

Abstract
The lower urinary tract’s virtually inevitable exposure to external microbial pathogens warrants efficient tissue-specialized defenses to maintain sterility. The observation that the bladder can become chronically infected in combination with clinical observations that antibody responses following bladder infections are not detectable, suggest defects in the formation of adaptive immunity and immunological memory. We have identified a broadly immunosuppressive transcriptional program specific to the bladder, but not the kidney, during infection of the urinary tract that is dependent on tissue-resident mast cells (MCs). This involves localized production of interleukin-10 and results in suppressed humoral and cell mediated responses and bacterial persistence. Therefore, in addition to the previously described role of MCs orchestrating the early innate immunity during bladder infection, they subsequently play a tissue-specific immunosuppressive role. These findings may explain the prevalent recurrence of bladder infections and suggest the bladder as a site exhibiting an intrinsic degree of MC-maintained immune privilege.
Introduction
Peripheral tolerance is rigorously maintained in certain tissue microenvironments, limiting the function and scope of immune responses and promoting relative immune privilege (Mellor, 2008). Immune-privileged sites, such as tumors, or organs, such as the gut and liver, have specialized strategies that increase the threshold for immune activation. Mechanisms that dampen immune responses are highly varied and can include the production of the predominately immunosuppressive cytokine, IL-10, activation of antigen-specific T regulatory (Treg) cells, and constraint of dendritic cell (DC) activation or function (Belkaid, 2010; Francisco, 2010; Steinbrink, 1997; Waldmann, 2006). Many questions remain regarding how a tolerogenic microenvironment affects immunity to pathogens but some evidence suggests that relative immune tolerance can be exploited by pathogens to maintain or initiate infection. For example, malarial parasites (genus Plasmodium) can remain latent in the liver, also a site of relative immune tolerance (Knolle, 2000) and subsequently restore fulminant infection (Prudencio, 2006). Certain anatomical locations are more susceptible to recurrent infections, including the bladder.
The bladder is a specialized mucosal tissue where the immune regulatory networks remain mostly unexplored. This organ has not traditionally been considered a functionally immune privileged site, particularly in light of a wealth of literature describing productive innate immune responses by the bladder to uropathogens (Haraoka, 1999; Mulvey, 2000; Song, 2007). Yet, the bladder also experiences a high level of recurrent infections, raising the question of where the balance lies for this organ with respect to the decision to maintain or break tolerance. With its unique physiology and function of storing host waste products, it is crucial for the bladder to balance host defense and the need to limit inappropriate responses to self-products and minimize tissue damage.
Mostly caused by uropathogenic E. coli (UPEC), urinary tract infections (UTIs) are the second most common bacterial infection in humans (Hagberg, 1981; Hooton, 1996) and many are recurrent (Foxman, 1990). Although the underlying basis is not known, the high frequency of recurring UTIs suggests a defect in immunological memory formation subsequent to bladder infection. UTIs can involve only the bladder, but a significant number also progress to the kidneys. Interestingly, clinical observations indicate that bladder infections, unlike kidney infections, fail to evoke detectable pathogen–specific antibodies in the serum and urine (Percival, 1964; Ratner, 1981; Rene, 1982; Sanford, 1978; Winberg, 1963). These observations seem counterintuitive since bladder infections are typically accompanied by a robust innate immune response involving vigorous IL-6 and IL-8 production and large numbers of neutrophils in the urine (Fihn, 2003; Nielubowicz, 2010; Stamm, 1983). Hence, there appears to be a disconnect between innate and adaptive immune responses in the bladder during infection. In this work, we sought to elucidate the underlying basis for the muted adaptive responses in the bladder and to explain the unique persistence of infection associated with this organ.
Results
UPEC persist in the bladder but not the kidneys
UTIs are typically accompanied by a robust innate inflammatory response involving neutrophil recruitment, which is capable of resolving the acute phase of bacterial infection (Haraoka, 1999; Mulvey, 2000; Nielubowicz, 2010). However, others and we have observed a remarkable persistence of E. coli within the bladder, a hallmark of UTI infection (Malaviya, 1999; Mulvey, 2001; Mysorekar, 2006). When UTIs were initiated with UPEC, high colony forming units (CFU) could be recovered from bladders and kidneys 12h post-infection. Complete clearance was achieved in the kidneys within 5d (Figure 1A). Yet, in the bladder, a population of bacteria persisted (Figure 1A) and could be viewed within or underneath the superficial bladder epithelium, weeks later (Figure 1B). It should be noted that acute infection within the bladder subsides by 3d, evidenced by the urine being sterile (Figure S1A) and the return of neutrophil detection to homeostatic levels (Figure S1B). Not only could persistent UPEC potentially act as a reservoir of bacteria for recurrent infections, but these findings also suggest there is a bladder specific shortfall in host immunity that is not applicable to the entirety of the urinary tract.
Figure 1. see also Figure S1. Bladders fail to eradicate persistent E. coli.
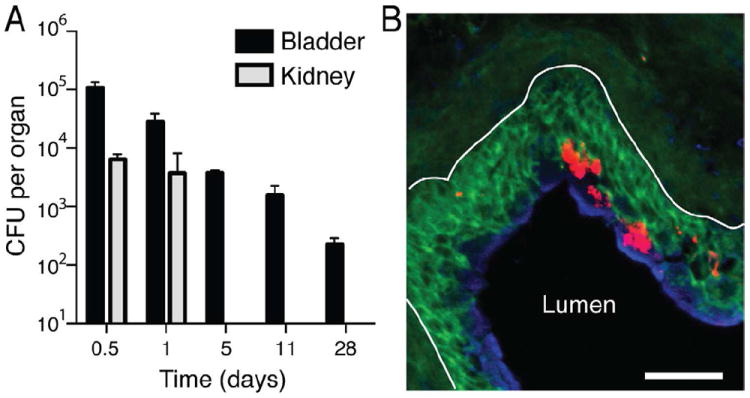
(A) Mice were infected either in the bladder or with concurrent bladder and kidney infections and the organs were homogenized and plated on bacterial agar for growth, to quantitate the CFUs of bacteria per organ. While the kidneys are able to completely clear infection within 3d, the UPEC in the bladder persist for over 4 weeks. All data are representative of three independent experiments, n≥3 per time point; error bars represent ±SEM. (B) E. coli (red) remain within the intermediate (green) and superficial bladder epithelium (blue) 2 weeks p.i. (basement membrane denoted by white line). Scale bar=50μm.
