

Abstract
Mitochondria are recognized as critical sites of localized injury in a number of chronic pathologies which has led to the development of organelle directed therapeutics. One of the approaches employed to target molecules to the mitochondrion is to conjugate a delocalized cation such as triphenylphosphonium (TPP+) to various redox active compounds. Mitochondrially targeted antioxidants have also been used in numerous cell culture based studies as probes of the contribution of the mitochondrial generation of reactive oxygen species on cell signaling events. However, concentrations used in vitro are typically 10–100 times greater than those generated from oral dosing in a wide range of animal models and in humans. In the present study, we determined the effects of mitochondrial targeted antioxidants, MitoQ, MitoTempol, and MitoE on cellular bioenergetics of mesangial cells in culture and compared these to TPP+ conjugated compounds which lack the antioxidant functional group. We found that all TPP+ compounds inhibited oxidative phosphorylation to different extents independent of the antioxidant functional groups. These findings show that the TPP+ moiety can disrupt mitochondrial function at concentrations frequently observed in cell culture and this behavior is dependent on the linker group and independent of antioxidant properties. Moreover, the TPP+ moiety alone is unlikely to achieve the concentrations needed to contribute to the protective mechanisms of the mitochondrially targeted compounds that have been reported in vivo.
Abbreviations: AA, Antimycin A; BTPP, butyl triphenylphosphonium; DTPP, decyl triphenylphosphonium; ECAR, extracellular acidification rate; FCCP, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone; MES-13, murine mesangial cells; MitoE, Mito Vitamin E; MitoQ, Mitoquinone; OCR, oxygen consumption rate; Oligo, Oligomycin; RNS, reactive nitrogen species; ROS, reactive oxygen species; TPP+, triphenylphosphonium; TPMP, triphenylmethylphosphonium
Keywords: Mitochondrial targeted compounds, Mitochondria, Respiration, Extracellular flux, MitoQ, TPP+ derivatives, Redox
Graphical abstract
Highlights
► Mitochondrial targeted antioxidants are conjugated to a triphenylphosphonium cation (TPP+) moiety that allows the compound to accumulate within the mitochondria. ► The effect of MitoQ, MitoTempol, and MitoE and their TPP+ carrier groups on oxidative phosphorylation was examined. ► Higher concentrations of TPP+ conjugated compounds alter cellular bioenergetics independent of the functional antioxidant group. ► Many of the effects ascribed to mitochondrial oxidant scavenging by these compounds in cell culture may be nonspecific effects of the carrier molecule.
Introduction
It is clear that the pathological mechanisms of several diseases are associated with increased mitochondrial oxidative stress and consequently bioenergetic dysfunction. This is expected since mitochondria are both producers and targets of reactive oxygen and nitrogen species (ROS/RNS). In many cases, mitochondrial dysfunction occurs due to oxidative modification of the respiratory chain complexes which then amplifies and promotes further oxidative damage. The precise mechanisms are still under active investigation but it appears that the mitochondrion is particularly vulnerable to reactive lipid species which are formed by lipid peroxidation [1]. Reactive lipid species appear to target the mitochondria by inducing the formation of superoxide and hydrogen peroxide which can ultimately result in bioenergetic dysfunction and mitophagy [2–4]. In particular, mitophagy is essential for maintaining mitochondrial quality control and cell survival and is largely dependent on mtDNA integrity, oxidative phosphorylation, and autophagy [5]. These pathways can be modified by ROS/RNS and are therefore potentially amenable to treatment with mitochondrially targeted antioxidants. Several strategies have been developed to target small molecule antioxidants to the mitochondria to assess their ability to suppress oxidative stress in various disease models [6,7]. For example, Mitoquinone (MitoQ) has been tested successfully in human diseases such as Hepatitis C induced liver disease and skin photo damage as well as a number of experimental animal models including ischemia-reperfusion, neurodegenerative diseases, diabetes, and alcohol-induced hepatosteatosis [8–14]. These findings support the concept that mitochondrially targeted antioxidants may be clinically beneficial by preventing aberrant redox signaling and oxidative stress in the mitochondrion.
There are several approaches to targeting molecules to the mitochondrion and one of the most versatile is to conjugate a pharmacophore to lipophilic cations such as triphenylphosphonium (TPP+) [6]. This delocalized cation is hydrophobic and can freely pass through the phospholipid bilayers of the plasma membrane and other organelles, without the requirement for a specific uptake mechanism. This is achieved because the cells’ negative plasma membrane potential (-30mV to -60mV) drives the movement of TPP+ from the extracellular space into the cytoplasmic compartment up to 10-fold. TPP+ can further accumulate within the mitochondria up to several hundred fold due to the higher mitochondrial membrane potential (−140 to −180 mV) [15,16]. It has been determined that there is an approximately tenfold accumulation of TPP+ within the mitochondria for every ∼60 mV increase in mitochondrial membrane potential [17]. Because the extent of uptake of a TPP+ conjugated compound depends on the plasma and mitochondrial membrane potential, the volume of the incubation, the cell volume, and the number of mitochondria within a given cell, the quantity of the compound found within mitochondria can vary substantially between different cell types [18]. Further, the therapeutic molecules employing the TPP+ moiety use varying carbon chain linkers to attach functional groups, it is important to understand how the cell responds to changes in the physical properties of TPP+ mitochondria-directed compounds. Once inside the mitochondrion, TPP+ molecules are primarily positioned on the mitochondrial matrix facing surface of the phospholipid bilayer, with the linker and functional group positioned within the inner membrane [19,20]. The extent of anchoring of the TPP+ molecules to the inner mitochondrial membrane is dependent upon the hydrophobicity, the length of the linker unit, and the functional group [17]. The membrane anchoring of the TPP+ molecules accumulated within mitochondria can elicit beneficial effects such as scavenging reactive radicals, preventing membrane lipid peroxidation, controlling mitochondrial redox signaling and/or modify any pharmacological effects of the TPP+ group.
