

Abstract
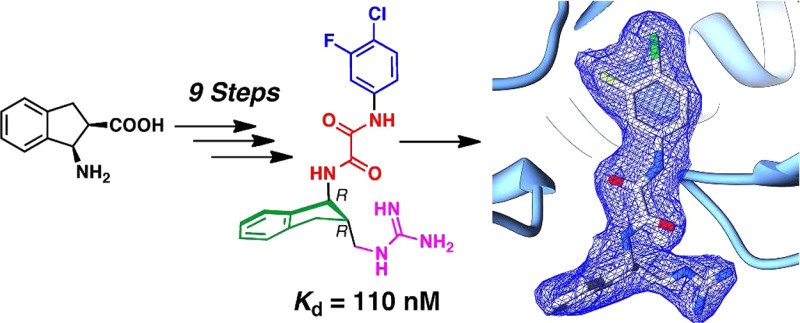
The design, synthesis, thermodynamic and crystallographic characterization of a potent, broad spectrum, second-generation HIV-1 entry inhibitor that engages conserved carbonyl hydrogen bonds within gp120 has been achieved. The optimized antagonist exhibits a submicromolar binding affinity (110 nM) and inhibits viral entry of clade B and C viruses (IC50 geometric mean titer of 1.7 and 14.0 μM, respectively), without promoting CD4-independent viral entry. The thermodynamic signatures indicate a binding preference for the (R,R)- over the (S,S)-enantiomer. The crystal structure of the small-molecule/gp120 complex reveals the displacement of crystallographic water and the formation of a hydrogen bond with a backbone carbonyl of the bridging sheet. Thus, structure-based design and synthesis targeting the highly conserved and structurally characterized CD4–gp120 interface is an effective tactic to enhance the neutralization potency of small-molecule HIV-1 entry inhibitors.
Keywords: HIV, gp120, CD4, entry inhibitor, structure-based drug design, thermodynamics, X-ray crystallography, viral inhibition, protein−protein interactions
The prevention of HIV-1 viral infection remains a worldwide public health objective.1 Despite the continued reduction in AIDS-related deaths, an estimated 2.7 million adults were newly infected in 2010 with HIV-1.2 Clinical trials of preventative approaches, including pre-exposure prophylaxis with oral antiretrovirals3 and/or microbiocide formulations,4 have demonstrated some success and, as such, continue to motivate the development of inhibitors that target alternative steps of the HIV-1 viral life cycle. Given that the initial step of viral infection comprises the attachment of gp120,5 the major surface-exposed protein of the trimeric viral envelope spike,6 to the human host cell receptor CD4,7 inhibition of this protein–protein interaction is attractive and would appear to be a viable prophylactic strategy to prevent HIV-1 transmission. The viral attachment process consists of three stages: (1) gp120 binding to CD4; (2) a conformational change within gp120 induced by the protein–protein interaction that exposes the binding site for the secondary transmembrane chemokine receptors CCR5 or CXCR4; and (3) gp120–chemokine receptor binding, which promotes formation of a six-helix bundle within the gp41 component of the viral envelope spike.5 This cascade of protein–protein interactions, triggered by conformational shifts,8 culminates in fusion of the viral and host cell membranes to permit cellular infection and viral propagation. The development of small-molecule viral entry antagonists that target well conserved hot spots within the viral Env complex and block CD4–gp120 binding continues to hold significant potential for augmenting pre-exposure prophylactic approaches to prevent HIV-1 transmission.4
Mutagenesis studies have revealed key hot spots within the CD4–gp120 interface,9 which were subsequently characterized structurally by a number of CD4/gp120 cocrystal structures.10,11 One important hot spot, the internal Phe43 cavity, is formed at the interface of the three gp120 domains (inner, outer, and bridging sheet) and is capped by the aromatic side chain of CD4 residue Phe43. In fact, the crystal structure of the small molecule NBD-55612 (1, Figure 1), first identified by Debnath and co-workers,13 reveals that 1 binds within the Phe43 cavity. Similarly, chemically derivatized CD4 constructs14 and a miniprotein CD4 mimetic possessing a biphenyl side chain15 form interactions deep within the Phe43 cavity. Subsequent synthesis and testing of NBD-based compounds by our group improved the interactions within the Phe43 cavity, affording the small molecule 2 (Figure 1).16 However, 2 and other region III congeners did not appear to mimic the second hot spot, consisting of an electrostatic interaction between Arg59CD4 and Asp368gp120. Moreover, 1 and 2 functionally mimic CD4 binding and thus behave as agonists of CD4-independent viral entry, leading to the enhancement of HIV-1 entry into cells lacking the CD4 receptor (CD4–CCR5+ Cf2Th cells),17 an undesired trait for the development of viral entry inhibitors.