A mouse model of bladder-restricted infection
To investigate the isolated influence of the bladder microenvironment on immunity, we developed a model where infection was restricted to this tissue. Although much mechanistic knowledge of UTIs has come from mouse models, these have rarely distinguished infection of the bladder alone, cystitis, from pyelonephritis, where pathogens also colonize the kidney via the ureter. Since evidence from human patients suggests that these two UTIs are highly divergent with regards to the profile of adaptive immune responses elicited (Percival, 1964; Ratner, 1981; Rene, 1982; Sanford, 1978; Winberg, 1963), we developed two models of experimental UTIs where infection was either exclusively contained in the bladder or also in the kidneys (replicating the clinical phenomena of cystitis and pyelonephritis, respectively). To induce pyelonephritis, we instilled UPEC in a larger volume with pressure so that a spontaneous retrograde flow (vesicoureteral reflux, VUR) from bladder to the kidneys would occur. Using Evans Blue Dye (EBD) to track our inoculum, we examined the lymph nodes (LNs) draining the bladder or kidney. In our pyelonephritis model, dye entered the iliac LNs (ILN), which are the draining LNs (DLNs) for the bladder, as well as those that drain the kidneys, the renal LNs (RLN) (Figure S2). However, in our cystitis-only model, dye was only present in the ILNs, and not the RLN (Figure S2), suggesting that the inoculum did not reflux into the kidneys. To support these visual observations, we determined the bacterial burden in the bladder and kidney for each model. CFUs were comparable in the bladders for both infectious models 12h post-infection, while bacteria were not present in the kidneys in our model of bladder-restricted infection (Figure 2A). These data confirm that we have a working model to examine bladder-specific immune responses in an infectious context.
Figure 2. see also Figure S2. A mouse model distinguishing between cystitis and pyelonephritis.
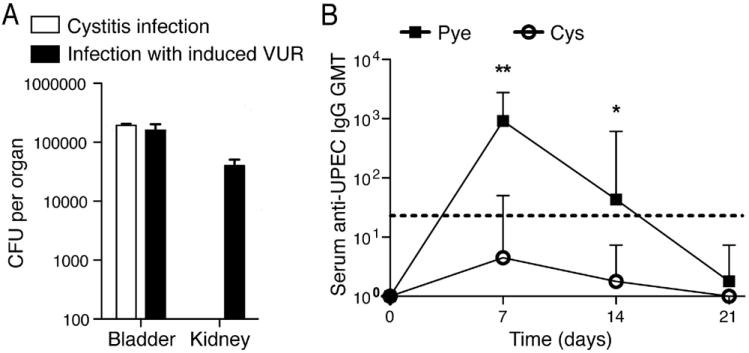
(A) Similar bacterial CFUs are obtained from bladders during both methods of infection; bacteria are only cultured from kidneys of pyelonephritis-infected mice. All data are representative of 2 independent experiments, n=3-4 per time point; error bars represent ±SEM. (B) Only mice with acute pyelonephritis show significant serum anti-UPEC IgG geometric mean titers (GMT) following infection. Dotted line denotes threshold of detection. *p<0.05; **p<0.01. Error bars represent the 95% confidence level; n=4-6.
Numerous reports examining human patients have showed that those with cystitis alone fail to evoke significant pathogen-specific antibody titers (Percival, 1964; Rene, 1982; Sanford, 1978; Winberg, 1963). This suggests that individuals with bladder-restricted infections fail to develop an observable adaptive immune response. To investigate this phenomenon in our two murine UTI models, we induced either cystitis or cystitis with pyelonephritis and tested for the presence of E. coli-specific antibodies in the sera from each group. In line with the clinical reports, mice with bladder-restricted infections fail to induce significant E. coli-specific antibodies over naïve mice (p >0.05); however, concurrent kidney infection resulted in a strong pathogen-specific serum IgG response (Figure 2B). Anti-E. coli IgA responses were also only detectable in mice with kidney infections (data not shown), supporting that mucosal responses are similarly divergent to serological responses. With this, we have developed infectious mouse models that distinguish between bladder infection alone or involving the kidneys, and identified site-specific defects in adaptive immunity similar to the human clinical picture.
Impaired immunological memory to bladder infection
Because persistent infection in the bladder is observed (Figure 1A-B) and recurrent infection is typical of human cystitis (Foxman, 2002), we sought to further define the host’s adaptive immune response and immunological memory formation after bladder infection, suspecting that bladder infections are unable to induce appropriate immunological memory. For these studies, either a bladder-restricted infection (cystitis) or combined bladder and kidney infection (pyelonephritis) were induced in mice on day 0 (Figure 3A, first arrow) serum antibodies against E. coli were measured every 7d post-infection (p.i.). Again, we observed a high anti-E. coli titers in mice with prior pyelonephritis and a lack of substantial anti-E. coli antibody titers in mice having had cystitis. On day 21 (Figure 3A, second arrow) both groups of mice (cystitis alone vs. pyelonephritis) were reinfected using the cystitis-only model with the same E. coli strain as the primary challenge. As expected, upon a second infection, mice with prior pyelonephritis showed a strong secondary antibody response to UPEC (Figure 3A). Although higher titers compared to the primary response (one hallmark of memory responses) were not observed at the time points assayed, this memory response was characterized by a prolonged duration of detectable antibody responses compared to the first infection (Figure 3A). However, mice that had a primary infection restricted to the bladder showed no significant (p > 0.05) memory recall response to cystitis re-challenge (Figure 3A), pointing to a tempering of the adaptive immunity in the bladder that does not follow the normal pattern for a secondary humoral response. These observations suggest that if adaptive immune responses have already been established, the bladder is immunologically competent to respond; yet, a primary infection of the bladder alone has limited ability to initiate productive immunological memory.
Figure 3. Bladder-contained infections fail to evoke significant antibody responses unless primary immunity was already established.
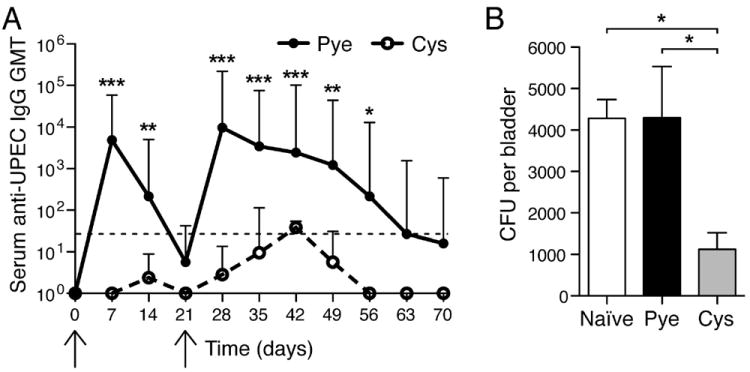
(A) Mice were infected transurethrally with 1×108 CFU to induce cystitis-only or pyelonephritis (first arrow) and serum GMT anti-UPEC antibodies were measured (x-axis shows days p.i.). On day 21 (second arrow) cystitis-only was induced in both groups. *p<0.05; **p<0.01; ***p<0.001, compared to naïve. Error bars represent the 95% confidence level with n=4-7. Dotted line denotes threshold of detection. (B) Naïve mice that were adoptively transferred splenocytes from mice that previously had acute pyelonephritis show enhanced bacterial clearance 5d post-cystitis challenge, compared to control naïve mice or mice adoptively transferred splenocytes from mice that previously had cystitis only. *p<0.05. Error bars represent ±SEM with n=4-6.
To investigate the functionality of memory lymphocytes and confirm this absence of protective immunological memory subsequent to bladder infection, we performed adoptive transfer experiments. Mice were infected to initiate bladder-restricted, or bladder combined with kidney infections. Splenocytes were isolated 21d p.i. and transferred i.v. into naïve hosts. Three days following transfer, mice were challenged with a bladder infection, after which the E. coli CFUs still present in the bladder 5d p.i. were analyzed. Naïve mice with splenocytes transferred from pyelonephritis-infected mice showed significantly increased bacterial clearance as compared to both naïve mice and mice given splenocytes from mice with bladder-restricted cystitis (p <0.05) (Figure 3B). Therefore, whereas the host is able to gain protective immunological memory in the context of a UTI involving the kidney, a primary infection of the bladder, alone, fails to generate a sufficient memory response to enhance bacterial clearance during re-infection.