It has been shown that the physical properties of the TPP+ conjugated compounds and the linker chain can also determine the extent of accumulation of the molecule within the mitochondria. For example, triphenylmethylphosphonium (TPMP), a one carbon methyl attached to TPP+, has relatively low hydrophobicity and slow rates of accumulation into mitochondria within cells compared to TPP+ molecules which are more hydrophobic due to longer carbon linkers [17,21]. The efficacy of the targeted antioxidant group also varies depending on its linker chain length owing to the differences in their physical properties and access to the active sites of activating and recycling enzymes such as complex II. The TPP+ moiety can also in principle have pharmacological or inhibitory properties due to its interactions with mitochondria which can potentially be modified by both the linker group and the functional pharmacophores possessing antioxidant properties [19].
Properties of TPP+ conjugated compounds have been extensively studied in cell culture systems. For example, high concentrations of TPP+ were shown to contribute to intra-mitochondrial calcium regulation in HELA cells stimulated with inositol 1,4,5-trisphosphate, where both MitoQ and TPP+ treatment significantly increased mitochondrial matrix calcium concentrations [22]. It was also shown that calcium efflux from the mitochondria was delayed after exposure to these targeted compounds [22]. This study indicated that TPP+ itself inhibits the Na+ (or H+)/Ca2+ exchanger and thus increases intra-mitochondrial calcium levels. The degree of inhibition for the mitochondrial Na+(or H+)/Ca2+ exchanger was greater with TPP+ compared to MitoQ [22]. Mitochondrial targeted antioxidants have also been used as probes to examine the contribution of mitochondrial ROS to cell signaling events. In human breast cancer cells, MitoQ was shown to inhibit cell proliferation through autophagy and apoptotic cell death mechanisms [23]. Specifically, MitoQ caused phosphorylation of checkpoint kinases and disassociation of Keap1 from Nrf2, leading to increased Nrf2 transcription activity [23]. In rat cardiac myoblasts, both MitoQ and MitoTempol were determined to inhibit muscle differentiation mediated by hydrogen peroxide signaling without altering ATP levels [24]. Most recently, it has been determined that MitoQ uncouples mitochondria when added to bovine aortic endothelial cells at concentrations of 150 nM and higher in a dose dependent fashion and simultaneously induces glycolysis [25]. In the studies mentioned above, the concentrations of all of the mitochondrial targeted antioxidants were higher than those that can be achieved pharmacologically and associated with protective effects in vivo [26].
Thus, it is increasingly clear that modifications to the linker, the mitochondrial targeting group, and the functional group are important in determining the biological properties of mitochondrially targeted compounds. Therefore, it is important to further assess these properties by carrying out a systematic investigation of the effects of TPP+ conjugated compounds over a concentration range which encompasses the low nM levels which are found pharmacologically. Here, we investigate the cellular bioenergetic response, specifically the oxygen consumption (OCR) and pH (ECAR) rates of MES-13 cells to selected pharmacophores attached to TPP+ using a Seahorse Biosciences XF-24 flux analyzer. Although effects on cellular bioenergetics are consistently observed with TPP+ conjugated compounds these typically occur at concentrations at least one order of magnitude or greater than the levels associated with beneficial effects in disease model and are unlikely to contribute to the mechanism of action in vivo. They also suggest that the use of mitochondrially targeted antioxidants in cell culture systems to test for the role of mitochondrially reactive oxygen species should be controlled for the non-specific effects of the TPP+ and linker groups.
Materials and methods
Reagents
Oligomycin, FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone), Antimycin A, TPMP (triphenylmethylphosphonium), BTPP (butyl triphenylphosphonium), and DTPP (decyl triphenylphosphonium) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). MitoQ (Mitoquinone), MitoTempol, and MitoE were provided by Drs. Michael P. Murphy and Balaraman Kalyanaraman.
Cell culture
Mouse kidney mesangial cells (MES-13) were purchased from ATCC (Manassas, VA) and maintained in a humidified incubator gassed with 5% CO2 at 37 °C. Cells were grown in Dulbecco's Modified Eagles Medium (DMEM, Cellgro) supplemented with D-Glucose (5.5 mM, Sigma-Aldrich), HAM's F-12 1X (Cellgro), L-Glutamine (4mM, Invitrogen), penicillin and streptomycin (100U/mL and 100 ng/mL, respectively, Invitrogen) and 2.5% fetal bovine serum (Atlanta Biologicals).