Figure 1.
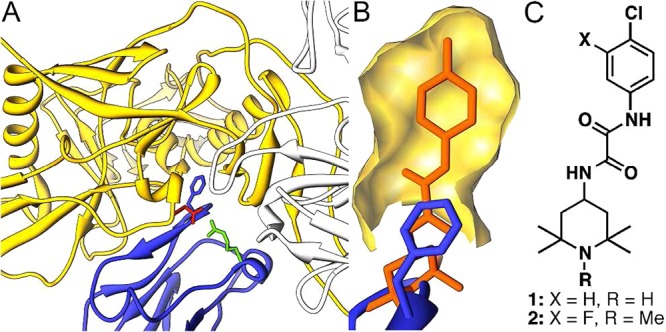
(A) CD4/gp120/17b structure (1GC1)13 in blue, yellow, and light gray, respectively. The Phe43 side chain, blue, emanates from the CD4 β-turn. A second critical CD4–gp120 interaction between Asp368gp120 and Arg59CD4 is highlighted in red and green, respectively. (B) The NBD-556/gp120 structure (3TGS)15 with similar orientation as part A. The surface of the Phe43 cavity is drawn in yellow with NBD-556 in orange. (C) NBD-556 (1) and TS-II-224 (2).
Measurement of the binding thermodynamics of sCD4,18,191, or 2(20,21) to full-length, monomeric gp120 demonstrates that gp120 undergoes a large conformational change upon ligand binding, manifested by a thermodynamic signature reminiscent of protein folding (large unfavorable binding entropy and large favorable binding enthalpy). Not only do 1 and 2 competitively inhibit CD4 binding, but both compounds also mimic CD4 by structuring gp120 and triggering the allosteric signal that enhances coreceptor binding and undesired enhancement of viral infectivity. Interestingly, thermodynamic analysis of a large number of NBD-like small molecules revealed a wide distribution of enthalpic and entropic contributions (ΔH and −TΔS terms vary by ±18.0 kcal/mol), even for compounds with similar binding affinities.22 These results revealed the possibility of designing compounds that do not elicit the unwanted conformational structuring, and delineated a thermodynamic-based strategy for the optimization of those compounds. This strategy implies improving binding affinity (ΔG), while simultaneously minimizing the unfavorable binding entropy.23 The optimization strategy essentially combines standard SAR with thermodynamic analysis by isothermal titration calorimetry (ITC). Importantly, thermodynamic-guided alanine scanning mutagenesis has recently demonstrated that gp120 residue contributions to allosteric structuring are not proportional to binding affinity, providing a structural foundation to the finding that not all NBD-like compounds elicit the unwanted allosteric structuring.24
We have previously described the design, synthesis, and characterization of guanidinium containing trans-1,2-indanes [(+)-3 and (−)-3; Table 1].21 Both (+)-3 and (−)-3 specifically inhibit viral infection of 42 Tier 2 clade B and C viruses, exhibiting geometric mean titers (GMT) IC50 of 7.9 μM and 8.9 μM, for the sensitive strains, respectively. We were pleased to observe that (+)-3 also displays an improved thermodynamic signature, with a reduced entropic penalty (−TΔS) upon gp120 binding, relative to 1 (ΔG, ΔH, and −TΔS values of −7.4, −24.5, and 17.1 kcal/mol, respectively).20 Furthermore, the undesired property of enhancing CD4 independent viral entry was essentially eliminated (Table 1). The cocrystal structure of (+)-3 bound to gp120 revealed specific interactions between the guanidinium moiety and a water mediated hydrogen-bonding network spanning both Asp368gp120 and Met426gp120. Thus, we concluded that incorporation of the guanidinium had converted the NBD congeners into functional antagonists. We therefore sought to optimize further the interactions between the guanidinium moiety of (+)-3 based on the cocrystal structure with residues Asp368gp120 and Met426gp120, an “affinity hot spot”24, in an effort to improve the functional antiviral potency.
