

Abstract
Tuberculosis (TB) is a reemerging disease. The only available vaccine, Mycobacterium bovis BCG, is delivered intradermally and confers highly variable efficacy against pulmonary disease. There is an urgent need for improved vaccination strategies. Murine studies suggest that immunizations delivered directly to the respiratory mucosa might be a more effective route of vaccination. This study compared the immunogenicity of a leading candidate tuberculosis (TB) vaccine, modified vaccinia virus Ankara expressing antigen 85A (MVA85A), in rhesus macaques, delivered either as an aerosol or as an intradermal boost immunization 12 weeks after an intradermal BCG prime vaccine. Aerosol vaccination was well tolerated. MVA85A delivered by aerosol or by intradermal injection induced antigen-specific immune responses in the periphery and the lung, with a trend toward the highest response when the compartment and route of delivery were matched. The ability of poxvirus-vectored vaccines delivered by the systemic route to induce responses in the mucosal immune compartment in macaques is in contrast to the independent compartmentalization of mucosal and systemic immune systems described in mice. Unlike intradermal vaccination, aerosol vaccination did not induce a detectable serum anti-vector antibody response. The delivery of vaccines to the lungs might provide an immunization strategy that limits the induction of systemic anti-vector immunity, which would be extremely useful in the development of improved vaccine strategies. This is the first study to show a recombinant MVA-vectored vaccine to be highly immunogenic when delivered by the aerosol route to nonhuman primates. These results provide important safety and proof-of-concept data for further evaluation of this route of immunization for use in human clinical trials.
INTRODUCTION
Tuberculosis (TB) in humans is caused by infection with Mycobacterium tuberculosis and is one of the leading global causes of death from a single infectious agent, with an estimated 8.7 million new cases worldwide and 1.4 million deaths in 2011 (1).
The only licensed vaccine against TB is bacillus Calmette-Guérin (BCG), a live attenuated strain of Mycobacterium bovis that was first introduced in 1921 and remains in routine use around the globe. During this time, it has been administered to several billion people, predominantly delivered by intradermal injection. While BCG is well tolerated, it is not effective in all populations (2), does not prevent infection in high-burden settings, and is contraindicated in immunocompromised patients (3). A more effective vaccine is urgently needed.
One leading approach in the development of a more effective vaccination regimen is to incorporate BCG or a BCG replacement vaccine into a heterologous prime-boost strategy with a subsequently administered booster vaccine. Modified vaccinia virus Ankara (MVA) is a safe (4) replication-defective (5, 6) viral vector, capable of inducing both cellular and antibody immunity to target antigens with protective potential (7, 8). The most clinically advanced “boost” candidate is the viral-vector subunit TB vaccine MVA85A (9), which expresses the highly conserved immunodominant mycobacterial antigen 85A (Ag85A).
Experiments in guinea pigs, cattle, and nonhuman primates have shown that a prime-boost schedule of vaccination with BCG followed by a systemically administered (intradermal) MVA85A boost induces cellular immunity and can improve protective efficacy against a subsequent challenge with M. tuberculosis or M. bovis, compared to the use of BCG alone (10, 11, 12). It is well tolerated and highly immunogenic in healthy adults, adolescents, children, and infants and in HIV- and M. tuberculosis-infected adults, and it is currently being evaluated in a BCG-prime with MVA85A-boost regimen in a large phase IIb efficacy trial in infants in South Africa.
Conventionally, both BCG and MVA85A have been administered by intradermal (i.d.) injection, a route of inoculation that has been shown to induce strong mycobacterium-specific immune responses in the periphery. However, the profile of responses induced at mucosal surfaces has yet to be defined. Furthermore, there is now a general recognition that the mucosa-associated lymphoid tissue (MALT) should be considered an immune compartment separate from the peripheral system and that triggering immune responses in the MALT might lead to increased protection against pulmonary TB. Vaccine delivery by aerosol directly to the lungs is advantageous because it is simple, quick, noninvasive, and painless.
The route of vaccine delivery determines the location of antigen-specific cells and thus the protective efficacy of the vaccine. Evidence from animal studies suggests that the prevalence of antigen-specific cells in the airway lumen is important for optimal protection against (in this case, M. tuberculosis) infection (13). Replication-deficient vaccinia virus and adenovirus vectors encoding antigen 85A demonstrated better protection when delivered directly to the murine respiratory mucosa than when administered systemically (13, 14).