TPP derivatives
TPMP, BTPP, and DTPP compounds contain side chains of varying lengths (methyl group, butyl group, and decyl group respectively) without antioxidant groups (Fig. 1A–C). MitoQ, MitoTempol, and MitoE are TPP derivatives that contain functional antioxidant groups (Fig. 1D–F). MitoQ was synthesized by the covalent addition of quinol to a lipophilic TPP+ moiety through an aliphatic carbon chain [27]. MitoTempol is comprised of the piperidine nitroxide TEMPOL conjugated to an aliphatic carbon chain terminating with a TPP+ moiety [28]. MitoE was generated by conjugating Vitamin E (α-tocopherol) to a lipophilic TPP+ cation [29].
Fig. 1.
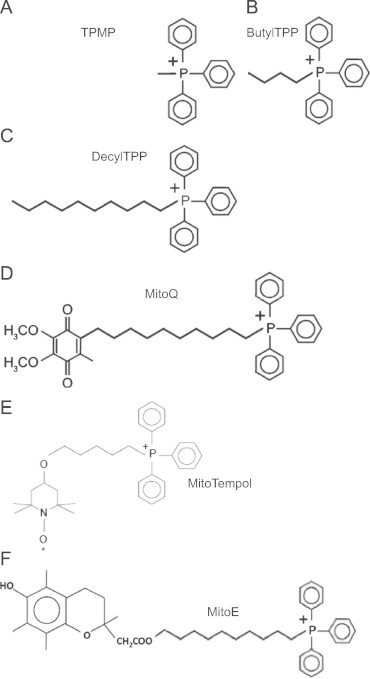
Chemical structures of TPP+ derivatives. (A–C) TPMP, BTPP, and DTPP are control TPP+ derivatives used to distinguish the effects of the antioxidant functional group versus the targeting moiety and the alkyl linker chain. (D–F) MitoQ, MitoTempol, and MitoE are TPP+ derivatives comprised of functional antioxidant groups.
Measurement of mitochondrial function using the Seahorse XF-24 extracellular flux analyzer
To test the effects of TPP derivatives on cellular bioenergetics in MES-13 cells, the Seahorse XF-24 analyzer from Seahorse Biosciences (North Billerica, MA) was utilized. MES-13 cells were seeded 30,000 cells per well in Seahorse XF-24 specialized cell culture plates. Approximately 24 h later, media was replaced with unbuffered DMEM for 1 h at 37 °C prior to assessing basal oxygen consumption rate (OCR) or extracellular acidification rate (ECAR) simultaneously in the Seahorse XF-24 extracellular flux analyzer. Thirty min later, all of the TPP+ derivatives were injected individually to MES-13 cells (0, 0.01, 0.1, or 1 μM). The effect of the TPP+ derivatives on bioenergetics was measured for two hours followed by evaluation of the metabolic profile using the mitochondrial function assay [30] (Fig. 2). Basal OCR and ECAR was measured over time followed by observing the effects of the mitochondrial inhibitors, oligomycin (1 μg/mL), FCCP (2 μM), and antimycin A (10 μM). The cellular bioenergetic parameters determined were ATP linked respiration, proton leak, maximal, and reserve capacity. ATP linked respiration was derived from the difference between OCR at baseline and respiration following oligomycin addition. The difference in OCR between antimycin A and oligomycin represented the amount of oxygen consumed that is due to proton leak. Maximal OCR was determined by subtracting the OCR after antimycin A addition from the OCR induced by FCCP. Lastly, the reserve capacity was calculated by the difference between maximal (FCCP) and basal respiration. All data is expressed as a percentage of the basal OCR or ECAR before TPP+ derivative injection to allow comparison between the different compounds.
Fig. 2.
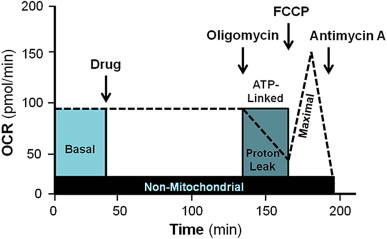
Schematic of bioenergetic profile used to evaluate the acute effects of mitochondrial targeted compounds on mitochondrial function.
Statistical analysis
All results are expressed as means±SEM (n=3–5 per group). An unpaired Student's t test was used to compare the control and treated groups. ANOVA and the Newman–Keuls test were used to compare the mean values for multiple group comparisons. All values were considered significant at P<0.05.
Results and discussion
The effects of lengthening the alkyl chain of TPP+ derivatives on cellular bioenergetics
TPMP is the methylated form of TPP+ and is the simplest derivative used in this study (Fig. 1A). It is a commonly used control for more complex derivatives containing an antioxidant or other functional groups to rule out a contribution of the TPP+ moiety to an observed biological effect. Fig. 3A shows the effects of TPMP (0.1 μM and 1 μM) on cellular bioenergetics. Cells were allowed to establish a stable baseline and then TPMP was injected into the cell media as shown. The lower concentration of TPMP (0.1 μM) had no significant affect on basal OCR whereas 1 μM TPMP progressively decreased OCR over a 2 h period. At this point a mitochondrial function assay was performed using mitochondrial inhibitors as described previously [30]. Oligomycin caused a substantial decrease in OCR in control cells which was significantly suppressed by TPMP. To determine the maximal respiratory capacity, FCCP was injected into the media and caused an increase in OCR measured relative to the point prior to the addition of oligomycin. Interestingly, the stimulation of respiration with FCCP after oligomycin was substantially greater in the presence of 1 μM TPMP. Lastly, injection of antimycin A significantly inhibited respiration in all groups with a significantly greater effect on the TPMP (1 μM) treated cells. The ECAR was measured as an index of glycolysis under the same conditions as shown in Fig. 4. Immediately upon addition of TPMP, ECAR increased such that the maximal level was reached approximately 1 h after its addition (Fig. 4A). In the control cells, the maximum ECAR was elicited upon the addition of FCCP and this was not further increased in the TPMP (1 μM) treated cells.