The bladder, unlike the kidney, maintains a tolerogenic environment during infection
To better understand the underlying differences in immunity between the bladder and kidney, we studied the cytokine profile of both organs during E. coli infection in the timeframe when adaptive immune responses should be induced and refined. As expected, a strong pro-inflammatory response, demonstrated by the drastic upregulation of cytokine expression, including Il6, KC (a functional homologue of Il8) and TNF-α (Figure 4A-C) was observed early in infection. However, 6h p.i., during both cystitis and pyelonephritis, there was upregulation in the bladder of a major inhibitor of pro-inflammatory immune responses, Il10, which was distinctly absent in the kidney (Figure 4D). Additional negative regulators of inflammation were also upregulated: TGF-β, a broadly-acting anti-inflammatory regulator, as well as suppressor of cytokine signaling 3 (SOCS3), a negative regulator of cytokine signaling that inhibits activation and/or differentiation pathways in macrophages, DC and T cells (Hagberg et al., 1983) (Figure 4E-F). However, both TGF-β and SOCS3 expression were not detected in the kidneys after infection. Unlike the pro-inflamatory responses in the bladder that begin within 2h, peak at 6h and begin resolving by 24h (Figure 4A-C), this secondary wave of enhanced expression of anti-inflammatory cytokines and signaling intermediaries persists throughout the first 3d of infection (Figure 4D-F), a critical time for the development of adaptive responses. These observations demonstrate that subsequent to strong proinflammatory innate responses, the bladder specifically responds to UPEC infection with a second, tolerogenic phase of response involving the induction of anti-inflammatory master regulators.
Figure 4. see also Figure S3. Enhanced Il10 transcription typifies the anti-inflammatory program upon infection, limits bacterial clearance and serum antibodies.
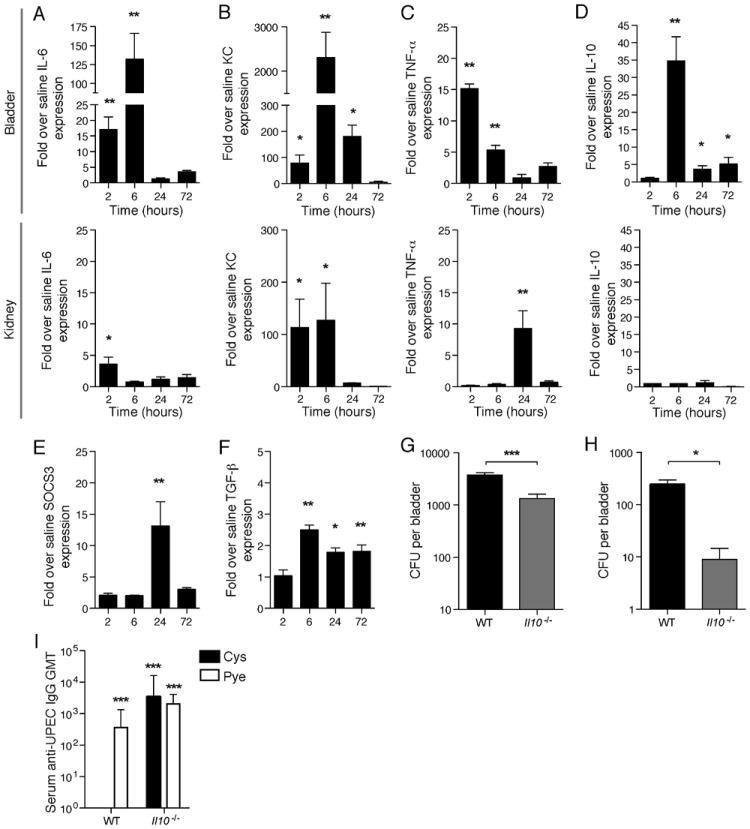
(A-D) Cytokine transcriptional profile in the bladder and kidney at various times p.i. determined by real time PCR for Il6, KC, TNF-α and Il10. (E-F) In addition to Il10 upregulation, significantly increased levels of other major anti-inflammatory mediators – TGF-β and SOCS3 – were also seen only in the bladder and not detected in the kidney. * p < 0.05, ** p ≤ 0.01; error bars represent ±SEM. All data are representative of two or more independent experiments, n ≥ 3. Il10-/- mice have better bacterial clearance in the bladder as compared to WT, (G) 5d and (H) 6 weeks after infection. * p<0.05, *** p<0.001; n=10-12 mice per group (5d) and n=5-6 mice per group (6 weeks) (I) Significant serum GMT anti-UPEC IgM antibodies are seen in both cystitis-only and mice with pyelonephritis in Il10-/- mice at comparable titers to mice with concurrent cystitis and pylenophritis infection at 7d p.i. There are no significant differences in antibody titers between WT mice with pyelonephritis and Il10-/- infected mice. UPEC-specfic IgM was not detected in WT mice with cystitis alone. Dotted line denotes threshold of detection. ND=not detected, *** p<0.001. Error bars represent the 95% confidence level with n=5-7.
In view of the relatively high degree of Il10 upregulation in the bladder, we focused on IL-10 as a target immunosuppressive cytokine that might alter the course of adaptive immunity to bladder infection. To discern the functional consequence of IL-10 during bladder infection, we infected Il10 deficient mice (Il10-/-) with E. coli, using our model of cystitis, and determined the load of persistent bacteria still present in the bladder 5d p.i. We saw that Il10-/- mice had significantly fewer residual bacteria in the bladder compared to wild-type (WT) mice after acute infection (5d) (p <0.001) (Figure 4G) as well as a lower number of persisting bacteria in the bladder at 6 weeks (Figure 4H), indicating that bacterial clearance is enhanced in the absence of IL-10. Both cystitis and pyelonephritis-infected mice had IgG titers against E. coli that were noticeably above that of naïve Il10-/- mouse sera, although, the titers of Il10-/- mice were still lower than those of WT mice (Figure S3). Studies have shown that IL-10 is important for mediating immunoglobulin class switching in B cells (Malisan, 1996; Vanderford, 2010) and this may explain why the detected IgG sera titers against E. coli were lower in Il10-/- mice than WT mice (Figure S3). IgM is the first antibody subclass that is produced in a naive host and, therefore, not likely to be greatly impacted by the class-switch defect in Il10-/- mice. In view of this, we assayed for E. coli-specific IgM antibodies in both WT and Il10-/- mice infected with cystitis alone or cystitis concurrent with pyelonephritis to provide a more accurate picture of the magnitude of antibody responses without the role of IL-10 in class-switching complicating the picture. We saw higher titers of IgM antibodies in sera collected from Il10-/- mice with a bladder-restricted infection, in contrast with WT mice (Figure 4I). These results indicate that, without IL-10, the bladder is able to initiate a functional adaptive immune response to cystitis.
