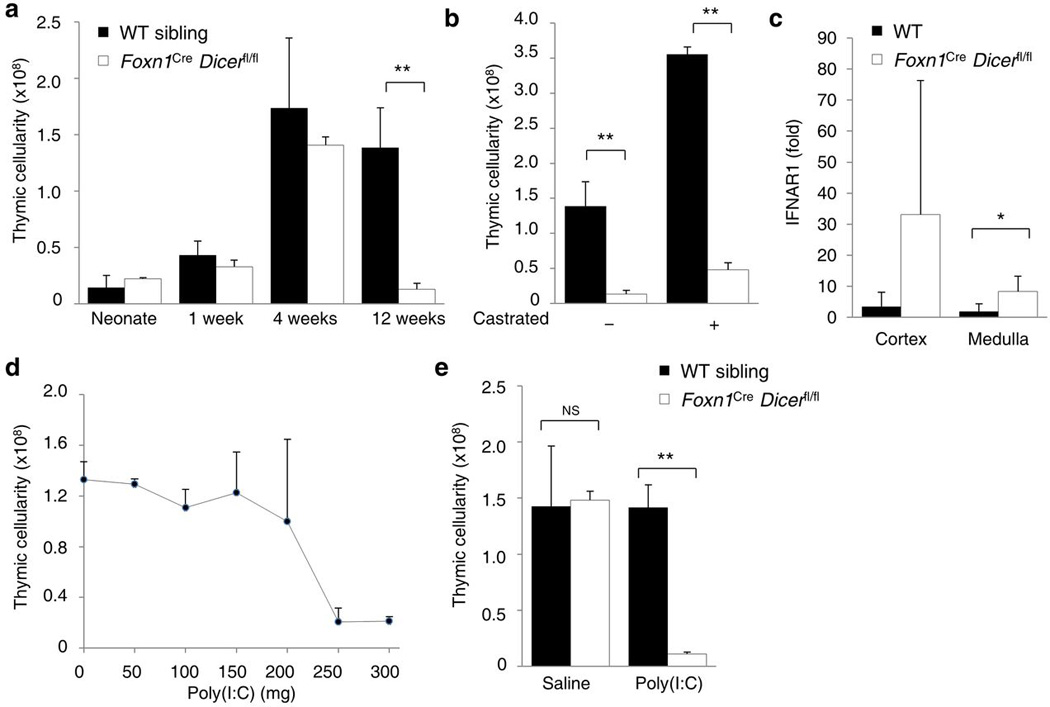
(a) Thymic cellularity of wild-type and Foxn1Cre Dicerfl/fl mice assessed at birth, 1, 4 and 12 weeks of age (n=3–17/group). (b) Thymic cellularity in wild-type and Foxn1Cre Dicerfl/fl surgically castrated at 7 weeks of age and assessed at 12 weeks (n=3–7/group). (c) Fold change of IFNAR1 mRNA expression in cortical (BP-1+UEA) and medullary (BP-1UEA+) thymic epithelium from wild-type and Foxn1Cre Dicerfl/fl mice at 6–8 weeks (n=5,4). Fold-change is relative to the average of wild-type cortex values. (d) Thymic cellularity in wild-type mice injected with 0–300µg of poly(I:C) exposure effect on thymic cellularity at 4 weeks of age (n=6,3,8,9,6,7,5/group). (e) Thymic cellularity of wild-type and Foxn1Cre Dicerfl/fl mice treated with 150µg dose of poly(I:C) at 4 weeks (n=3–5/group). *p< 0.05, **p< 0.0001.
The second major pathway involved in thymic involution is the IFN-α-mediated pathway. In a bioinformatic discovery approach, IFNAR1 was the only component of the IFN-α signaling pathway to show increased expression in Dicer-deficient thymic epithelium (S.Z., G.A.H., manuscript in preparation). To test if miRNA regulate TECs sensitivity to IFN-α signaling, cortical (BP-1+UEA−) and medullary (BP-1−UEA+) thymic epithelium were purified from wild-type and Dicer-deficient mice. Compared to housekeeping genes, IFN-α receptor 1 mRNA was upregulated ~10-fold in the cortex and ~5-fold in the medulla of Dicer-deficient mice (Fig. 4c). IFNAR1 expression on the thymic stroma was shown to be critical for poly(I:C)-mediated thymic involution 4, suggesting that Dicer-deficiency may drive inappropriate thymic involution through enhanced sensitivity of thymic epithelial cells to PAMP signals. To directly test this hypothesis, poly(I:C) exposure was titrated in wild-type mice to determine the concentration of poly(I:C) unable to induce thymic involution. Doses of 250µg and above induced thymic involution, while doses of 200µg and below were unable to drive involution in wild-type mice (Fig. 4d). When exposed to sub-optimal doses of poly(I:C), Dicer-deficient mice, unlike wild-type mice, were hyper-sensitive to low-dose poly(I:C) and underwent rapid involution (Fig. 4e). These data demonstrated that the thymic epithelial miRNA network was important for protecting the thymus from inappropriate involution at low PAMP levels.