Fig. 3.
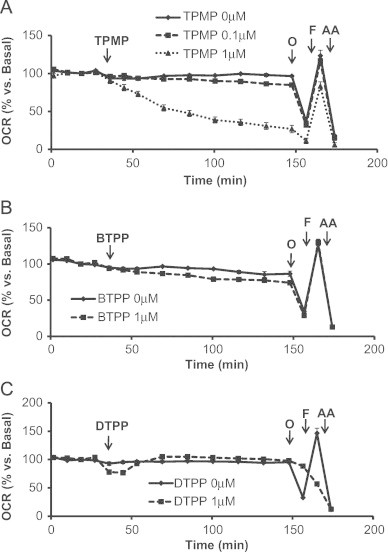
Acute effects of TPMP, BTPP, and DTPP on oxygen consumption rate (OCR). MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. Cellular media was replaced with low-buffered DMEM media prior to the assay and allowed to equilibrate for 1 h at 37 °C. Basal OCR measurements were made followed by injection of (A) TPMP (0.1 μM or 1 μM), (B) BTPP (1 μM), or (C) DTPP (1 μM). After assessing the acute effects of the TPP+ controls, oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) were injected. Data is expressed as a percent relative to the basal OCR rate before TPMP, BTPP, or DTPP injection. All data shown are mean±SEM, n=3–5 group.
Fig. 4.
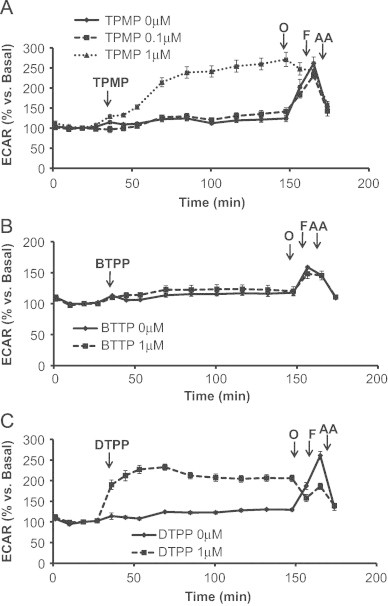
Acute effects of TPMP, BTPP, and DTPP on extracellular acidification rate (ECAR). MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. The following day basal ECAR was determined prior to injecting (A) TPMP (0.1 μM or 1 μM), (B) BTPP (1 μM), or (C) DTPP (1 μM) into the cellular medium. Subsequently, oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) was injected to further evaluate glycolysis. Data is expressed as a percent relative to the basal ECAR rate before TPMP, BTPP, or DTPP injection. All data shown are mean±SEM, n=3–5 group.
We next examined the effects of lengthening the alkyl chain on cellular bioenergetics using two additional compounds, BTPP and DTPP using the protocol established for TPMP (Fig. 3B, C). In contrast to TPMP, cells exposed to 1 μM BTPP showed no changes in any of the bioenergetic parameters measured compared to control cells (Fig. 3B). DTPP (1 μM) caused a transient decrease in basal OCR which reverted back to control levels within 20 min (Fig. 3C). However, addition of oligomycin did not elicit the anticipated decrease in OCR and further addition of FCCP resulted in an inhibition of respiration rather than a stimulatory response. Antimycin A inhibited OCR to the same extent as controls indicating that these responses are mitochondrial in origin. As shown in Fig. 4B, BTPP had no effect on the cellular basal glycolytic rate. However, DTPP induced a rapid increase in basal ECAR and this was not further stimulated upon addition of oligomycin or FCCP (Fig. 4C).
It is clear from these data that the TPP+ group has a functional impact on cellular bioenergetics which is highly sensitive to the alkyl linker groups which have previously been assumed to be largely inert. Much of this effect may be due to the increased extent of uptake of the TPP molecules when conjugated to a more hydrophobic carbon chain. The effects of TPMP are to inhibit basal respiration but have a minimal effect on the maximal respiration. Since FCCP will result in depolarization of the mitochondrial inner membrane and loss of TPMP from the mitochondrial matrix, it suggests the effects of TPMP on oxidative phosphorylation are reversible. The rapid increase in ECAR by the cells exposed to TPMP indicates activation of glycolysis to compensate for loss of mitochondrial ATP production. In contrast, DTPP behaves quite differently in that oligomycin sensitivity is now completely diminished which is consistent with a rapid cycling of DTPP between the mitochondrial matrix and cytosol thereby decreasing the control of respiration by the proton electrochemical potential gradient, effectively uncoupling mitochondria.