Table 1. Antagonists of CD4-gp120 Binding and HIV-1 Entry.
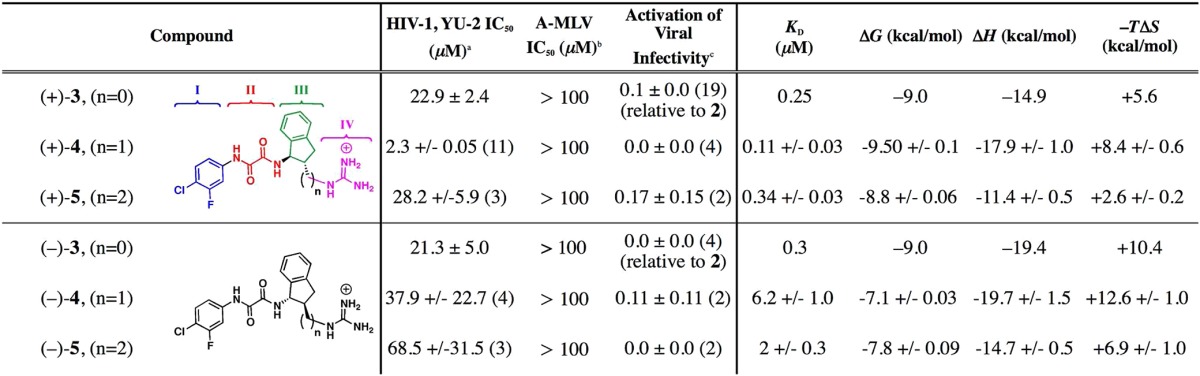
The IC50 was determined in Cf2Th-CD4/CCR5 cells infected with HIV-1 YU2 virus.
The IC50 in cells infected with amphotropic murine leukemia virus (A-MLV).
The relative activation of viral infectivity in CD4 negative Cf2Th-CCR5 cells infected with HIV-1 YU2 virus normalized to that of 1. Data for (+)-3 and (−)-3 have been published.22 See experimental details in the Supporting Information.
To improve these interactions, we chose to vary the distance between the trans indane ring system (region III) and the guanidinium functionality (region IV; Table 1). Hence, the binding properties of the methylene and ethylene congeners of (+)-3 were evaluated by docking (see Supporting Information). These results led to selection of 4 as an initial synthetic target (Table 1). Initially, (±)-4 was constructed (see Supporting Information). When assessed in a single-round viral infection assay, (±)-4 demonstrated a 2-fold improvement of the IC50 value (10.3 ± 3.2 μM) relative to (+)-3 (22.9 ± 2.4 μM). Titration of gp120 with (±)-4, employing ITC, resulted in a complex binding curve that suggested more than one binding event (Figure 2). We reasoned that this observation was related to one enantiomer having a higher affinity within the mixture of (±)-4.
Figure 2.

ITC titrations of gp120 with (A) (+)-4 and (B) (−)-4 at 25 °C. The titration with (±)-4 (inset) resulted in a complex binding curve (see text).
We turned next to X-ray crystallography to investigate the interactions between antagonist (±)-4 and gp120 and to define the enantiomer that preferentially binds to the gp120 core. The formate salt of (±)-4 was soaked into preformed crystals of gp120 from Clade C1086,12 and diffraction data were obtained to 2.5 Å Bragg spacings (Supporting Information Table S1). The observed electron density for each of the two 4/gp120 complexes in the asymmetric unit clearly revealed preferential binding of the (R,R)-4 enantiomer to gp120 during the soaking process (Supporting Information Figure S1). Interestingly, the (R,R)-4/gp120 crystal structure revealed that the guanidinium moiety did not directly interact with Asp368gp120.