The miR-29a cluster controls thymic involution
A likely miRNA candidate for hyper-sensitivity to PAMP-mediated thymic involution was miR-29a. Among the miRNA expressed in the thymic epithelium (S.Z., G.A.H., manuscript in preparation), miR-29a was the highest expressed miR predicted to show multiple interactions with IFNAR1 22. In order to test whether miR-29a targeted Ifnar as predicted, we cloned the 3’ untranslated region of Ifnar into the 3’ region of a luciferase reporter. The addition of a miR-29a mimic, but not that of a scrambled mimic, to HeLa-M cells demonstrated the specific capacity of miR-29a to destabilize mRNA containing the Ifnar 3’ UTR region (Fig. 5a). Due to potential redundancy in function by miR-29a, miR-29b and miR-29c we looked at the expression of all three miR through in situ hybridization. Unlike the brain, where all three miR are expressed at high levels (Fig. 5b), only miR-29a was detected at substantial levels in the thymus (Fig. 5c). The detection of only miR-29a and not miR-29b in the thymus, despite cotranscription from the bicistronic miR-29a cluster, suggested independent regulation of expression, a feature previously described in the miR-29a locus, with the presence of a 3’ hexanucleotide sequence in miR-29b driving nuclear localization and accelerated turnover in the non-mitotic portions of the cell cycle 23. The in situ data indicating poor miR-29b expression in thymic epithelial cells relative to miR-29a was supported by microarray analysis, where miR-29a was expressed at high levels in the cortex and medulla, while miR-29b was low to undetectable (S.Z., G.A.H., manuscript in preparation), and by qPCR analysis, in which miR-29a was strongly expressed while miR-29b was undetectable in half the samples and very poorly expressed in others (Supplementary Fig. 8).
Figure 5. IFNAR is a direct target of miR-29a.
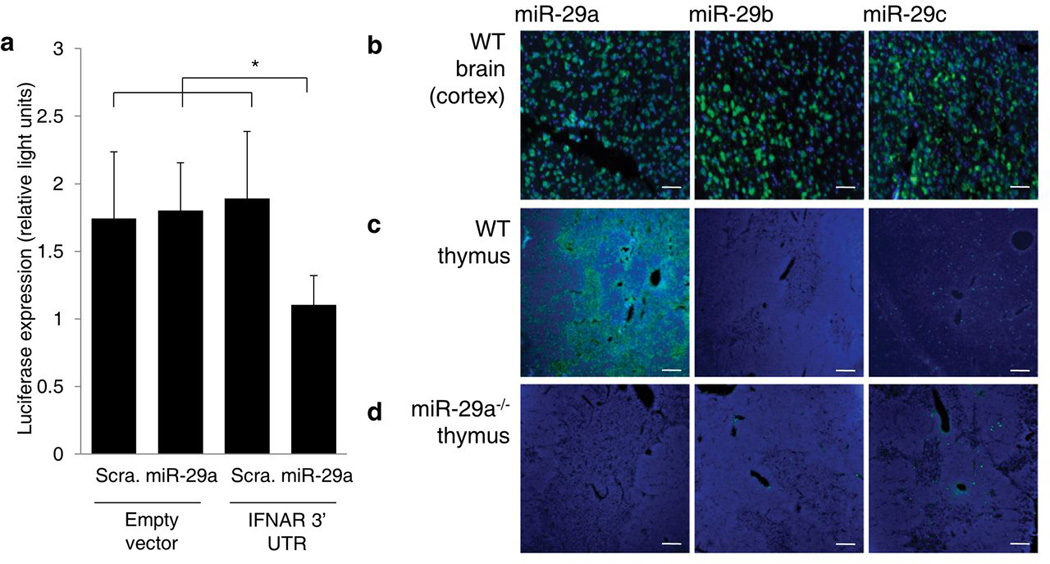
(a) Luciferase expression in HeLa-M cells transfected with empty PsiCheck2 or PsiCheck2 containing the Ifnar1 3’UTR and treated with miR-29a mimic or a scrambled mimic (n=5/group, representative of three independent experiments). *p< 0.05. (b) In situ hybridization for miR-29a, miR-29b and miR-29c in the brain of wild-type. Probe (green) and DAPI (blue). (c) In situ hybridization of thymus of wild-type mice, as in b. (d) In situ hybridization of thymus of miR-29a−/− mice, as in b. Representative pictures from two experiments are shown. Scale bar represents 100µm.
To test the function of miR-29a in the thymus, we generated a targeted deletion of the miR-29a/b-1 locus (hereafter called “miR-29a KO mice”). A ~1.7kb region spanning the entire miR-29a/b-1 cluster precursor sequence was removed through homologous recombination (Supplementary Fig. 9a) and loss of the miR-29a/b-1 gene was demonstrated through Southern Blotting (Supplementary Fig. 9b). In situ hybridization of the thymus of mir29a−/− mice demonstrated complete loss of miR-29a expression, without substantial compensatory upregulation of miR-29b or miR-29c (Fig. 5d). Initial characterization of these mice indicated no major developmental defects, with birth in a normal Mendelian ratio and no noticeable abnormalities in major organs, such as the brain (Supplementary Fig. 10, Supplementary Table 3).