To investigate the mechanism underlying IL-10-mediated suppression of responses in the bladder, we examined the initial events in the induction of adaptive immunity. We suspected that DC migration might be differentially affected during cystitis with and without pyelonephritis but observed no difference in the numbers DCs within the ILN or RLN upon bladder or kidney infection, respectively, in the initial hours of infection (Figure 5A). Migration of DCs is only one aspect required for efficient activation of adaptive immunity within DLNs; antigen presenting cell (APCs) activation, including co-stimulatory molecule expression, is also important for activation of naïve T cells. Along these lines, we observed significantly greater numbers of activated DCs, expressing the co-stimulatory molecule CD86, in the RLN of WT mice during kidney infection, but not in the ILNs draining the bladder (p <0.05) (Figure 5B). Since we observed that Il10-/- mice respond to bladder infection with adaptive responses of greater magnitude than WT mice, and the upregulation of costimulatory molecules on DCs has been established to be influenced by IL-10 (Enk, 1993; Steinbrink, 1999, 1997), we next investigated the influence of IL-10 on the process of DC activation during cystitis. Il10-/- mice showed significantly greater numbers of CD86+ DCs in the ILN as compared to ILN from WT mice during cystitis (p <0.01) (Figure 5B). Likely as a consequence of having fewer activated DCs draining to the ILN, WT mice with cystitis had significantly reduced numbers of CD4+ T helper cells (TH cells) (Figure 5C) and reduced numbers of germinal center B cells (CD19+GL7+) 7d p.i. (Figure 5D). Therefore, increase of IL-10 in the bladder upon infection influences the activation of LN-draining DCs and may explain the uniquely dampened adaptive immune responses to bladder infection.
Figure 5. see also Figure S4. Suppression of DC activation in ILN during cystitis.
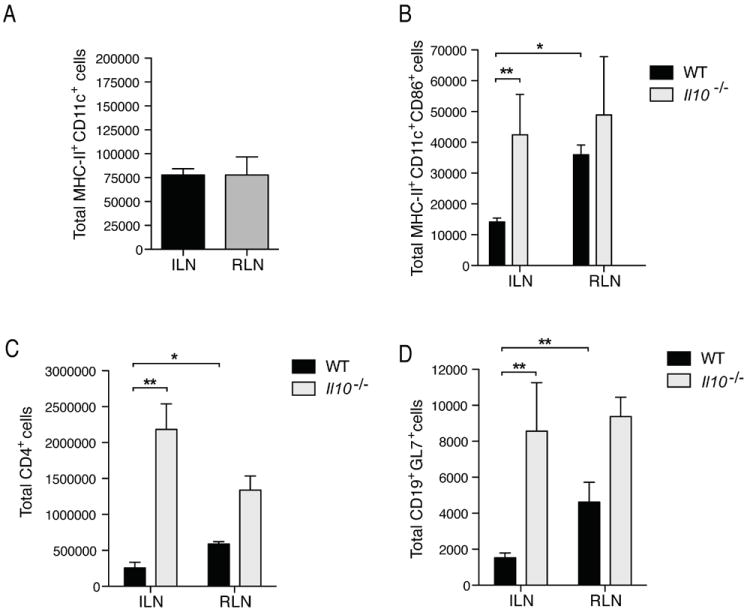
(A) Numbers of DCs draining to the ILN and RLN upon cystitis or pyelonephritis were determined by flow cytometry by staining for MHC-II and CD11c, 24h after infection with 1×108 UPEC. Data does not differ significantly. (B) Increased numbers of activated DCs (MHCII+ CD11c+ CD86+) were present in the RLNs of WT mice and in ILN of Il10-/- mice, similarly infected. WT mice have fewer (C) TH cells (CD4+) and (D) germinal center B cells (CD19+GL7+) in LNs 7d p.i. * p<0.05, ** p<0.01; error bars represent ±SEM. All data are representative of 2 or more experiments with n=3-7. Representative flow cytometry plots are shown in Figure S4.
MCs mediate IL-10-dependent suppression of adaptive immunity in the bladder
With the observation that IL-10 expression increased in the bladder 6h p.i., we sought to find the source of this anti-inflammatory cytokine. One immune cell that has been shown to produce IL-10 during inflammation is the MC (Grimbaldeston, 2007). Although better known for their roles in exacerbating asthma (Kobayashi, 2000; Williams, 2000) and in providing immunosurveillance for pathogens (Abraham, 2010; Chan, 2012; Kalesnikoff, 2008), MCs also appear to have roles in maintaining tissue homeostasis. For example, they have been shown to be promote the maintenance of Treg cell tolerance to allographs (Lu, 2006) and it has been reported that MCs limit inflammation in an IL-10-dependent manner during contact hypersensitivity (Grimbaldeston, 2007). Although these latter results from the context of cutaneous sterile inflammation have recently been contested (Dudeck, 2011), they raise the question of whether MCs could also temper inflammation in an infectious context within the bladder. MCs are also well positioned, physically, to serve this role in the bladder, being located in high abundance beneath the uroepithelium at the host-environment interface (Figure 6A). However, we have previously reported that MCs can respond to infection of the bladder and play a crucial proinflammatory role in recruitment of neutrophils for bacterial clearance (Malaviya, 2004), as well as recruitment of APCs to the site of infection (Shelburne, 2009). Therefore, we next investigated if, contrasting their function initiating pro-inflammatory responses (Abraham, 2010; Chan, 2012; Malaviya, 2004; Shelburne, 2009), MCs play a secondary function in the bladder by promoting IL-10-mediated suppression of adaptive immunity. We observed that basal Il10 expression in the bladder was lower in MC-deficient (Wsh) mice compared to WT mice (Figure 6B). Therefore, even under homeostatic conditions, MCs appear to contribute to IL-10 production in the bladder. Six hours p.i., WT mice had increased IL-10 expression in the bladder, which was not seen in Wsh mice (Figure 6C). The kidneys also have resident MCs but, in contrast to the observation that bladder MCs contribute to basal IL-10 levels (Figure 6B), the basal level of IL-10 was higher in kidneys of Wsh mice (Figure 6D). In Wsh mice, IL-10 levels also dropped dramatically during infection (Figure 6E). To determine if bladder MCs, themselves, were a significant source of IL-10, we stained bladders 24h after infection for IL-10 and the MC-specific protein MMCP6 (Figure 6E). Although our real time PCR data suggests that MCs contribute to maintaining the basal levels of IL-10 expression (Figure 6B), staining for this cytokine in the tissues of saline control mice did not reveal striking levels of IL-10 (Figure 6F). Background staining for IL-10 also could not be detected in Il10-/- mice (data not shown), yet infected IL-10-sufficient animals showed strong co-localization of IL-10 with MMCP6 (Figure 6F). In addition to MCs, other cell populations such as Treg cells and macrophages have the ability to produce and contribute to IL-10 levels in the bladder (Couper, 2008). To confirm that MCs are the major source of IL-10 upon infection, we stained single cell suspensions of infected bladders intracellularly for IL-10 and analyzed the IL-10 producing cell populations by flow cytometry. Although the Treg cell and macrophage compartments contribute to the pool of IL-10+ cells in the bladder (Figure 6G), the majority of IL-10+ cells were CD117+ MCs (Figure 6F). We also detected increased secreted IL-10 protein in the urine of infected WT, but not Wsh mice (Figure S5A-C), supporting that MCs are the predominate contributors of IL-10 to the bladder microenvironment.
Figure 6. see also Figure S5. MCs in the bladder promote Il10 expression.