With the crystal structure suggesting that the (R,R)-4 enantiomer possesses higher affinity for gp120, a synthetic route to the single (R,R)-enantiomer was developed (Scheme 1). Toward this end, [2 + 2] cycloaddition of indene (6) and chlorosulfonyl isocyanate (7) furnished the racemic β-lactam (±)-8. A kinetic resolution employing lipase B was carried out to yield a separable mixture of β-amino acid (−)-9 and β-lactam (+)-8, both in 99% ee.25−27 The enantiomeric excess was determined by supercritical fluid chromatography (SFC), as described in the Supporting Information. The β-amino acid (−)-9 was next converted to the cis indanol (+)-11 in two steps.21 Although the initial synthetic plan to incorporate a primary amine via oxidation to the aldehyde, followed by reductive animation, proved unsuccessful, we were pleased to find that mildly acidic conditions led to epimerization of the α stereocenter. Subsequent reduction with sodium borohydride established the desired trans stereochemical relationship [cf. (+)-12]. A three-step sequence involving mesylation, displacement of the mesylate with sodium azide, and reduction of the azide led to amine (+)-13. Finally, installation of the guanidinium functionality employing 1H-pyrazol-1-carboxamidine monohydrochloride (14) furnished (+)-4 (99% ee by chiral SFC).21 An identical synthetic sequence was employed to furnish (−)-4 following opening of the β-lactam (+)-8.
Scheme 1. Synthetic Route to Single Enantiomer (+)-4.

Antiviral assays revealed that (+)-4 inhibits viral entry of the YU-2 primary HIV-1 isolate with an IC50 value of 3.1 ± 0.6 μM, while the (−)-4 antipode exhibits a 10-fold reduction in antiviral activity, with an observed IC50 = 37.9 ± 22.7 μM (Table 1). To assess further the HIV-1 neutralization breadth and potency, we assayed 1, (+)-3, and (+)-4 against 42 diverse strains of clades B and C Env-pseudoviruses (see Table S2 in the Supporting Information). As previously observed for 1 and (+)-3,21 (+)-4 also neutralized clade B viruses better than clade C viruses, with 100% breadth and an IC50 GMT of 1.7 μM against clade B viruses, compared to 59% breadth and an IC50 GMT of 14.0 μM against the sensitive clade C viruses. Moreover, (+)-4 demonstrated a 60% improvement over (+)-3 based on IC50 titers in clade B viruses and a 1.5-fold improvement based on IC80 titers. In addition, ITC measurements found that (+)-4 binds full-length gp120 with Kd = 110 nM (Table 1 and Figure 2). In contrast, (−)-4 has a significantly reduced binding affinity of 6200 nM. These results are consistent with the gp120-bound cocrystal structure derived from (±)-4, suggesting the (R,R)-enantiomer preferentially binds to the monomer gp120 core.
Given that inclusion of the methylene spacer between regions III and IV led to significant improvements in both binding affinity and functional antagonism of HIV-1 viral entry into target cells, we constructed (±)-5, containing an additional methylene spacer between the indane scaffold and the guanidinium moiety (see Supporting Information). Semipreparative chiral SFC furnished samples of (+)-5 and (−)-5 for biological evaluation. Assessment of the functional antagonist activity of (+)-5 and (−)-5 revealed that both were less potent than (+)-4 (Table 1). Evaluation of compounds (+)-4, (−)-4, (+)-5, and (−)-5 by ITC (Table 1 and Figure 2) when compared to (+)-3 and (−)-3 demonstrates that (+)-4 exhibits the best submicromolar binding affinity observed thus far for a small-molecule/gp120 complex. Moreover, (+)-3, (+)-4, and (+)-5 all exhibit improved binding affinities when compared to the corresponding (−)-antipodes. Examination of the thermodynamic signatures (Table 1) reveals that (−)-3, (−)-4, and (−)-5 exhibit a larger favorable enthalpy coupled to a larger compensatory unfavorable entropy, suggesting that these compounds trigger a more significant structuring in gp120 than the (+)-3, (+)-4, and (+)-5 compounds. The smaller entropic penalty for (+)-3, (+)-4, and (+)-5 indicates that the (R,R)-configuration possesses the greatest potential for improving enthalpic contributions to binding that are not related to gp120 structuring. In general, improving inhibitor–gp120 interactions should be reflected in better enthalpic interactions not correlated to the structuring of gp120.22
Crystallography was once more employed to ascertain the specific binding interactions between gp120 and (+)-4. Cocrystallization of (+)-4 with the clade A/E93TH057 extended gp120(H375S) core12 produced crystals that diffracted to 2.5 Å spacings (crystallographic data are summarized in Table S1 in the Supporting Information). There are two complexes in the asymmetric unit of these crystals, and each (+)-4 molecule in both complexes has similar conformations that closely resemble those observed in the (R,R)-4/gp120 structure obtained from (±)-4 (Supporting Information Figure S1 and Table S3). As expected, the previously observed hydrogen bonds between the oxalamide linker and the Asn425gp120 and Gly473gp120 amide nitrogen atoms12,21 are preserved in the (+)-4/gp120 complex (Figure 3). Surprisingly, as noted for (R,R)-4/gp120 (vide supra), the guanidinium moiety did not directly interact with Asp368gp120. Instead, a hydrogen bond is formed between one guanidinium nitrogen and the bridging sheet backbone carbonyl of Met426gp120. Importantly, the crystallographic water molecules necessary for the indirect interaction with Met426gp120 in the (+)-3/gp120 structure are now displaced by the extended guanidinium of the (+)-4/gp120 (Figure 3), allowing for direct hydrogen bonding to the carbonyl of Met426gp120. The direct hydrogen bond and the displacement of crystallographic water molecules, as well as the improved binding affinity of (+)-4 over the previous described (+)-3, provide additional motivation to modify region III moieties. Further modification of the guanidinium-Asp368gp120 contact, as previously observed in the (+)-3/gp120 complex with the newly revealed guanidinum–Met426gp120 interaction exhibited in the (+)-4/gp120 structure, will in the future yield even more potent inhibitors of viral entry.