Analysis of the miR-29a-deficient thymus demonstrated that the thymic architecture and thymic involution phenotypes of the Dicer-deficient mice segregated in the miR-29a-deficient mouse. Unlike Foxn1Cre Dicerfl/fl mice, mir29a−/− mice demonstrated no major changes to thymic architecture, with normal development of the cortical and medullary thymic epithelial populations and no progressive decay or epithelial void formation (Fig. 6, Supplementary Fig. 11, Supplementary Fig. 12). Likewise, thymic function appeared intact, with normal thymocyte and peripheral T cell differentiation (Supplementary Fig. 13, Supplementary Table 4). miR-29a-deficient mice did, however, undergo premature thymic involution after four weeks of age (Fig. 6c), at levels indistinguishable from Foxn1Cre Dicerfl/fl mice (Supplementary Fig. 14a,b). As with Foxn1Cre Dicerfl/fl mice, this premature involution was independent of sex hormone levels, as it was not reversed by chemical castration (Supplementary Fig. 14c). Unlike Foxn1Cre Dicerfl/fl mice, loss of miR-29a was not restricted to the thymic epithelium, raising the possibility that the same phenotype was being generated through alterations in other anatomical compartments, such as T cell precursors in the bone-marrow. We therefore used bone-marrow chimeras and thymic transplants to test compartmentalization. Wild-type recipients of either miR-29a-deficient or wild-type bone marrow exhibited normal thymic size. By contrast, miR-29a-deficient recipients of either mir29a−/− or wild-type bone marrow had severely (90%) reduced thymic size (Fig. 6d). When neonatal thymus from mir29a−/− mice or wild-type siblings were transplanted into wild-type hosts, wild-type thymi grafted well and grew in size, while mir29a−/− thymi involuted (Fig. 6e). These results conclusively demonstrate that the premature thymic involution observed in miR-29a-deficient mice is due to loss of miR-29a in the thymic epithelial compartment, and is not secondary to changes occurring in other cell types.
Figure 6. Thymic involution and architecture alterations segregate in miR-29a−/− mice.

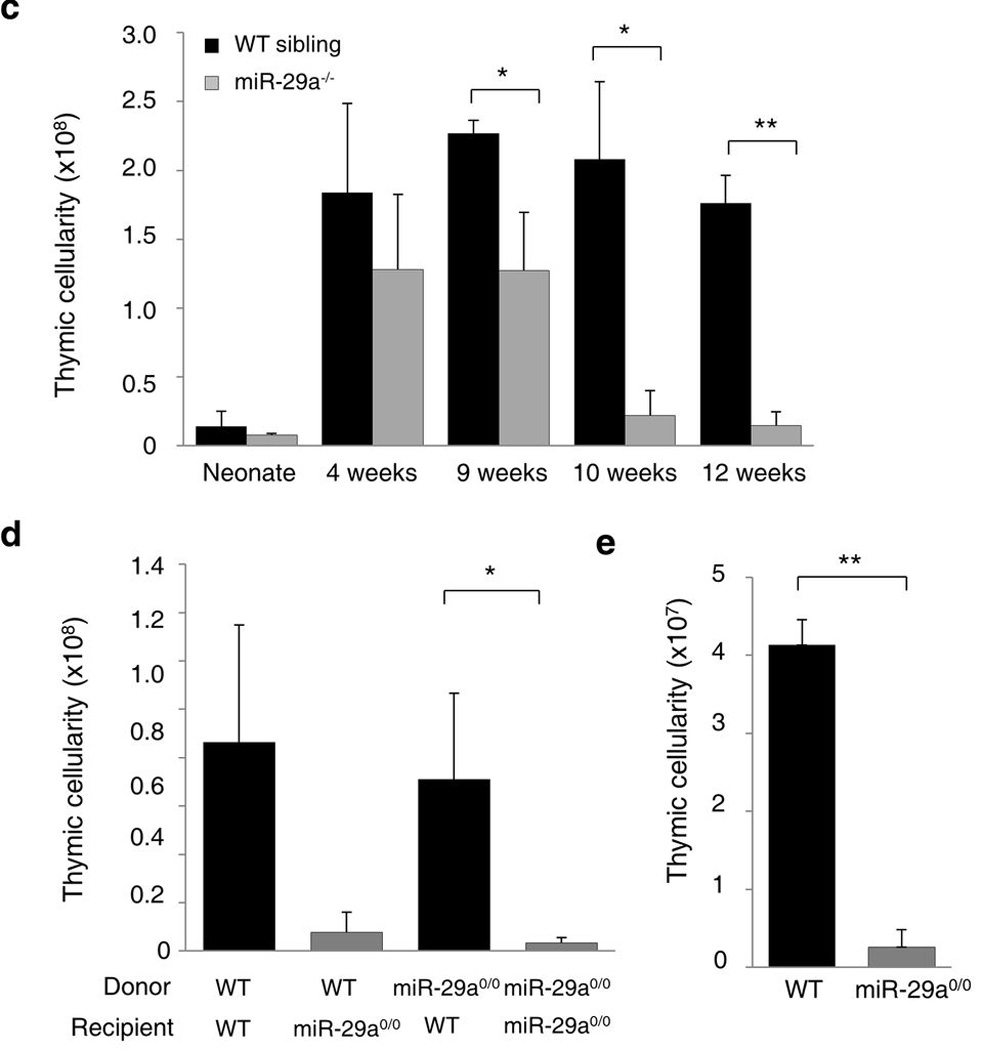
(a) Immunofluorescent staining of the thymus of wild-type and miR-29a−/− mice assessed at 1, 6 and 10 weeks of age. Representative pictures from five experiments are shown. UEA (red), Keratin 14 (green). Scale bar represents 100µm. (b) Immunofluorescent staining of the thymus as in a. Keratin 14 (red) and G8.8 (EPCAM; green). (c) Thymic cellularity of wild-type and miR-29a−/− mice at birth and 4, 9, 10 and 12 weeks of age (n=2–7/group). (d) Thymic cellularity of bone-marrow chimeric mice, indicating the genotypes of the bone-marrow donor and the recipients (n=13,10,11,4). Results pooled from three independent experiments. (e) Thymic cellularity of wild-type mice transplanted with wild-type (n=3) or miR-29a0/0 (n=4) thymi, assessed at 6 weeks post-transplant. *p< 0.05, **p< 0.0001
The miR-29a cluster suppresses IFNAR1
To determine whether inappropriate thymic involution in the miR-29a-deficient mouse was due to PAMP hyper-sensitivity, Ifnar1 mRNA was measured in purified cortical and medullary epithelium in the pre-involution thymus. As befitting a bona fide direct target, expression was increased ~8-fold, a similar scale to that observed in Dicer-deficient epithelium (Fig. 7a). Increases in Ifnar1 mRNA expression were greater in the medulla following thymic involution, suggestive of initiation of a positive feedback loop (Supplementary Fig. 15). To directly test whether this heightened expression recapitulated the Dicer-deficiency phenotype of hyper-sensitivity to PAMP signals, wild-type and miR-29a-deficient mice were exposed to sub-optimal levels of poly(I:C). Unlike wild-type mice, but like Dicer-deficient mice, miR-29a-deficient mice were hyper-sensitive to low-dose poly(I:C) and underwent rapid involution (Fig. 7b). This hyper-sensitivity was dependent on IFNAR1, as coinjection of anti-IFNAR1, but not isotype control, restored ~60% of thymic cellularity, in line with the ~60% neutralization capacity of