The effects of adding an antioxidant functional group to alkyl TPP+ chain on cellular bioenergetics
To assess the impact of coupling a functional group to the alkyl TPP+ moiety, we investigated the effects of MitoQ, MitoTempol, and MitoE (Fig. 1D–F). The bioenergetic response of cells to 1 μM MitoQ is shown in Fig. 5A. After the cells established a stable baseline, MitoQ was injected into the cellular media and caused a steady decline in oxygen consumption over 2 h. Surprisingly, oligomycin did not elicit a pronounced decrease in cellular respiration as expected nor did FCCP stimulate respiration. However, antimycin A inhibited respiration to levels similar to control cells. The effects of MitoQ on glycolysis (Fig. 6A) were determined based on the parameters measured in Fig. 5A. MitoQ caused an initial transient increase in ECAR that returned back to basal levels within 2 h (Fig. 6A). Glycolysis increased minimally following oligomycin injection and was not further stimulated with FCCP or antimycin A treatment (Fig. 6A) suggesting that MitoQ is disrupting the ability of the respiratory chain to transfer electrons to oxygen.
Fig. 5.
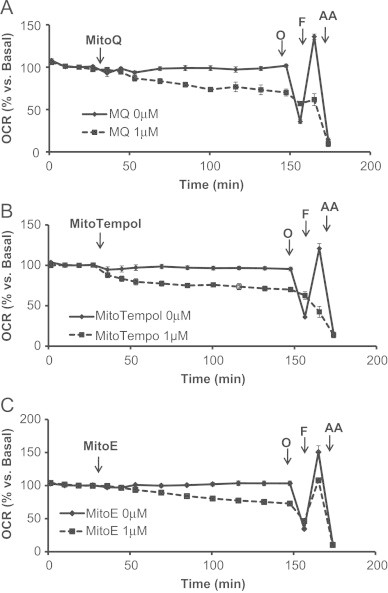
Acute effects of MitoQ, MitoTempol, and MitoE on oxygen consumption rate (OCR). MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. Cellular media was replaced with low-buffered DMEM media prior to the assay and allowed to equilibrate for 1 h at 37 °C. Basal OCR measurements were made for approximately 30 min followed by injection of (A) MitoQ (1 μM), (B) MitoTempol (1 μM), or (C) MitoE (1 μM). Two hours later, the effects of oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) treatment were determined. Data is expressed as a percent relative to the basal OCR rate before MitoQ, MitoTempol, or MitoE injection. All data shown are mean±SEM, n=3–5 group.
Fig. 6.
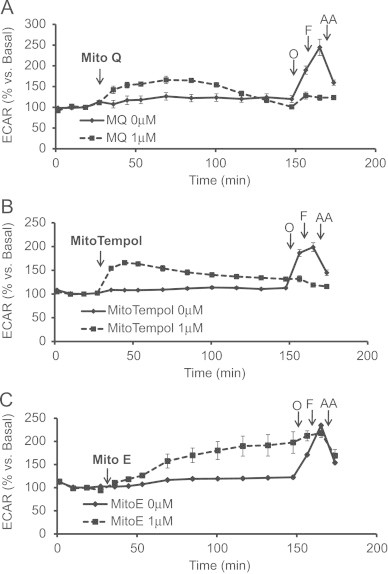
Acute effects of MitoQ, MitoTempol, and MitoE on extracellular acidification rate (ECAR). Basal ECAR was measured before evaluating the effects of acute (A) MitoQ (1 μM), (B) MitoTempol (1 μM), or (C) MitoE (1 μM) treatment. After a two hour period, oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) was injected. Data is expressed as a percent relative to the basal OCR rate before MitoQ, MitoTempol, or MitoE injection. All data shown are mean±SEM, n=3–5 group.
The effects of MitoTempol (1 μM) on cellular oxygen consumption are shown in Fig. 5B. Following basal OCR readings, MitoTempol was added to the cells and caused a progressive decline in mitochondrial respiration, which was later shown to be exacerbated with oligomycin and FCCP treatment. Mitochondrial respiration was further inhibited to control levels with antimycin A injection. Interestingly, the respiratory response to the mitochondrial assay in MitoTempol treated cells was analogous to the response mediated by DTPP (Fig. 3B). These findings suggest that MitoTempol may be decreasing respiration in a similar manner as DTPP. As shown in Fig. 6B, MitoTempol caused a steady increase in ECAR for 20 min before continuously declining over a 2 h period.
Finally, MitoE was tested and was found to cause a progressive decrease in respiration following injection similar to TPMP (Figs. 5C, 3A). Consistent with inhibition of oxidative phosphorylation, MitoE caused an increase in glycolysis which was not further stimulated by oligomycin or FCCP injection (Fig. 6C). Further analysis of the effects of MitoQ, MitoTempol, and MitoE compared to DTPP was also assessed and is shown in Fig. 7. In a separate series of experiments, it was determined that non-mitochondrially targeted compounds, Coenzyme Q10, Tempol, and Vitamin E had no affect on mitochondrial respiration (Table 1). However, the effects of the mitochondrially targeted antioxidants at higher concentrations (1 μM) were significantly different than untreated controls (Table 2).
Fig. 7.