Figure 3.
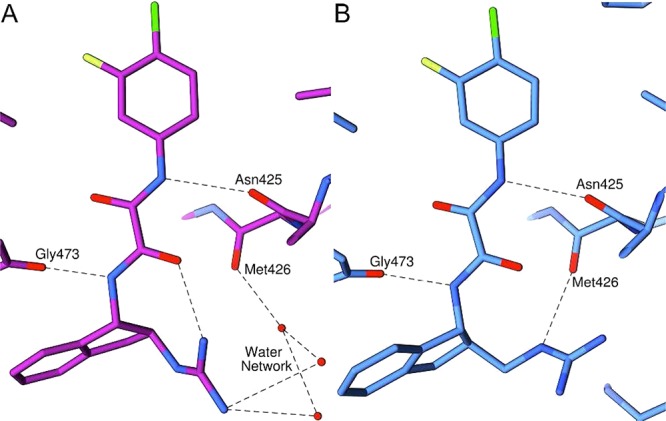
Comparison of the structures of (A) (+)-3/gp120 (4DKQ)21 and (B) (+)-4/gp120 (Clade E, copy A) indicates that (+)-3 interacts with Met426gp120 via a network of water molecules whereas the guanidinium group of (+)-4 hydrogen bonds directly to the backbone carbonyl of Met426gp120. Coloring is by atom type: nitrogen (blue), oxygen (red), chlorine (neon-green), fluorine (neon-yellow-green), and carbon [magenta for (+)-3 and sky blue for (+)-4]. Hydrogen bonds are represented by dashed lines.
In summary, we have employed the crystal structures of antagonist/gp120 complexes to achieve an increase of both binding affinity and antiviral potency for HIV-1 entry inhibitors. The crystal structure of this new compound, (+)-4, bound to gp120, reveals a newly formed hydrogen bond to Met426gp120 and the displacement of crystallographic water derived from the extension of the guanidinium group via a methylene spacer. Whereas the parent compounds (−)-3 and (+)-3 had comparable potency and bound alternatively to gp120, the (R,R)-enantiomer (+)-4 is strongly preferred and binds uniquely. On the basis of the antiviral potency and binding affinity, the methylene spacer between regions III and IV of (+)-4 is favored compared to the ethylene spacer of (+)-5. Furthermore, the thermodynamic signatures indicate that the (R,R)-stereochemical configuration encounters less entropic penalty than the (S,S) one when binding to gp120. The design, development, and synthesis of small-molecule viral entry antagonist (+)-4 thus validates the application of static structures of ligands bound to the conserved monomeric core of gp120 to improve antagonist potency. Structural information from small-molecule/gp120 complexes, however, must be combined with thermodynamic information, as allosteric structuring and solvation effects are not revealed in the X-ray structures. Moreover, the improved antiviral potency and structural characterization of (+)-4 provides an important contribution to small-molecule viral entry antagonists for prophylactic strategies for the prevention of HIV-1 transmission.