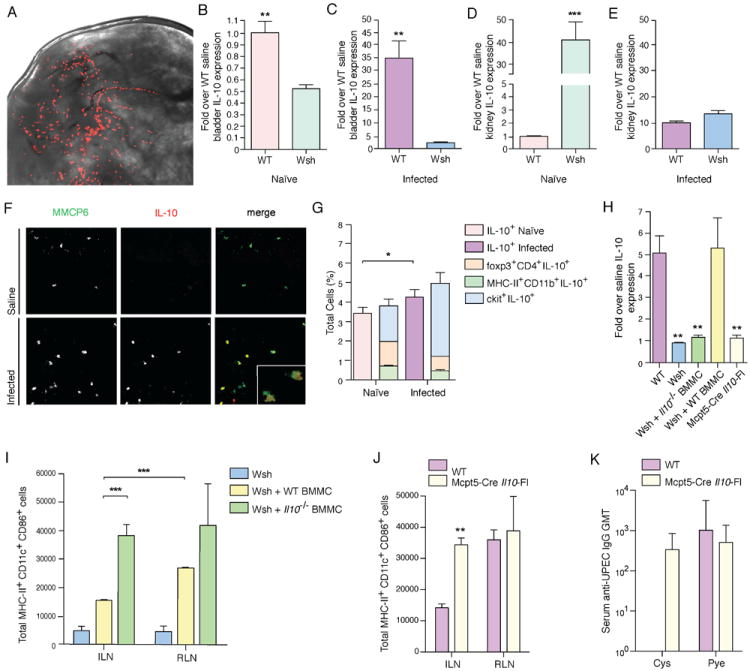
(A) A whole mount picture of the luminal side of a murine bladder, showing the abundance of MCs (red, avidin-TRITC stained) under the epithelium. (B) Wsh mice, even at basal levels, have lower expression of Il10 in the bladder as compared to WT bladders, determined by real time PCR. (C) With infection, at 6h after bacterial instillation, Il10 expression increases in bladders of WT but not Wsh mice. (D) Il10 is expressed at higher basal levels in the kidneys of Wsh compared to WT mice, as determined by real time PCR. (E) However, upon infection, the high Il10 levels of expression in Wsh kidneys drop dramatically to be not significantly different from those of WT kidneys at 6h. (F) Image shows a bladder section stained for MCs (MMCP6, green), and for IL-10 (red) 24h after UPEC infection. (G) Intracellular staining for IL-10 reveals IL-10+ cells in the bladder 24h p.i. Though Treg cells (foxp3+CD4+) and macrophages (MHC-II+CD11b+) contribute to the IL-10 in the bladder, the majority of cells that are IL-10+ are MCs (ckit+). (H) Rescue of increased Il10 expression is seen in infected bladders of Wsh mice reconstituted with WT BMMCs but not Il10-/- BMMCs at 24h p.i. (I) Wsh mice reconstituted with WT BMMCs have significantly greater activated DCs draining to the RLNs, compared to ILNs 24h p.i. Reconstitution with BMMC from Il10-/- mice results in comparable numbers of activated DCs in both LNs. *p<0.05, **p ≤0.01, ***p≤0.001; data are representative of ≥3 independent experiments with n≥3. Error bars represent ±SEM. (J) Mcpt5-Cre Il10Fl mice showed the same phenotype as Il10-/- mice and Il10-/- BMMC-reconstituted Wsh mice: significantly greater numbers of mature DCs present in both ILN and RLN as compared to WT ILN 24h p.i. (K) Serum UPEC-specific IgG was detected in Mcpt5-Cre Il10Fl mice 7d p.i. Error bars represent the 95% confidence level with n=5.
To verify that enhanced cellular trafficking during infection is MC-dependent, we reconstituted Wsh mice with bone-marrow derived MCs (BMMC) (Figure S5D-E), resulting in rescue of LN hypertrophy (Figure S5F) and influx of neutrophils (Figure S5G). We also saw an increase in Il10 expression in bladders of infected but not IL10-/- BMMC-reconstituted Wsh mice (Figure 6H) and comparable to WT mice, there were greater numbers of activated DCs draining to the RLN as compared to the ILN (Figure 6I). More importantly, when Wsh mice were reconstituted with Il10-/- BMMCs, the pro-inflammatory responses were restored, with abundant activated DCs in DLNs and equivalent cell numbers in the ILN and RLN (Figure 6I). This observation was similar to that seen, phenotypically, in Il10-/- mice (Figure 5B). Since the proposed role of MC-derived IL-10 in sterile inflammatory responses (Grimbaldeston, 2007) has been recently contested and was alternatively explained to be potentially due to the neutrophilia phenotype of the Wsh model (Dudeck, 2011), we verified that neutrophil depletion did not affect the augmented production of IL-10 by MCs or activation of DCs (Supplemental Text and Figure S5H-K). Additionally, to definitively show the contributing role of MC-derived IL-10 in the tolerogenic bladder microenvironment, we used an established Cre-Lox recombination model with the Cre gene under the regulation of the MC protease-5 (Mcpt5) promoter and the Il10 gene flanked with loxP-sites (Dudeck, 2011). In these mice only MCs are deficient in IL-10 (Mcpt-Cre Il10Fl). In Mcpt-Cre Il10Fl mice, we observed similar phenotype to Il10-/- mice: significantly greater numbers of activated mature DCs draining to the RLNs during infection (p <0.01) (Figure 6J). Without the effects of MC IL-10, UPEC-specific antibodies were detectable in the sera of cystitis-infected Mcpt-Cre Il10fl mice (Figure 6K). It should also be noted that the levels of Il10 expression in the bladder 24h p.i. were comparable between Mcpt-Cre Il10Fl mice, Wsh mice, and Wsh mice reconstituted with Il10-/- BMMC (Figure 6H). These data point to MC-derived IL-10 as a major factor constricting DC activation upon infection, specifically within the ILN but not RLN. These studies show that the adaptive immune response initiated from the DLN of the bladder is lower than that of the kidneys and, furthermore, suggest that the muted initiation of adaptive immunity observed in cystitis, but not pyelonephritis, is due to the immunosuppressive effects of predominantly MC-derived IL-10 in the bladder.
MC-derived IL10 Promotes Bladder Bacterial Persistence
In spite of the role we observe for IL-10 in permitting long-term bacterial persistence (Figure 4H) and the critical role MCs play in contributing to IL-10 production in the bladder during infection (Figure 6B-C), these cells are not dispensable during the acute phase of infection. Indeed, the first and primary response of MCs is pro-inflammatory and, perhaps, it is as a result of this lack of pro-inflammatory response that Wsh mice have a greater bacterial burden in the bladder compared to WT mice 6 weeks p.i. (Figure S6A) despite that initial intracellular bacterial burden is similar only 4h p.i. (Figure S6B). Wsh mice also exhibited lower antibody titers upon pyelonephritis infection (Figure S6C) and these defects in adaptive responses likely underlie the long-term elevated bacterial burden in Wsh mice.