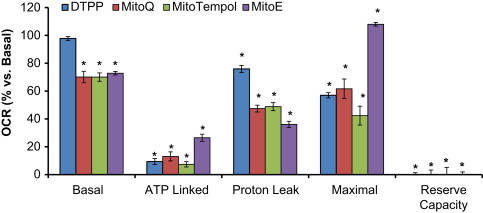
Acute effects of DTPP, MitoQ, MitoTempol, and MitoE on mitochondrial bioenergetics. MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. Cellular media was replaced with low-buffered DMEM media prior to the assay and allowed to equilibrate for 1 h at 37 °C. Basal OCR measurements were made for approximately 30 min followed by injection of DTPP (1 μM), MitoQ (1 μM), MitoTempol (1 μM), or MitoE (1 μM). Two hours later, the effects of oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) treatment were determined. Basal OCR is calculated as the difference in OCR before compound injection and after two hours of exposure. ATP linked is the difference in OCR before and after Oligomycin. Proton Leak is the difference in OCR after Oligomycin injection and Antimycin A. Maximal is the OCR after FCCP injection. Reserve capacity is the difference between basal and Maximal OCR. Data is expressed as a percent relative to the basal OCR rate before compound injection. All data shown are mean±SEM, n=3–5 group. ⁎p<0.05 vs. untreated controls, statistical comparison performed using ANOVA and Newman–Keuls.
Table 1.
Bioenergetic profiles of non-mitochondrially targeted antioxidants. MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. Cellular media was replaced with low-buffered DMEM media prior to the assay and allowed to equilibrate for 1 h at 37 °C. Basal OCR measurements were made for approximately 30 min followed by injection of Coenzyme Q10 (1 μM), Tempol (1 μM), or Vitamin E (1 μM). Two hours later, the effects of oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) treatment were determined. Basal OCR is calculated as the difference in OCR before compound injection and after two hours of exposure. ATP linked is the difference in OCR before and after Oligomycin. Proton Leak is the difference in OCR after oligomycin injection and antimycin A. Maximal is the OCR following FCCP injection. Data is expressed as a percent relative to the basal OCR rate before compound injection. All data shown are mean±SEM, n=3–5 group. *, p<0.05 vs. untreated controls, statistical comparison performed using ANOVA and Newman–Keuls.
| Bioenergetic profiles of Non-mitochondrially targeted antioxidants | ||
|---|---|---|
| Basal OCR (%) | ||
| Coenzyme Q10 | 1 μM | 99±1.2 |
| Tempol | 1 μM | 89±1.7 |
| Vitamin E | 1 μM | 103±2.4 |
| ATP linked OCR (%) | ||
| Coenzyme Q10 | 1 μM | 72±2.5 |
| Tempol | 1 μM | 65±1.6 |
| Vitamin E | 1 μM | 72±3.7 |
| Proton leak OCR (%) | ||
| Coenzyme Q10 | 1 μM | 20±0.5 |
| Tempol | 1 μM | 19±0.8 |
| Vitamin E | 1 μM | 19±1.6 |
| Maximal OCR (%) | ||
| Coenzyme Q10 | 1 μM | 143±7.1 |
| Tempol | 1 μM | 146±9.1 |
| Vitamin E | 1 μM | 149±6.2 |
Table 2.
Bioenergetic profiles of mitochondrially targeted antioxidants. MES-13 cells were seeded in a Seahorse XF-24 plate at 30,000 cells per well for 24 h. Cellular media was replaced with low-buffered DMEM media prior to the assay and allowed to equilibrate for 1 h at 37 °C. Basal OCR measurements were made for approximately 30 min followed by injection of MitoQ, MitoTempol, or MitoE. Two hours later, the effects of oligomycin (1 μg/mL), FCCP (2 μM), and AA (10 μM) treatment were determined. Basal OCR is calculated as the difference in OCR before compound injection and after two hours of exposure. ATP linked is the difference in OCR before and after Oligomycin. Proton Leak is the difference in OCR after Oligomycin injection and Antimycin A. Maximal is the OCR after FCCP injection. Data is expressed as a percent relative to the basal OCR rate before compound injection. All data shown are mean±SEM, n=3–5 group. ⁎p<0.05 vs. all groups, statistical comparison performed using ANOVA and Newman–Keuls.