this antibody 24. The ability of miR-29a-deficient mice to recover from poly(I:C) appeared intact, with ~50% cellular recovery 10 days post-treatment (Supplementary Fig. 16). Even when not treated with low-dose poly(I:C), by 12 weeks of age mir29a−/− mice showed elevated phosphorylation of the direct downstream target of IFNAR1, STAT1, evidence that the increase in Ifnar1 mRNA has a biologically relevant effect. Together, these results demonstrate that loss of miR-29a in the thymic stroma results in premature thymic involution and hyper-sensitivity to pathogen-associated signals, indistinguishable from complete loss of the microRNA network in thymic epithelium.
Figure 7. Thymic epithelial defects in miR-29a result in PAMP hyper-sensitivity and inappropriate thymic involution.
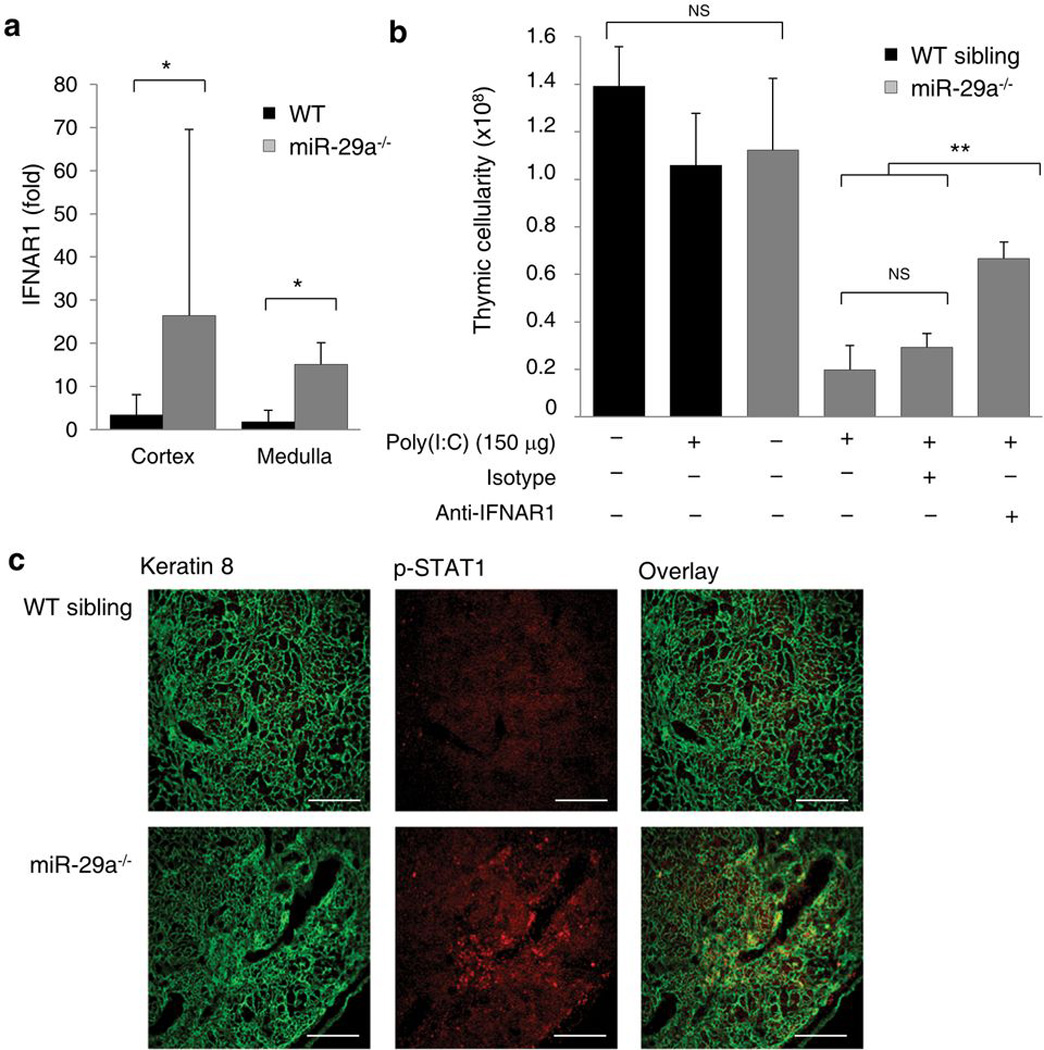
(a) Fold change of IFNAR1 mRNA expression in cortical (BP-1+UEA) and medullary (BP-1UEA+) thymic epithelium from wild-type and miR-29a−/− mice at 6–8 weeks (n=5,3). Fold-change is relative to the average of wild-type cortex values. (b) Thymic cellularity of 4 week-old wildtype and miR-29a−/− mice treated with 150µg dose of poly(I:C) at 4 weeks, with or without coinjection of anti-IFNAR1 antibody or isotype control (n=3–10/group). (c) Immunofluorescent staining of the thymus of wild-type and miR-29a−/− mice assessed at 12 weeks of age. Representative pictures of two experiments are shown. Keratin 8 (green, left), phosphorylated STAT1 (red, centre) and colocalization (yellow, right). Scale bar represents 100µm. *p< 0.05, **p< 0.0001
Discussion
The role of miRNA in the adaptive immune system has been investigated through systematic removal of the miRNA network in T cells 25, 26, B cells 27 and key accessory cell lineages such as dendritic cells 28. While many functions of the adaptive immune system appear to be independent of the miRNA network, several key aspects are under tight miRNA-mediated control, including regulatory T cell lineage stability 29, immunoglobulin repertoire generation 27 and PAMP responsiveness 28. One of the major accessory cell types in the adaptive immune system which has not yet been profiled for miRNA function is the thymic epithelial cell. Our study used the deletion of a conditional Dicer allele to show that loss of the miRNA network within thymic epithelial cells led to a decay of epithelial architecture, reduced thymocyte positive selection, an increased B cell presence, increased susceptibility to induced autoimmunity and inappropriate chronic thymic involution. This diverse array of phenotypes can be explained through two distinct cellular processes. Firstly, a miR-29a-independent increase in epithelial cell apoptosis, which gives a parsimonious account for the majority of observed phenotypes, and secondly a miR-29a-dependent sensitivity to thymic involution.
The root driver of the miR-29a-independent phenotypes is likely to be the changes in thymic architecture. The progressive loss of thymic epithelium is the first phenotype to chronologically appear, with ‘epithelial voids’ apparent by one week of age. It is likely that these ‘epithelial voids’ are formed by enhanced rates of apoptosis in Dicer-deficient epithelium, with the increase in epithelial cell proliferation and immature cells suggestive of a compensatory process. Two separate explanations can be postulated for why the enhanced apoptosis results in epithelial voids rather than a generalized decrease in epithelial density across the thymus. Firstly, the death may be synchronized among expanses of the thymus populated by clonal epithelial cells 30, where an intrinsic sensitivity could create a large void through synchronous death. Secondly, the epithelial void may progressively form due to extrinsic effects resulting from localization along the void boundary. As thymic epithelial cells are strongly reliant on the three dimensional network 31, 32, loss of interactions at the edge of the void may be sufficient to provoke apoptosis. Alternatively, proliferation of cells at the edge of the void in compensation may place them at elevated risk of apoptosis, as Dicer-deficient cells are especially vulnerable to apoptosis during proliferation 33. The presence of apoptotic epithelial cells along the void perimeter may support the second explanation, but does not rule out the first. Why the medulla shows a chronological primacy in void formation remains an open question, but may reflect an enhanced sensitivity to one of the models described above.