To investigate the contributions of the IL-10-mediated immunosuppression by MCs to UPEC clearance in the bladder, while retaining the immunesurveillance capacity of MCs within the bladder and their contributions to promoting adaptive immune responses, we reconstituted mice with Il10-/- BMMCs. This experiment revealed that the elevated level of MC-derived IL-10 in the bladders of WT mice during infection promotes the persistence of bacteria, since these Il10-/- BMMC reconstituted mice had a greatly reduced intracellular bacterial reservoir, compared to both Wsh, MC-sufficient WT, and WT BMMC-reconstituted mice (Figure 7A). It is of note that the same phenotype was observed in Mcpt5-Cre Il10Fl mice and Il10-/- mice, while WT bladders retained greater numbers of intracellular bacteria (Figure 7B). Interestingly, we also observed that the numbers of MCs within bladders were augmented by 24h in infected mice by ~30%, compared to controls (Figure 7C). This augmented MC population persists in the bladder, since the MC numbers at 6 weeks p.i. were still significantly increased (p <0.05) (Figure 7D). Therefore, it appears that MCs in the bladder serve a unique function of maintaining IL-10 levels in the resting state and promoting IL-10 production after the initial pro-inflammatory response by MCs to cystitis, thus resulting in a localized immunosuppressive environment in the bladder. This inhibitory response may be further augmented during infection through the recruitment of additional MCs into the bladder, which persists alongside the hidden intracellular bacteria population.
Figure 7. see also Figure S6. MC-derived IL-10 promotes bacterial persistence in bladder.

(A) Wsh mice reconstituted with Il10-/- BMMCs had significantly lower persistent bacteria in bladders compared to both WT mice and Wsh mice reconstituted with WT BMMCs. Bladders were harvested 2 weeks p.i. (1×108 UPEC) for quantification of bacterial persistence by CFU. (B) This phenotype was also observed in Il10-/- and Mcpt5-Cre Il10Fl mice. The bladders of WT mice retained significantly greater numbers of intracellular bacteria 6 weeks p.i. (C) Increased numbers of MCs are present in the bladder at 24h, as determined by quantification from tissue sections. (D) This increase of MCs persisted in the bladder 6 weeks p.i. *p<0.05, **p≤0.01; n=3. Error bars represent ±SEM.
Discussion
The bladder’s primary task is to store excreted products in the form of urine. However, like many other mucosal surfaces, its virtually inevitable exposure to external microbial pathogens (which make their way up the urethra to colonize urine) warrants efficient tissue-specialized defenses to maintain sterility. The initial barriers to pathogens within the bladder are physical, such as the highly impermeable urothelium and the flushing actions of urine flow. Should pathogens, such as UPEC, breach this layer and initiate infection, the bladder is also equipped to respond with a vigorous pro-inflammatory primary immune response including the production of cytokines and a substantial neutrophil influx. However, during bladder infection, we and others (Malaviya, 1999; Mulvey, 2001; Mysorekar, 2006) observe persistence of infection and notable defects in the ability of the host to mount an adaptive immune response (Percival, 1964; Ratner, 1981; Rene, 1982; Sanford, 1978; Winberg, 1963). These unexpected limitations are characteristic of a broadly tolerogenic transcriptional program (including the upregulation of TGF-β and SOCS3) in the bladder upon infection and dependent on the specific upregulation within the bladder of the anti-inflammatory cytokine, IL-10. This upregulation is not seen in infected kidneys, leading us to conclude that the tempering of adaptive immune responses and associated increase of Il10 expression is a bladder-specific phenomenon to promote a tolerogenic microenvironment. Using mice deficient in IL-10, we examined various facets of the adaptive immune response to cystitis alone or pyelonephritis and observed that IL-10 competency significantly decreases DC activation and numbers of CD4 T cells and germinal center B cells in DLNs. Since Il10-/- animals are also capable of producing comparable humoral responses after cystitis to mice with pyelonephritis, we expect that the uniquely elevated production of IL-10 by infected bladders but not kidneys explains the divergent outcomes that we and others (Percival, 1964; Ratner, 1981; Rene, 1982; Sanford, 1978) observe with regards to adaptive immunity during cystitis versus pyelonephritis.
Subsequent investigations revealed that MCs, which produce IL-10 in infected bladders, are a major driving force behind this immunosuppressive response in the bladder site, since infected MC-deficient bladders have reduced levels of Il10 compared to controls. Grimbaldeston et al. have reported that that MC-derived IL-10 suppresses inflammatory responses in a cutaneous sterile inflammation model (Grimbaldeston, 2007); however, recently, Dudeck et al. using a novel MC-deficient model, found that IL-10 from MCs is not relevant for suppression of contact hypersensitivity (CHS) and attributed the increased inflammation in Wsh mice to the kit mutation (Dudeck, 2011). It should be noted that the cutaneous sterile inflammation protocols employed in these two models differ significantly and it has been demonstrated that the sensitization dose can be critical for MC-dependent outcomes during CHS (Norman, 2008). In view of the discordance between these reports we sought to verify findings obtained here using the Wsh mouse/reconstitution model by employing a c-kit-independent model where only MCs were Il10-deficient. Using consistent methods, these two models produced consistent results, supporting a role of MC-derived IL-10 in the tolerogenic microenvironment of the bladder. These findings also indicate that with regards to bladder infections, the role of MCs remained the same regardless of whether c-kit-independent or -dependent MC-targeting mouse models are used.
We have previously shown that MCs play a vital role in orchestrating the immune response during bacterial infection (Malaviya, 1996; McLachlan, 2003; Shelburne, 2009). Here, the observation that MCs also express substantial amounts of IL-10 during bladder infection suggests that MCs in the bladder play a dual role: first, promoting inflammation to infection and, later, limiting it. In contrast to the bladder, Il10 levels are increased at basal levels in the kidneys of mice lacking MCs. Therefore, just as the bladder and the kidney have different responses to infection, the MCs in these two tissues appear to have differing regulatory roles in tuning the inflammatory profile of the tissues in the resting state. This could be due to tissue-specific heterogeneity in MC function (Bienenstock, 1985; Kitamura, 1989; Rao, 2008), a topic that requires further investigation.
One can speculate that MC-driven production of IL-10 in the bladder may relate to its unique storage function. Urine contains many intact host proteins filtered by the kidneys (Adachi, 2006; Barratt, 2007) to which immunological tolerance must be maintained. It is interesting to note that the delayed MC mediated Il10 expression coincides with the timeframe when the bladder sheds its superficial epithelium in the early hours of infection in an attempt to reduce bacterial burden (Mulvey, 2000). The induction of proinflammatory cytokines in the bladder is much higher than those seen in the kidneys, and the contraction of the inflammatory response within the bladder may be for the purposes of tempering harmful immune responses to the many antigens in urine when the protective epithelial coating is lost or to facilitate regeneration of the superficial epithelium and restore the bladder’s critical barrier function. It is notable that IL-10 has been associated with wound repair in multiple contexts (Carrington, 2006; Liechty, 2000; Sawa, 1997). Abrogation of adaptive immune responses in the bladder might not be detrimental to a host that is capable of clearing infection the vast majority of the time with its vigorous innate immune response. In this case, the risk of breaking tolerance to an associated self- or modified self-antigen could be more detrimental than the risk of not fully eliminating a largely contained infection. In this way, the bladder, like the gut, may have highly organ-specialized strategies for balancing the need for tolerance to its contents and the imperative of resisting microbial exploitation.