| Bioenergetic profiles of mitochondrially targeted antioxidants | |||||
|---|---|---|---|---|---|
| Basal OCR (%) | Proton leak OCR (%) | ||||
| MitoQ | 0.01 μM | 94±2.2 | MitoQ | 0.01 μM | 14±5.3 |
| 0.1 μM | 97±1.3 | 0.1 μM | 25±2.1 | ||
| 1 μM | 70±4.1⁎ | 1 μM | 47±2.4⁎ | ||
| MitoTempol | 0.01 μM | 101±1.5 | MitoTempol | 0.01 μM | 21±1.4 |
| 0.1 μM | 91±2.8 | 0.1 μM | 19±0.7 | ||
| 1 μM | 70±3.0⁎ | 1 μM | 49±2.9⁎ | ||
| MitoE | 0.01 μM | 100±5.4 | MitoE | 0.01 μM | 23±1.5 |
| 0.1 μM | 103±1.6 | 0.1 μM | 22±3.1 | ||
| 1 μM | 73±1.2⁎ | 1 μM | 36±2.2⁎ | ||
| ATP linked OCR (%) | Maximal OCR (%) | ||||
| MitoQ | 0.01 μM | 66±1.7 | MitoQ | 0.01 μM | 136±5.8 |
| 0.1 μM | 72±3.3 | 0.1 μM | 130±3.3 | ||
| 1 μM | 13±3.2⁎ | 1 μM | 62±7.0⁎ | ||
| MitoTempol | 0.01 μM | 63±2.2 | MitoTempol | 0.01 μM | 119±6.2 |
| 0.1 μM | 57±2.7 | 0.1 μM | 124±4.5 | ||
| 1 μM | 7±1.9⁎ | 1 μM | 42±6.8⁎ | ||
| MitoE | 0.01 μM | 72±2.9 | MitoE | 0.01 μM | 170±9.0 |
| 0.1 μM | 74±6.3 | 0.1 μM | 150±3.8 | ||
| 1 μM | 26±2.6⁎ | 1 μM | 108±1.3⁎ | ||
Conclusion
Taken together, these data suggest that the alkyl TPP+ chain elicits cellular bioenergetic responses which are consistent with inhibition of oxidative phosphorylation and dependent on the length of the alkyl chain. Previous studies have shown that the effects of different carbon chain lengths which link the ubiquinol moiety to the TPP+ group showed that retention and accumulation of the compounds is dependent on the alkyl chain length [17,21,31]. The hydrophobicity of the TPP+ compounds is an important factor controlling accumulation of the compounds. For instance, TPMP (octan-1-ol/PBS partition coefficient of 0.35) slowly accumulates into mitochondria within cells before reaching a steady state which is consistent with the slow onset of inhibition of respiration shown in Fig. 4 [17]. However, a TPP+ compound with a longer carbon chain linker such as DTPP or MitoQ, (octan-1-ol/PBS partition coefficient of 3000) accumulates faster in mitochondria and reaches a steady state much more rapidly and can then effectively uncouple the mitochondria perhaps by cycling between the matrix and inter-membrane space or possibly by interacting with proteins in the mitochondrial inner membrane to increase their catalysis of proton leak across the mitochondrial inner membrane (Figs. 6 and 7) [17]. In bovine aortic endothelial cell isolated mitochondria, MitoQ was shown to induce superoxide, apoptosis, and redox cycling at complex I in a concentration and time dependent manner [32]. Based on our findings, it is likely that the majority of these effects can be ascribed to the non-specific effects of the TPP+ group on cellular bioenergetics.
Importantly, the inhibition of oxidative phosphorylation achieved in cell culture by TPP+ conjugated compounds under conditions where metabolism and clearance of the drugs is minimal are extremely unlikely to occur with oral dosing in either animal models or human subjects. The effects of TPP+ compounds is also likely to be dependent on the cell type since the bioenergetics of cells show a wide range of responses depending on their number of mitochondria and regulation [30]. In cell culture systems some of the bioenergetic and redox effects ascribed to the effects of mitochondrially targeted antioxidants are more likely due to the effects of the linker group and are evident only at concentrations which are not achieved in therapeutic dosing.
Author contribution
Victor Darley-Usmar and Michael P. Murphy designed the research. Colin Reily, Balu K. Chacko, and Gloria Benavides performed seahorse experiments and data analysis; Michael P. Murphy, Tanecia Mitchell, Colin Reily, and Victor Darley-Usmar wrote the manuscript and received feedback on the manuscript from all authors.
Funding
This work was supported by NIH grants: DK007545 (TM) and AA13395, DK079337 and DK075865 (VDU).
Disclosure of potential conflicts of interest
VDU is a member of the Seahorse Biosciences Scientific Advisory Board.
Acknowledgments
We would like to thank Dr. B. Kalyanaraman and Dr. Joy Joseph (Medical College of Wisconsin) for providing MitoE and MitoTempol.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Higdon A., Diers A.R., Oh J.Y., Landar A., Darley-Usmar V.M. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. The Biochemical Journal. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higdon A.N., Benavides G.A., Chacko B.K., Ouyang X., Johnson M.S., Landar A., Zhang J., Darley-Usmar V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. American Journal of Physiology—Heart and Circulatory Physiology. 2012;302:H1394–1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landar A., Zmijewski J.W., Dickinson D.A., Le Goffe C., Johnson M.S., Milne G.L., Zanoni G., Vidari G., Morrow J.D., Darley-Usmar V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. American Journal of Physiology—Heart and Circulatory Physiology. 2006;290:H1777–1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- 4.Diers A.R., Higdon A.N., Ricart K.C., Johnson M.S., Agarwal A., Kalyanaraman B., Landar A., Darley-Usmar V.M. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. The Biochemical Journal. 2010;426:31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill B.G., Benavides G.A., Lancaster J.R., Ballinger S., Dell’italia L., Zhang J., Darley-Usmar V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biological Chemistry. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy M.P. Targeting lipophilic cations to mitochondria. Biochimica Biophysica Acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 7.Horton K.L., Pereira M.P., Stewart K.M., Fonseca S.B., Kelley S.O. Tuning the activity of mitochondria-penetrating peptides for delivery or disruption. Chembiochem. 2012;13:476–485. doi: 10.1002/cbic.201100415. [DOI] [PubMed] [Google Scholar]