The remaining thymic phenotypes can be explained as secondary effects resulting from the primary decay in thymic architecture. The defects in positive selection and the increase in B cell number were not observed before four weeks, suggesting that these changes result not from abnormal regulation of key epithelial proteins, but rather from the ‘anatomical traps’ formed by epithelial cell decay. Early thymocytes precursors that entered the thymus and found themselves in the absence of Notch ligand expression from thymic epithelial cells may default into B cell differentiation 34, 35. Likewise, DP thymocytes that migrated into epithelial cell voids would have reduced interaction with cortical epithelial cells, likely causing reduced positive selection. It is notable that the defect in positive selection is solely observed in the lineage differentiating to CD4+ SP cells, as CD8+ SP selection can occur with reduced efficiency through interaction with non-epithelial cells 36. Increased susceptibility of Dicer-deficient mice to autoimmunity is also supported by the ‘anatomical trap’ model, as SP thymocytes in Dicer-deficient mice have very low exposure to medullary epithelium and Aire+ epithelial cells for efficient negative selection. The progressive decay observed here is unlikely to be amenable to dissection to a single miRNA or mRNA target. While individual miRNA have been shown to induce 18 or suppress 37 apoptosis, the observation of enhanced apoptosis in Dicer-deficient cell-types of diverse origin 27, 38, 39 suggests that this is an emergent feature of regulatory disruption of the whole-network knockout achieved by Dicer excision.
By contrast, the most striking feature of the Dicer-deficient thymus, that of inappropriate chronic involution, can be accounted for by a single miRNA-mRNA interaction. It is notable that both the Dicer-deficient and the mir29a−/− thymus develop to a normal size at birth and expand during growth to 4 weeks, before collapsing in size between 9 and 12 weeks of age. This observation demonstrates that the phenomenon is involution and not a failure in thymic growth. The inappropriate involution caused by loss of miR-29a expression can be explained due to the loss of tight regulation of IFNAR1 expression. We demonstrated here that ifnar1 is a bona fide target of miR-29a and that expression of IFNAR1 is upregulated in thymic epithelium following the loss of Dicer or miR-29a. Furthermore, IFNAR1 upregulation enhanced the sensitivity of the thymic epithelium to PAMP signals, driving thymic involution at sub-optimal levels of PAMP exposure. The spontaneous involution that occurs in older miR-29a-deficient mice is accompanied by activation of STAT1, and thus elevated sensitivity to baseline levels of IFN-α is the parsimonious explanation for the observed chronic involution. In this model, miR-29a plays a key role in the thymic epithelium in limiting the expression of IFNAR1 and hence reducing the threshold at which thymic epithelial cells become triggered to undergo involution. In the absence of epithelial miRNA, baseline PAMP exposure is sufficient to trigger the chronic involution observed. By contrast, in the presence of epithelial miRNA, the thymus is buffered against low amounts of IFN-α, permitting involution to be triggered only upon an adequate infectious trigger. The prior observation that both IFN-α and PAMP exposure drive the downregulation of Dicer 40 raises the possibility that positive feedback loops may be induced by pathogen exposure, where amounts of IFN-α sufficient to drive Dicer downregulation result in upregulation of IFNAR1, initiating the involution cascade.