Protection from TB is critically dependent on the cellular immune response, in particular CD4+, and probably also CD8+, T-cell-mediated cellular responses, the mechanisms of which are not fully elucidated (15, 16). The main immunological readout used in TB vaccine studies to date has been the measurement of antigen-specific gamma interferon (IFN-γ) in one of several immunological assays (17). While there is strong evidence to support an essential role for IFN-γ in the protective immune response against M. tuberculosis (18, 19, 20), it might not be sufficient alone. The presence of antigen-specific polyfunctional CD4 and CD8 T cells, expressing combinations of IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin 2 (IL-2), has been correlated to a positive clinical outcome in HIV plus TB coinfection, and these polyfunctional cells are induced following vaccination with BCG and the novel candidate TB vaccine, MVA85A (21).
It is widely accepted that nonhuman primates (NHPs) are the most relevant species to predict the safety, immunogenicity, and protective efficacy of vaccines in humans (22, 23). Macaques are naturally susceptible to infection with M. tuberculosis via the respiratory route and develop a disease that closely mimics the equivalent disease in humans. The similarities to humans in the physiology, anatomy, and most importantly immune function of NHPs means that they are potentially the most reliable models in which to identify relevant correlates of protection and/or disease. BCG vaccination of NHPs provides a limited level of protection against M. tuberculosis that can be quantified through a variety of clinical and nonclinical parameters (24, 25, 26, 27, 28, 29).
In this study, we evaluated the safety and immunogenicity of MVA85A, administered by nebulizer to the respiratory mucosa of BCG-vaccinated rhesus macaques, and compared the findings to those found through intradermal administration of the vaccines.
MATERIALS AND METHODS
Verification of viral viability following aerosolization by mesh nebulizer.
Studies were performed to assess the viability of MVA85A following aerosolization with the Omron Micro Air handheld vibrating mesh nebulizer. The mouthpiece attached to the nebulizer was connected by a sealed system to a collection vessel in which aerosolized MVA85A was condensed. The viability of the batch of clinical-grade MVA85A selected for use in the macaque study after aerosolization was compared alongside an identical nonaerosolized control sample. MVA85A, in a quantity of 2.7 × 107 PFU, was diluted in 1 ml phosphate-buffered saline (PBS), nebulized using a Micro Air device, and collected using an impinger. The viability of MVA85A after aerosolization was evaluated by plaque assay using chicken embryo fibroblast (CEF) cells.
The reproducibility of different mesh nebulizers was determined by comparing the time taken to aerosolize 1 ml of sterile PBS.
Plaque assay for MVA85A PFU determination method for biodistribution and in vitro studies.
Briefly, tissue samples were thawed and homogenized in 2 ml 2% fetal calf serum (FCS)–Dulbecco's modified Eagle's medium (DMEM) (D6546, Invitrogen) using gentleMACS M tubes on a Dispomix instrument (program 14), followed by 2 cycles of snap-freeze/thaw in dry ice-isopropanol to release any viral particles from within the organ tissue. Samples were spun to remove as much tissue debris as possible before being plated out on subconfluent low-passage (P3) CEF cells in a 96-well plate and were incubated for 96 h. Supernatant was then plated onto a second set of CEF cells in polylysine-coated 96-well plates to allow further viral replication for 2 days before fixing cells with methanol. Cells were immunostained with rabbit anti-vaccinia virus primary antibody (ISL 126-1063) and anti-rabbit horseradish peroxidase (HRP) conjugated secondary antibody (donkey anti-rabbit-HRP conjugated antibody, Amersham NA934V) for the presence of MVA. Detection was by 3,3′-diaminobenzidine (DAB) chromogenic staining (ImmPACT DAB chromogenic substrate, catalog no. SK-4105; Vector Laboratories). A reference MVA85A virus at a low PFU concentration (2 × 103 PFU/ml) was used as a positive control in both rounds of amplification, with noninfected cell supernatants and cells used as negative controls. An additional control of inguinal lymph node material from each test subject, spiked with a known amount of MVA85A (80 μl at 2 × 104 PFU/ml added to 720 μl inguinal lymph node [ILN] sample, 100 μl per well) was also used. This assay has a limit of detection of 2 × 102 PFU.