Acknowledgments
We thank Irwin Chaiken and all the members of the PO1 Consortium Structure-Based Antagonism of HIV-1 Envelope Function in Cell Entry. J.M.L. thanks the Pittsburgh Supercomputing Center for an allocation for computing resources #MCB090108. M.L. and W.A.H. thank Young Do Kwon and Peter Kwong of the Vaccine Research Center of NIAID for transferring clade C and clade A/E gp120 crystallization technology.
Glossary
Abbreviations
- (HIV-1)
Human immunodeficiency virus type 1
- (SIV)
simian immunodeficiency virus
- (sCD4)
soluble CD4
- (ITC)
isothermal titration calorimetry
- (A-MLV)
amphotropic murine leukemia virus
- (GMT)
geometric mean titer
- (GA)
genetic algorithm
- (HRMS)
high-resolution mass spectroscopy
- (DMEM)
Dulbecco’s Modified Eagle Medium
- (TsCl)
tosyl chloride
- (DMAP)
4-dimethylaminopyridine
Supporting Information Available
Synthesis, experimental methods, and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: 4I53 and 4I54.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding was provided by NIH GM 56550 to J.M.L., E.F., W.A.H., A.B.S., and J.S. and by NIH Intramural IATAP and NIAID programs to J.R.M. and J.S. Funding to N.M. was provided by NIH AI090682-01. N.M. and J.S. were also supported by Ragon Institute of MGH, MIT, and Harvard.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Global HIV/AIDS response—epidemic update and health sector progress towards universal access; Progress report 2011; WHO, UNAID, UNICEF: 2011.
- Global report, UNAIDS report on the global aids epidemic; Joint United Nations Programme on HIV/AIDS (UNAIDS); 2010; ISBN 978-92-9173-871-7.
- Cohen M. S.; Chen Y. Q.; McCauley M.; Gamble T.; Hosseinipour M. C.; Kumarasamy N.; Hakim J. G.; Kumwenda J.; Grinsztejn B.; Pilotto J. H. S.; Godbole S. V.; Mehendale S.; Chariyalertsak S.; Santos B. R.; Mayer K. H.; Hoffman I. F.; Eshleman S. H.; Piwowar-Manning E.; Wang L.; Makhema J.; Mills L. A.; de Bruyn G.; Sanne I.; Eron J.; Gallant J.; Havlir D.; Swindells S.; Ribaudo H.; Elharrar V.; Burns D.; Taha T. E.; Nielsen-Saines K.; Celentano D.; Essex M.; Fleming T. R. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 2011, 365, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdool Karim Q.; Abdool Karim S. S.; Frohlich J. A.; Grobler A. C.; Baxter C.; Mansoor L. E.; Kharsany A. B. M.; Sibeko S.; Mlisana K. P.; Omar Z.; Gengiah T. N.; Maarschalk S.; Arulappan N.; Mlotshwa M.; Morris L.; Taylor D. Effectiveness and safety of Tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010, 329, 1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt R.; Sodroski J. The HIV-1 envelope glycoproteins fusogens antigens and immunogens. Science 1998, 280, 1884–1888. [DOI] [PubMed] [Google Scholar]
- Lu M.; Blacklow S. C.; Kim P. S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [DOI] [PubMed] [Google Scholar]
- Dalgleish A. G.; Beverley P. C.; Clapham P. R.; Crawford D. H.; Greaves M. F.; Weiss R. A. The CD4 (t4) antigen is an essential component of the receptor for the aids retrovirus. Nature 1984, 312, 763–767. [DOI] [PubMed] [Google Scholar]
- Sattentau Q. J.; Moore J. P.; Vignaux F.; Traincard F.; Poignard P. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J. Virol. 1993, 67, 7383–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moebius U.; Clayton L. K.; Abraham S.; Harrison S. C.; Reinherz E. L. The human immunodeficiency virus gp120 binding site on CD4: Delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J. Exp. Med. 1992, 176, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P. D.; Wyatt R.; Robinson J.; Sweet R. W.; Sodroski J.; Hendrickson W. A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-C.; Lam S. N.; Acharya P.; Tang M.; Xiang S.-H.; Shahzad-ul Hussan S.; Stanfield R. L.; Robinson S.; Sodroski J.; Wilson I. A.; Wyatt R.; Bewley C. A.; Kwong P. D. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 2007, 319, 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. D.; Finzi A.; Wu X.; Dogo-isonagie C.; Lee L. K.; Moore L. R.; Schmidt S. R.; Stuckey J.; Yang Y.; Zhou T.; Zhu J.; Vicice D. A.; Debnath A. K.; Shapiro L.; Bewley C. A.; Mascola J. R.; Sodroski J.; Kwong P. D. Unliganded HIV-1 gp120 structures reveal propensity of core to assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5663–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ma L.; Jiang S.; Lu H.; Liu S.; He Y.; Strick N.; Neamati N.; Debnath A. K. Identification of n-phenyl-n’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [DOI] [PubMed] [Google Scholar]