Methods
Mice
Foxn1cre mice 41 were intercrossed with Dicerfl mice 21 to produce mice with thymic epithelial cell-specific excision of Dicer on the C57BL/6 background. miR-29a knockout mice were generated as described below. Chemical castration was performed at 4 months, through the intramusclar injection of 0.4mg Leuprorelin Acetate (Lupron®), with analysis at 6 months. Surgical castration was performed at 7 weeks, and assessed at 12 weeks 9. Bone-marrow chimeras were constructed using 1×106 complement-treated (Low-Tox M Rabbit complement, Cedarlane) bone-marrow cells/recipient, injected into irradiated (9.5 Gy) hosts. Transplants of neonatal thymus under the kidney capsule were performed at 6 weeks under sterile conditions. High molecular weight poly(I:C) (Invivogen) was injected i.p. on day 0 and 3, with cellularity measured on day 4. In neutralization experiments, 0.75mg of anti-IFNAR1 antibody (MAR1-5A3) or isotype control antibody (GIR-208) were injected i.v on days 0 and 3, 1 hour prior to poly(I:C). Histology was performed by Histology Consultation Services and pathology reports were generated by BioGenetics. Experimental mice were housed under specific pathogen–free conditions and were used in accordance with the University of Leuven Animal Ethics Committee.
Generation of miR-29a knockout mice
BAC clone RPCIB731L15459Q2 was used as the source for the 10.9kb region surrounding the miR-29a/b-1 gene. A 1.7kb region between the 5’ Afel site and the 3’ Sal1 site was deleted through the introduction of the hygromycin resistance gene, and the linearized pBluescript II KSM vector was electroporated into E14 129/Ola embryonic stem cells. Clones that underwent homologous recombination events were identified through hygromycin selection and screening by Southern blotting. Genomic DNA was digested with either ECoRI, ECoRV or NcoI and hybridised with gDNA external 5´probe (PCR product using primers FW: 5´-accctgacattgacacagc-3´ and RV: 5´-aaaggggtcttagcatcca-3´), gDNA external 3´probe (PCR product using primers FW: 5´-ctagagccaaggatagctgtgt-3´ and RV: 5´-ccttcatgagcgtgtaagaca-3´) and internal hygromycin DNA probe (PCR product using primers FW: 5´-cagcgagagcctgacctattgc −3´ and RV: 5´-cgatcctgcaagctccggatg −3´). Embryonic stem cell clones were microinjected into C57BL/6 bastocysts for the generation of chimeras. The chimeras then were back-crossed with C57BL/6 mice for more than seven generations. Genotyping of knockout mice was performed through PCR for the wildtype (561bp, FW: cttaattcttacctgtggctccaacg, RV: gaactattgcacggacttcaccttcc) and knockout bands. (717bp, FW: aaatggttcaaacgctccac, RV: cagaaagcgaaggagcaaag).
Arthritis induction
Collagen-induced arthritis was induced through intradermal injection of chicken collagen type II (Sigma-Aldrich) in Freund’s complete adjuvant, as previously described 42. Clinical severity of disease was monitored for 40 days post-induction, using the previously described scoring system 43. Arthritis susceptibility was tested as the incidence at completion, with four independent groups (two male and two female) treated as paired values in a two-tailed t-test. Arthritis severity among affected mice showed no significant difference across sex or replicate, thus groups were merged and differences in severity were tested with unpaired t-tests for each time point and maximum disease score.
Flow Cytometry and immunofluorescence
Mice were analyzed using the following antibodies: CD69 (FITC), CD25 (FITC), IgM (FITC), CD8 (PE), CD19 (PE), CD3 (PerCP), B220 (PerCP), Foxp3 (APC), CD44 (APC), CD45 (APC), CD43 (APC), CD4 (PE-Cy7), CD23 (PE-Cy7), Ki67 (APC), Active Caspase 3 (biotin, followed by streptavidin 647), G8.8 (PE-Cy7), M5/114 (PerCP/Cy5.5), BP-1 (PE) and UEA (FITC) (Pharmingen, Biolegend, Vector Labs and eBioscience). Intracellular staining was performed following fixation and permeabilization using the reagents from the eBiosciences Foxp3 staining kit.