The host’s muted adaptive immune response to cystitis could also explain why the bladder is unable to fully eradicate the persistent population of bacteria that can be detected for weeks after infection. Remarkably, bacterial persistence occurs in parallel to elevated MC numbers in the bladder, implying a role for these recruited MCs in promoting bacterial persistence. Residual intracellular E. coli could contribute to the clinical phenomenon of frequent cystitis recurrences, since ~1/3 of recurrent cystitis is caused by the original infecting strain (Ikäheimo, 1996). Many studies have revealed bacteria-associated determinants of pathogenesis can influence infection (Connell, 1996; Johnson, 1991; Nielubowicz, 2010), but this propensity towards recurrence could also be attributable to the failure of the host to form protective immunological memory after primary cystitis, which we observe in our murine model. Although some groups have observed that bladder infection can generate productive primary adaptive immune responses (Thumbikat, 2006), and others have emphasized the importance of bacteria-associated factors to disease progression (Connell, 1996; Johnson, 1991; Nielubowicz, 2010), our work may explain why bladder-confined infections are less frequently cleared than those that progress to pyelonephritis. However, the results from this study also highlight the potential of cystitis to elicit an effective secondary immune response once immunological memory has been established, since animals that have previously experienced pyelonephritis have significantly increased UPEC-specific antibody responses during cystitis compared to those having had cystitis previously. Therefore, while emphasizing the unique microenvironment of the bladder as tailored to suppress primary adaptive responses, our findings inform us that UTI treatment and vaccination strategies are plausible since, particularly when immunological memory has already been formed, adaptive immune responses can be elicited by bladder infection.
Materials and Methods
Mice and UTI Infections
6-8 week old C57BL/6 female mice obtained from NCI were used for most animal studies. MC-deficient (Wsh), Il10-deficient (Il10-/-) mice, and Mcpt-5Cre Il10(Fl/Fl) mice, all on C57BL/6 backgrounds, were bred at the Duke University Medical Center animal care facility. Wsh mice were reconstituted by i.v. injection of 1×107 BMMCs and allowed 15 weeks for MC maturation in tissues. All animal experiments were approved by the Duke University IACUC and Division of Laboratory Animal Resources. Mice were catheterized for inoculation of 108 E,coli transurethrally using clinical isolate UPEC strains J96 (Normark, 1983) or CI5 (Abraham, 1998; Mulvey, 2001), under pentobarbital anesthesia. To exclusively initiate cystitis, the bladder was inoculated with 30μl at a slow rate (2.5μl/sec). For combined bladder and kidney infection, 50μl was given at a fast rate (10μl/sec).
Flow Cytometry from LN, Bladder and Kidney suspensions
To quantitate DCs, single cell suspensions of lymphoid tissues were stained with the following antibodies: α-MHCII-FITC (eBioscience), α-CD11c-PE-Cy5.5 (eBioscience), and for detection of activation, α-CD86-PE (eBioscience). To stain germinal center B cells and T-helper cells, we used α-CD19-PE (eBioscience) and α-GL7-FITC (BD Pharmingen) or α-CD4-PE-Cy5.5 (eBioscience). For intracellular staining of Foxp3 (eBioscience) and IL-10 (eBioscience), cells were permeabilized with 0.1% saponin. Cells were fixed in 4% paraformaldhyde prior flow cytometry using a FACscaliber (BD Biosciences) and analysis by CellQuest software.
Statistical analysis
We determined significance by unpaired two-tailed Student’s t-test to assess data with two groups, or by one or two-way analysis of variance and Tukey’s multiple comparison or Bonferroni posttests, as appropriate, for all other experiments. All graphs and analyses were done with Prism (Graphpad) software.
Supplementary Material
Acknowledgments
We thank P. Seed, H. Staats, M. Kuehn, and L. Hale for discussions and J. Wright for the use of equipment. E. coli J96 was a gift from S. Normark (Umea University). Special thanks to A. Roers (University of Technology, Dresden) for Mcpt5-Cre(+/+) Il10(Fl/Fl) mice and T. Haering for assisting with animal import. We also thank Zachary Swan for acquiring the image in Figure 1B and W.X.G. Ang for critical manuscript reading. This work was supported by US-NIH grants R01 A135678, R01 DK077159, R01 A150021, R37 DK50814 and R21 A1056101.
Footnotes
Supplemental Methods Accompany this Manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham SN, Arock M. Mast cells and basophils in innate immunity. Seminars in immunology. 1998;10:373–381. doi: 10.1006/smim.1998.0140. [DOI] [PubMed] [Google Scholar]
- Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome biology. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt J, Topham P. Urine proteomics: the present and future of measuring urinary protein components in disease. CMAJ. 2007;177:361–368. doi: 10.1503/cmaj.061590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Chen W. Regulatory ripples. Nature immunology. 2010;11:1077–1078. doi: 10.1038/ni1210-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienenstock J, Befus D, Denburg J, Goto T, Lee T, Otsuka H, Shanahan F. Comparative aspects of mast cell heterogeneity in different species and sites. International archives of allergy and applied immunology. 1985;77:126–129. doi: 10.1159/000233766. [DOI] [PubMed] [Google Scholar]
- Carrington LM, Albon J, Anderson I, Kamma C, Boulton M. Differential regulation of key states in early corneal wound healing by TGF-beta isoforms and their inhibitors. Invest Ophthalmol Vis Sci. 2006;47:1886–1894. doi: 10.1167/iovs.05-0635. [DOI] [PubMed] [Google Scholar]
- Chan CY, St John AL, Abraham SN. Plasticity in mast cell responses during bacterial infections. Current opinion in microbiology. 2012;15:78–84. doi: 10.1016/j.mib.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell I, Agace W, Klemm P, Schembri M, Marild S, Svanborg C. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couper KN, Blount DG, Riley EM. IL10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Köhler A, Peschke K, Vöhringer D, Waskow C, Krieg T, Müller W, Waisman A, Hartmann K, Gunzer M, Roers A. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 2011;34:973–984. doi: 10.1016/j.immuni.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Enk AH, Angeloni VL, Udey MC, Katz SI. Inhibition of Langerhans cell antigen-presenting function by IL10. A role for IL10 in induction of tolerance. J Immunol. 1993;151:2390–2398. [PubMed] [Google Scholar]
- Fihn SD. Clinical practice. Acute uncomplicated urinary tract infection in women. N Engl J Med. 2003;349:259–266. doi: 10.1056/NEJMcp030027. [DOI] [PubMed] [Google Scholar]
- Foxman B. Recurring urinary tract infection: incidence and risk factors. Am J Public Health. 1990;80:331–333. doi: 10.2105/ajph.80.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113(Suppl 1A):5S–13S. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunological reviews. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- Hagberg L, Engberg I, Freter R, Lam J, Olling S, Svanborg Eden C. Ascending, unobstructed urinary tract infection in mice caused by pyelonephritogenic Escherichia coli of human origin. Infection and immunity. 1983;40:273–283. doi: 10.1128/iai.40.1.273-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg L, Jodal U, Korhonen TK, Lidin-Janson G, Lindberg U, Svanborg Eden C. Adhesion, hemagglutination, and virulence of Escherichia coli causing urinary tract infections. Infect Immun. 1981;31:564–570. doi: 10.1128/iai.31.2.564-570.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraoka M, H L, Frendéus B, Godaly G, Burdick M, Strieter R, Svanborg C. Neutrophil recruitment and resistance to urinary tract infection. J Infect Dis. 1999;180:1220–1229. doi: 10.1086/315006. [DOI] [PubMed] [Google Scholar]