- 8.Gane E.J., Orr D.W., Weilert F., Keogh G.F., Gibson M., Murphy M.P., Smith R.A., Lockhart M.M., Frampton C.M., Taylor K.M. 847 phase II study of the mitochondrial antioxidant mitoquinone for hepatitis C. Journal of Hepatology. 2008;48:S318. [Google Scholar]
- 9.Fisher G.J., Quan T., Purohit T., Shao Y., Cho M.K., He T., Varani J., Kang S., Voorhees J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. American Journal of Pathology. 2009;174:101–114. doi: 10.2353/ajpath.2009.080599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A.J., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB Journal. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell T., Rotaru D., Saba H., Smith R.A., Murphy M.P., MacMillan-Crow L.A. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. Journal of Pharmacology and Experimental Therapeutics. 2011;336:682–692. doi: 10.1124/jpet.110.176743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orsucci D., Mancuso M., Ienco E.C., LoGerfo A., Siciliano G. Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Current Medicinal Chemistry. 2011;18:4053–4064. doi: 10.2174/092986711796957257. [DOI] [PubMed] [Google Scholar]
- 13.Chacko B.K., Reily C., Srivastava A., Johnson M.S., Ye Y., Ulasova E., Agarwal A., Zinn K.R., Murphy M.P., Kalyanaraman B., Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(þ/)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochemical Journal. 2010;432:9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chacko B.K., Srivastava A., Johnson M.S., Benavides G.A., Chang M.J., Ye Y., Jhala N., Murphy M.P., Kalyanaraman B., Darley-Usmar V.M. Mitochondria-targeted ubiquinone (MitoQ) decreases ethanol-dependent micro and macro hepatosteatosis. Hepatology. 2011;54:153–163. doi: 10.1002/hep.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholls D.G., Ward M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends in Neuroscience. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- 16.Nicholls D.G. Commentary on: ‘old and new data, new issues: the mitochondrial Deltapsi’ by H. Tedeschi. Biochimica Biophysica Acta. 2005;1710:63–65. doi: 10.1016/j.bbabio.2005.09.002. discussion 66. [DOI] [PubMed] [Google Scholar]
- 17.Ross M.F., Prime T.A., Abakumova I., James A.M., Porteous C.M., Smith R.A.J., Murphy M.P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochemical Journal. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- 18.Smith R.A.J., Porteous C.M., Gane A.M., Murphy M.P. Delivery of bioactive molecules to mitochondria invivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith R.A., Kelso G.F., James A.M., Murphy M.P. Targeting coenzyme Q derivatives to mitochondria. Methods in Enzymology. 2004;382:45–67. doi: 10.1016/S0076-6879(04)82003-2. [DOI] [PubMed] [Google Scholar]
- 20.Flewelling R.F., Hubbell W.L. The membrane dipole potential in a total membrane potential model. Applications to hydrophobic ion interactions with membranes. Biophysical Journal. 1986;49:541–552. doi: 10.1016/S0006-3495(86)83664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Millard M., Pathania D., Shabaik Y., Taheri L., Deng J., Neamati N. Preclinical evaluation of novel triphenylphosphonium salts with broad-spectrum activity. PLoS One. 2010;5:e13131. doi: 10.1371/journal.pone.0013131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leo S., Szabadkai G., Rizzuto R. The mitochondrial antioxidants MitoE(2) and MitoQ(10) increase mitochondrial Ca(2+) load upon cell stimulation by inhibiting Ca(2+) efflux from the organelle. Annals of the New York Academy of Sciences. 2008;1147:264–274. doi: 10.1196/annals.1427.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao V.A., Klein S.R., Bonar S.J., Zielonka J., Mizuno N., Dickey J.S., Keller P.W., Joseph J., Kalyanaraman B., Shacter E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. Journal of Biological Chemistry. 2010;285:34447–34459. doi: 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S., Tak E., Lee J., Rashid M.A., Murphy M.P., Ha J., Kim S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Research. 2011;21:817–834. doi: 10.1038/cr.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fink B.D., Herlein J.A., Yorek M.A., Fenner A.M., Kerns R.J., Sivitz W.I. Bioenergetic effects of mitochondrial-targeted coenzyme q analogs in endothelial cells. Journal of Pharmacology and Experimental Therapeutics. 2012;342:709–719. doi: 10.1124/jpet.112.195586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Cuenca S., Cocheme H.M., Logan A., Abakumova I., Prime T.A., Rose C., Vidal-Puig A., Smith A.C., Rubinsztein D.C., Fearnley I.M., Jones B.A., Pope S., Heales S.J., Lam B.Y., Neogi S.G., McFarlane I., James A.M., Smith R.A., Murphy M.P. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radical Biology and Medicine. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 27.Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. Journal of Biological Chemistry. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 28.Trnka J., Blaikie F.H., Smith R.A., Murphy M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radical Biology and Medicine. 2008;44:1406–1419. doi: 10.1016/j.freeradbiomed.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 29.Murphy M.P., Smith R.A. Drug delivery to mitochondria: the key to mitochondrial medicine. Advanced Drug Delivery Reviews. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 30.Dranka B.P., Benavides G.A., Diers A.R., Giordano S., Zelickson B.R., Reily C., Zou L., Chatham J.C., Hill B.G., Zhang J., Landar A., Darley-Usmar V.M. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biology and Medicine. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asin-Cayuela J., Manas A.R., James A.M., Smith R.A., Murphy M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Letters. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 32.Doughan A.K., Dikalov S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxidants and Redox Signalling. 2007;9:1825–1836. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]