Thymus sections were prepared and stained as previously described 44. Brain tissues were fixed in 4% paraformaldehyde, permeabilized in 0.25% Tritox-X100 and blocked with 1% Blocking reagent (Roche), 0.2% FCS. Sections were stained using the monoclonal antibodies K8 (Troma-1 DSHB), Meca-32 (ATCC), CD4 (clone GK1.5), CD8 (clone 2.43), CD19 (clone 1D3), G8.8, F4/80, N418 (ebioscience), ERTR7 and NeuN (clone A60, Millipore), and the polyclonal antibodies K5 (Covance), K14 (Covance), Aire (D-17 Santa Cruz), p63 (poly6190, Biolegend), Phospho-Stat1 (Ser727, Cell Signaling technology) and UEA1 (Vector laboratories). For immunofluorescence the following detection antibodies were used: Goat anti-Mouse 594 (Invitrogen), Donkey anti-Rat 488, Donkey anti-Rabbit 488, Donkey anti-Goat 546, Donkey anti-Rabbit 555, Chicken anti-Rat 647, streptavidin 488 and streptavidin 546 (all from Molecular Probes). Images were acquired using a Ziess LSM 510 meta confocal microscope.
In situ hybridization
In situ hybridization for miR-29 was performed as previously described 45. LNA probes with 2´O Methyl modifications were used for hybridization to the miRNA. Fresh frozen 10 µm sections of the thymus were thawed and dried and fixed with 4% PFA for 10 minutes. After washing, brief sinking of slides in acetic anhydrite/TEA solution and washing, prehybridisation took place for 1hour, followed by hybridization for 30 minutes. Different stringency washes followed and incubation with 3%H2O2 occurred. After washing and blocking, anti-FITC/HRP (Roche) was applied to the slides and the signal was amplified after appropriate washings with the TSA system (Perkin Elmer). Sections were mounted with Prolong Gold Antifade reagent (Invitrogen).
Luciferase assay
The Ifnar1 3’ UTR was amplified from genomic DNA, inserted into the PsiCheck2 renilla luciferase reporter plasmid (Promega) and confirmed by sequencing. Reporter assays were performed in HeLa-M cells cotransfected in triplicate with Lipofectamine 2000 (Invitrogen) with empty PsiCheck2 or PsiCheck2 containing the Ifnar1 3’UTR in conjunction with either murine miR-29a mimic or a scrambled mimic control (Dharmacon) at a final concentration of 40 nM. Firefly luciferase was used as a normalization control. Reporter activity was detected 24 hr after transfection with the Dual-Glo Luciferase Assay System (Promega).
Thymic epithelial expression analysis
Thymic stroma preparations were enriched from 3–6 pooled thymi 46. Cells were stained with UEA (FITC, Vector labs), BP-1 (PE, eBioscience), M5/114 (PerCP-Cy5.5, Biolegend), CD45 (APC, eBiosciences), G8.8 (PE-Cy7, Biolegend) and sorted into cortical (CD45−MHC-II+ G8.8+BP-1+UEA−) and medullary (CD45−MHC-II+G8.8+BP-1−UEA+) epithelium. Total RNA was isolated using the miRVana PARIS kit™ (Ambion). cDNA was produced using the “High Capacity cDNA Reverse Transcription Kit” (Applied Biosystems) according to the manufacturer´s protocol. Quantitative RT-PCR was performed on a Roche LightCycler 480 Sequence Detector based on the SYBR green I fluorescence (qPCR MasterMix Plus for SYBR Green I, Eurogentec) and analyzed by the 2−ΔΔCT method 47. All mRNA values are normalized to β-actin, while miRNA are normalized to RNU19.
Supplementary Material
Acknowledgements
We thank A. Tarakhovsky for providing Dicerflox mice, N. Manley for providing Foxn1Cre mice, A. Farr for providing hybridomas and S. Schonefeldt for mouse colony support. D. Anz provided much fruitful discussion. This work was supported by grants from the VIB and FWO (A.L.), Methusalem financing (KULeuven and Flemish government) and ERC (B.D.S), IWT (W.P.), the Australian NH&MRC (D.H.D.G.), EU consortium “Tolerage” and the DKFZ (B.K.).
Footnotes
Author contributions. A.S.P., J.D., M.A.L., W.P. and O.U. performed the experiments. B.K., S.Z., G.A.H. and D.H.G. shared data for study design. J.D., P.M., B.D.S and A.L. designed the study. J.D. and A.L. wrote the manuscript. All authors read and approved the manuscript.