- Hooton TM, Scholes D, Hughes JP, Winter C, Roberts PL, Stapleton AE, Stergachis A, Stamm WE. A prospective study of risk factors for symptomatic urinary tract infection in young women. N Engl J Med. 1996;335:468–474. doi: 10.1056/NEJM199608153350703. [DOI] [PubMed] [Google Scholar]
- Ikäheimo R, Siitonen A, Heiskanen T, Kärkkäinen U, Kuosmanen P, Lipponen P, Mäkelä PH. Recurrence of urinary tract infection in a primary care setting: analysis of a 1-year follow-up of 179 women. Clin Infect Dis. 1996;22:91–99. doi: 10.1093/clinids/22.1.91. [DOI] [PubMed] [Google Scholar]
- Johnson JR. Virulence factors in Escherichia coli urinary tract infection. Clinical microbiology reviews. 1991;4:80–128. doi: 10.1128/cmr.4.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol. 1989;7 doi: 10.1146/annurev.iy.07.040189.000423. [DOI] [PubMed] [Google Scholar]
- Knolle PA, Gerken G. Local control of the immune response in the liver. Immunological reviews. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Miura T, Haba T, Sato M, Serizawa I, Nagai H, Ishizaka K. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J Immunol. 2000;164:3855–3861. doi: 10.4049/jimmunol.164.7.3855. [DOI] [PubMed] [Google Scholar]
- Liechty KW, Kim HB, Adzick NS, Crombleholme TM. Fetal wound repair results in scar formation in interleukin-10-deficient mice in a syngeneic murine model of scarless fetal wound repair. J Pediatr Surg. 2000;35:866–873. doi: 10.1053/jpsu.2000.6868. [DOI] [PubMed] [Google Scholar]
- Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, Strom TB, Zheng XX, Noelle RJ. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- Malaviya R, Gao Z, Thankavel K, van der Merwe PA, Abraham SN. The mast cell tumor necrosis factor alpha response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8110–8115. doi: 10.1073/pnas.96.14.8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Ikeda T, Abraham SN, Malaviya R. Contribution of mast cells to bacterial clearance and their proliferation during experimental cystitis induced by type 1 fimbriated E. coli Immunol Lett. 2004;91:103–111. doi: 10.1016/j.imlet.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- Malisan F, Briere F, Bridon JM, Harindranath N, Mills FC, Max EE, Banchereau J, Martinez-Valdez H. Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. The Journal of experimental medicine. 1996;183:937–947. doi: 10.1084/jem.183.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, Abraham SN. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol. 2003;4:1199–1205. doi: 10.1038/ni1005. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nature reviews Immunology. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infection and immunity. 2001;69:4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci. 2000;97:8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14170–14175. doi: 10.1073/pnas.0602136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielubowicz GR, Mobley HL. Host-pathogen interactions in urinary tract infection. Nat Rev Urol. 2010;7:430–441. doi: 10.1038/nrurol.2010.101. [DOI] [PubMed] [Google Scholar]
- Norman MU, Hwang J, Hulliger S, Bonder CS, Yamanouchi J, Santamaria P, Kubes P. Mast cells regulate the magnitude and the cytokine microenvironment of the contact hypersensitivity response. Am J Pathol. 2008;172:1638–1649. doi: 10.2353/ajpath.2008.070559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark S, Lark D, Hull R, Norgren M, Båga M, O’Hanley P, Schoolnik G, Falkow S. Genetics of digalactoside-binding adhesin from a uropathogenic Escherichia coli strain. Infect Immun. 1983;41:942–949. doi: 10.1128/iai.41.3.942-949.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival A, Birumfitt W, Delouvois J. Serum-antibody leves as an indication of clinically inapparent pyelonephritis. Lancet. 1964;2:1027–1033. doi: 10.1016/s0140-6736(64)90988-2. [DOI] [PubMed] [Google Scholar]
- Prudencio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nature reviews Microbiology. 2006;4:849–856. doi: 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci. 2008;1143:83–104. doi: 10.1196/annals.1443.023. [DOI] [PubMed] [Google Scholar]
- Ratner JJ, Thomas VL, Sanford BA, Forland M. Bacteria-specific antibody in the urine of patients with acute pyelonephritis and cystitis. J Infect Dis. 1981;143:404–412. doi: 10.1093/infdis/143.3.404. [DOI] [PubMed] [Google Scholar]
- Rene P, Silverblatt FJ. Serological response to Escherichia coli pili in pyelonephritis. Infect Immun. 1982;37:749–754. doi: 10.1128/iai.37.2.749-754.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford BA, Thomas VL, Forland M, Carson S, Shelokov A. Immune response in urinary tract infection determined by radioimmunoassay and immunofluorescence: serum antibody levels against infecting bacterium and Enterobacteriaceae common antigen. J Clin Microbiol. 1978;8:757–579. doi: 10.1128/jcm.8.5.575-579.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa T, Corry DB, Gropper MA, Ohara M, Kurahashi K, Wiener-Kronish JP. IL10 improves lung injury and survival in Pseudomonas aeruginosa pneumonia. J Immunol. 1997;159:2858–2866. [PubMed] [Google Scholar]
- Shelburne CP, Nakano H, St John AL, Chan C, McLachlan JB, Gunn MD, Staats HF, Abraham SN. Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe. 2009;6:331–342. doi: 10.1016/j.chom.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Duncan MJ, Li G, Chan C, Grady R, Stapleton A, Abraham SN. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Pathog. 2007;3:541–552. doi: 10.1371/journal.ppat.0030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm WE. Measurement of pyuria and its relation to bacteriuria. Am J Med. 1983;75:53–58. doi: 10.1016/0002-9343(83)90073-6. [DOI] [PubMed] [Google Scholar]
- Steinbrink K, Jonuleit H, Müller G, Schuler G, Knop J, Enk AH. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood. 1999;93:1634–1642. [PubMed] [Google Scholar]
- Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL10-treated dendritic cells. J Immunol. 1997;159:4772–4780. [PubMed] [Google Scholar]
- Thumbikat P, Waltenbaugh C, Schaeffer AJ, Klumpp DJ. Antigen-specific responses accelerate bacterial clearance in the bladder. J Immunol. 2006;176:3080–3086. doi: 10.4049/jimmunol.176.5.3080. [DOI] [PubMed] [Google Scholar]
- Vanderford DA, Greer PK, Sharp JM, Chichlowski M, Rouse DC, Selim MA, Hale LP. Alopecia in IL10-deficient mouse pups is c-kit-dependent and can be triggered by iron deficiency. Experimental dermatology. 2010;19:518–526. doi: 10.1111/j.1600-0625.2009.01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann H. Immunology: protection and privilege. Nature. 2006;442:987–988. doi: 10.1038/nature05165. [DOI] [PubMed] [Google Scholar]
- Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg J, Andersen HJ, Hanson LA, Lincoln K. Studies of urinary tract infections in infancy and childhood. I. Antibody response in different types of urinary tract infections caused by coliform bacteria. Br Med J. 1963;2:524–527. doi: 10.1136/bmj.2.5356.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.