Experimental animals.
The animals used in this study were rhesus macaques of Indian origin obtained from an established United Kingdom breeding colony. All animals were 4.5 to 5 years old at the time of vaccination and naïve in terms of prior exposure to mycobacterial antigens (M. tuberculosis infection or environmental mycobacteria), as demonstrated by a negative tuberculin test while in their original breeding colony and by the IFN-γ-based Primagam test kit (Biocor, CSL) just prior to the study start. Monkeys were housed according to the Home Office (United Kingdom) Code of Practice for the Housing and Care of Animals Used in Scientific Procedures (1989) and the National Committee for Refinement, Reduction, and Replacement (NC3Rs) Guidelines on Primate Accommodation, Care, and Use, August 2006. They were sedated by intramuscular (i.m.) injection with ketamine hydrochloride (10 mg/kg) (Ketaset; Fort Dodge Animal Health Ltd., Southampton, United Kingdom) for all procedures requiring removal from their cages. All protocols involving animals were approved by the Ethical Review Committee of the Health Protection Agency, Porton, United Kingdom. None of the animals had been used previously for experimental procedures.
Vaccination.
Eight animals (divided into groups A and B) were immunized intradermally in the upper left arm with 100 μl BCG, Danish strain 1331 (SSI, Copenhagen, Denmark). The BCG vaccine was prepared and administered according to the manufacturer's instructions for the preparation of vaccine for administration to human adults; this was done by adding 1 ml Saunton's medium as the diluent to a vial of vaccine to give a suspension of BCG at an estimated concentration of 2 × 106 to 8 × 106 CFU/ml. Twelve weeks after immunization with BCG, four animals (group A) were immunized intradermally in the upper right arm with 100 μl (1 × 108 PFU) MVA85A. Four other animals (group B) were exposed to an aerosol containing MVA85A created by the nebulization of 108 PFU in 1 ml sterile PBS using an Omron Micro Air mesh nebulizer (Omron Healthcare UK, Ltd., Milton Keynes, United Kingdom), and it was delivered via the nose of each sedated primate using a modified pediatric anesthesia mask.
Skin vaccination sites were monitored and assessed for local reactions after vaccination with BCG and MVA85A.
Clinical assessment.
The primary purpose of the study was to assess tolerability using the following readouts. Animal behavior was observed daily throughout the study for contraindications, such as depression, withdrawal from the group, aggression, reduced food and water intake, changes in respiration rate, or cough. Animals were sedated every 2 weeks to measure weight, body temperature, blood hemoglobin levels, and erythrocyte sedimentation rate (ESR) and to collect blood samples for immunology. Blood cell hemoglobin levels were measured using a hemoglobinometer (HemoCue Ltd., Dronfield, United Kingdom), and ESRs were measured using the Sediplast system (Guest Medical, Edenbridge, United Kingdom). Chest X rays were collected prior to BCG vaccination, 2 weeks prior to vaccination with MVA85A, and 1 and 8 weeks after MVA85A vaccination.
Bronchoalveolar lavage.
To determine the immune response in the lungs, bronchoalveolar lavage (BAL) fluid samples were collected using a bronchoscope (Allscope XE30 4-mm flexible bronchoscope; VES, Essex, United Kingdom) on two occasions 4 weeks apart prior to BCG vaccination, at 4 and 8 weeks after BCG vaccination, and also 1, 3, and 7 weeks after the MVA85A booster vaccination. On each occasion, three consecutive washes were performed, each using 20-ml volumes of Hanks' balanced salts (Sigma-Aldrich, Dorset, United Kingdom), which was instilled in the lungs and collected.
IFN-γ ELISpot assay.