References
- 1.Miller JF. Immunological function of the thymus. Lancet. 1961;2:748–749. doi: 10.1016/s0140-6736(61)90693-6. [DOI] [PubMed] [Google Scholar]
- 2.Itoh M, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. 1999;162:5317–5326. [PubMed] [Google Scholar]
- 3.King CC, et al. Viral infection of the thymus. J Virol. 1992;66:3155–3160. doi: 10.1128/jvi.66.5.3155-3160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anz D, et al. Activation of melanoma differentiation-associated gene 5 causes rapid involution of the thymus. J Immunol. 2009;182:6044–6050. doi: 10.4049/jimmunol.0803809. [DOI] [PubMed] [Google Scholar]
- 5.Aronson M. Hypothesis: involution of the thymus with aging--programmed and beneficial. Thymus. 1991;18:7–13. [PubMed] [Google Scholar]
- 6.Dowling MR, Hodgkin PD. Why does the thymus involute? A selection-based hypothesis. Trends Immunol. 2009;30:295–300. doi: 10.1016/j.it.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Fu Y, Paul RD, Wang Y, Lopez DM. Thymic involution and thymocyte phenotypic alterations induced by murine mammary adenocarcinomas. J Immunol. 1989;143:4300–4307. [PubMed] [Google Scholar]
- 8.Olsen NJ, Olson G, Viselli SM, Gu X, Kovacs WJ. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology. 2001;142:1278–1283. doi: 10.1210/endo.142.3.8032. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland JS, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J Immunol. 2005;175:2741–2753. doi: 10.4049/jimmunol.175.4.2741. [DOI] [PubMed] [Google Scholar]
- 10.Dominguez-Gerpe L, Rey-Mendez M. Time-course of the murine lymphoid tissue involution during and following stressor exposure. Life Sci. 1997;61:1019–1027. doi: 10.1016/s0024-3205(97)00606-1. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki S, Ishida Y, Nishio N, Ito S, Isobe K. Thymic involution correlates with severe ulcerative colitis induced by oral administration of dextran sulphate sodium in C57BL/6 mice but not in BALB/c mice. Inflammation. 2008;31:319–328. doi: 10.1007/s10753-008-9081-3. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) J Biol Chem. 2009;284:16334–16342. doi: 10.1074/jbc.M109.011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, et al. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheedy FJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 17.Androulidaki A, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liston A, Linterman M, Lu LF. MicroRNA in the adaptive immune system, in sickness and in health. J Clin Immunol. 2010;30:339–346. doi: 10.1007/s10875-010-9378-5. [DOI] [PubMed] [Google Scholar]
- 19.Hou J, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 20.Liston A, et al. Lack of Foxp3 function and expression in the thymic epithelium. J Exp Med. 2007;204:475–480. doi: 10.1084/jem.20062465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi R, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet. 2006;38:356–362. doi: 10.1038/ng1744. [DOI] [PubMed] [Google Scholar]
- 22.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 24.Sheehan KCF, et al. Blocking Monoclonal Antibodies Specific for Mouse IFN-a/b Receptor Subunit 1 (IFNAR-1) from Mice Immunized by In Vivo Hydrodynamic Transfection. Journal of Interferon and Cytokine Research. 2006;26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 25.Muljo SA, et al. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cobb BS, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koralov SB, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 28.Kuipers H, Schnorfeil FM, Fehling HJ, Bartels H, Brocker T. Dicer-dependent microRNAs control maturation, function, and maintenance of Langerhans cells in vivo. J Immunol. 2010;185:400–409. doi: 10.4049/jimmunol.0903912. [DOI] [PubMed] [Google Scholar]
- 29.Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodewald HR, Paul S, Haller C, Bluethmann H, Blum C. Thymus medulla consisting of epithelial islets each derived from a single progenitor. Nature. 2001;414:763–768. doi: 10.1038/414763a. [DOI] [PubMed] [Google Scholar]
- 31.Sato T, et al. Surface molecules essential for positive selection are retained but interfered in thymic epithelial cells after monolayer culture. Cell Immunol. 2001;211:71–79. doi: 10.1006/cimm.2001.1823. [DOI] [PubMed] [Google Scholar]
- 32.Flomerfelt FA, Kim MG, Schwartz RH. Spatial, a gene expressed in thymic stromal cells, depends on three-dimensional thymus organization for its expression. Genes Immun. 2000;1:391–401. doi: 10.1038/sj.gene.6363695. [DOI] [PubMed] [Google Scholar]
- 33.Fukagawa T, et al. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol. 2004;6:784–791. doi: 10.1038/ncb1155. [DOI] [PubMed] [Google Scholar]
- 34.Anderson G, Pongracz J, Parnell S, Jenkinson EJ. Notch ligand-bearing thymic epithelial cells initiate and sustain Notch signaling in thymocytes independently of T cell receptor signaling. Eur J Immunol. 2001;31:3349–3354. doi: 10.1002/1521-4141(200111)31:11<3349::aid-immu3349>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 35.Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med. 2001;194:1003–1012. doi: 10.1084/jem.194.7.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bix M, Raulet D. Inefficient positive selection of T cells directed by haematopoietic cells. Nature. 1992;359:330–333. doi: 10.1038/359330a0. [DOI] [PubMed] [Google Scholar]
- 37.Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–298. doi: 10.1111/j.1365-2567.2009.03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fedeli M, et al. Dicer-dependent microRNA pathway controls invariant NKT cell development. J Immunol. 2009;183:2506–2512. doi: 10.4049/jimmunol.0901361. [DOI] [PubMed] [Google Scholar]
- 39.Campbell-Yeo ML, et al. Effect of domperidone on the composition of preterm human breast milk. Pediatrics. 125:e107–e114. doi: 10.1542/peds.2008-3441. [DOI] [PubMed] [Google Scholar]
- 40.Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46:1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon J, et al. Specific expression of lacZ and cre recombinase in fetal thymic epithelial cells by multiplex gene targeting at the Foxn1 locus. BMC Dev Biol. 2007;7:69. doi: 10.1186/1471-213X-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geboes L, et al. Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum. 2009;60:390–395. doi: 10.1002/art.24220. [DOI] [PubMed] [Google Scholar]
- 43.Kelchtermans H, et al. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther. 2005;7:R402–R415. doi: 10.1186/ar1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dooley J, Erickson M, Farr AG. Alterations of the medullary epithelial compartment in the Aire-deficient thymus: implications for programs of thymic epithelial differentiation. J Immunol. 2008;181:5225–5232. doi: 10.4049/jimmunol.181.8.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silahtaroglu AN, et al. Detection of microRNAs in frozen tissue sections by fluorescence in situ hybridization using locked nucleic acid probes and tyramide signal amplification. Nat Protoc. 2007;2:2520–2528. doi: 10.1038/nprot.2007.313. [DOI] [PubMed] [Google Scholar]
- 46.Dooley J, Erickson M, Farr AG. An organized medullary epithelial structure in the normal thymus expresses molecules of respiratory epithelium and resembles the epithelial thymic rudiment of nude mice. J Immunol. 2005;175:4331–4337. doi: 10.4049/jimmunol.175.7.4331. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.