- Xie H.; Ng D.; Savinov S. N.; Dey B.; Kwong P. D.; Wyatt R.; Smith A. B. 3rd; Hendrickson W. A. Structure–activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J. Med. Chem. 2007, 50, 4898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. C.; Stricher F.; Martin L.; Decker J. M.; Majeed S.; Barthe P.; Hendrickson W. A.; Robinson J.; Roumestand C.; Sodroski J.; Wyatt R.; Shaw G. M.; Vita C.; Kwong P. D. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure 2005, 13, 755–768. [DOI] [PubMed] [Google Scholar]
- LaLonde J. M.; Elban M. A.; Courter J. R.; Sugawara A.; Soeta T.; Madani N.; Princiotto A.; Kwon Y. D.; Kwong P. D.; Schön A.; Freire E.; Sodroski J.; Smith A. B. III Design, synthesis and biological evaluation of small molecule inhibitors of CD4–gp120 binding based on virtual screening. Bioorg. Med. Chem. 2011, 19, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani N.; Schön A.; Princiotto A. M.; Lalonde J. M.; Courter J. R.; Soeta T.; Ng D.; Wang L.; Brower E. T.; Xiang S. H.; Kwon Y. D.; Huang C. C.; Wyatt R.; Kwong P. D.; Freire E.; Smith A. B. 3rd; Sodroski J. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 2008, 16, 1689–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myszka D. G.; Sweet R. W.; Hensley P.; Brigham-Burke M.; Kwong P. D.; Hendrickson W. A.; Wyatt R.; Sodroski J.; Doyle M. L. Energetics of the HIV gp120–CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9026–9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey B.; Pancera M.; Svehla K.; Shu Y.; Xiang S. H.; Vainshtein J.; Li Y.; Sodroski J.; Kwong P. D.; Mascola J. R.; Wyatt R. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: Antigenicity, biophysics, and immunogenicity. J. Virol. 2007, 81, 5579–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön A.; Madani N.; Klein J. C.; Hubicki A.; Ng D.; Yang X.; Smith A. B. 3rd; Sodroski J.; Freire E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 2006, 45, 10973–10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J. M.; Kwon Y. D.; Jones D. M.; Sun A. W.; Courter J. R.; Soeta T.; Kobayashi T.; Princiotto A. M.; Wu X.; Schön A.; Freire E.; Kwong P. D.; Mascola J. R.; Sodroski J.; Madani N.; Smith A. B. III Structure-Based Design, Synthesis, and Characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J. Med. Chem. 2012, 55, 4382–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön A.; Madani N.; Smith A. B. III; LaLonde J. M.; Freire E. Some drug properties are dependent on thermodynamic signature. Chem. Biol. Drug. Des. 2011, 77, 161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön A.; Lam S. Y.; Freire E. Thermodynamics-based drug design: Strategies for inhibiting protein–protein interactions. Future Med. Chem. 2011, 3, 1129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Schön A.; Freire E. Optimization of CD4/gp120 inhibitors by thermodynamic guided alanine scanning mutagenesis. Chem. Biol. Drug Des. 2012, 15, 12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülöp F.; Palkó M.; Kámán J.; Lázár L.; Sillanpää R. Synthesis of all four enantiomers of 1-aminoindane-2-carboxylic acid, a new cispentacin benzologue. Tetrahedron: Asymmetry 2000, 11, 4179–4187. [Google Scholar]
- Fülöp F.; Forró E.; Toth M. K. A new strategy to produce beta-peptides: Use of alicyclic beta-lactams. Org. Lett. 2004, 6, 4239–4241. [DOI] [PubMed] [Google Scholar]
- Forró E.; Fülöp F. An efficient enzymatic synthesis of benzocispentacin and its new six- and seven membered homologues. Chem.—Eur. J. 2006, 12, 2587–2592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.