Peripheral blood mononuclear cells (PBMCs) were isolated from heparin-anticoagulated blood, and lymphoid mononuclear cells from tissue samples, by Ficoll-Hypaque Plus (GE Healthcare, Buckinghamshire, United Kingdom) density-gradient separation using standard procedures. An IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay was used to estimate the numbers and the IFN-γ production capacity of mycobacterium-specific T cells in PBMCs using a human/monkey IFN-γ kit (Mabtech, Nacka, Sweden) as described previously (29). In brief, PBMCs were cultured with 10 μg/ml purified protein derivative (PPD) (SSI, Copenhagen, Denmark), with a pool containing overlapping 15-mer peptides spanning antigen 85A (Ag85A) (Peptide Protein Research Ltd., Wickham, United Kingdom), or without antigen, in triplicate, and incubated for 18 h. Phorbol 12-myristate (100 ng/ml) (Sigma-Aldrich, Dorset, United Kingdom) and ionomycin (1 μg/ml) (CN Biosciences, Nottingham, United Kingdom) were used as a positive control. After culture, spots were developed according to the manufacturer's instructions. Determinations from triplicate tests were averaged. Data were analyzed by subtracting the mean number of spots in the cells for medium-only control wells from the mean counts of spots in wells with cells and antigen or peptide pools.
Intracellular cytokine staining.
PBMCs were thawed, washed, resuspended in R10 medium (consisting of RPMI 1640 supplemented with l-glutamine [2 mM], penicillin [50 U/ml]-streptomycin [50 μg/ml], and 10% heat-inactivated fetal bovine serum with 1 U/ml of DNase [Sigma, Poole, United Kingdom]), and incubated at 37°C for 2 h. BAL cells were isolated from BAL fluid by centrifugation (400 × g for 5 min) and were assessed directly following isolation. Cell concentrations were adjusted to 1 × 106 cells/ml in R10 and stimulated with 10 μg/ml PPD (SSI, Copenhagen, Denmark). Intracellular cytokine staining to evaluate the production of the cytokines IFN-γ, TNF-α, and IL-2 was performed as described previously (29).
Flow cytometric acquisition and analysis.
Cells were analyzed using a 4b SORP LSRII instrument (BD Biosciences, Oxford, United Kingdom). Cytokine-secreting T cells were identified using a forward scatter-height (FSC-H) versus side scatter-area (SSC-A) dot plot to identify the lymphocyte population, to which appropriate gating strategies were applied to exclude doublet events, nonviable cells, monocytes (CD14+), and B cells (CD20+) prior to sequential gating through CD3+, CD8−, and CD4+ versus IFN-γ, and CD3+, CD8+, and CD4− versus IFN-γ, histograms. Data were analyzed using FlowJo v8.8.6 before further manipulation using the programs PESTLE v1.6.2 and SPICE v5.0 to generate graphical representations of T-cell responses using background-subtracted flow data (Mario Roederer, Vaccine Research Centre, National Institute of Allergy and Infectious Diseases [NIAID], NIH).
Vaccinia virus antibody ELISA.
Anti-vector IgG titers were measured in serum samples by a vaccinia virus-specific direct enzyme-linked immunosorbent assay (ELISA). In brief, Nunc MaxiSorp 96-well plates were coated with inactivated Lister strain vaccinia virus (Autogen Bioclear). After blocking, samples were diluted 1:25 in assay diluent to blocking buffer (PBS plus 5% skim milk powder plus 0.1% Tween 20) and added in duplicate. A sample dilution series was created by a 4-fold serial dilution. A reference curve was created by the inclusion of a polyclonal anti-vaccinia virus human immunoglobulin (VIG) control sample (NR-650, Biodefense and Emerging Infections Research Resource Repositories), serially diluted in parallel. Serum and VIG control samples were incubated at room temperature for 2 h. After washing, plates were incubated for 2 h with anti-monkey IgG peroxidase (Insight Biotechnology), washed, and then developed using a 2,2′-azinobis(3-ethylbenzthiazoline sulfonic acid) (ABTS) peroxidase substrate system (Insight Biotechnology). Absorbance at 405 nm was then measured using a VersaMax plate reader with SoftMax Pro software. A VIG control curve was plotted for each plate and the inflection point was used to interpret the serum sample IgG titer.
Necropsy.
Before necropsy, animals were sedated with ketamine (15 mg/ml, i.m. administration), weighed, and photographed, chest X rays were taken, clinical data were collected, and exsanguination was effected via the heart before termination by the injection of a lethal dose of anesthetic (140 mg/kg) (Dolethal, Vétoquinol UK Ltd.). A full necropsy was performed and gross pathology assessed. Samples of spleen, liver, kidneys and hilar, inguinal and axillary lymph nodes, tonsil, brain and olfactory bulb, heart and pericardium, the small intestine (ileum, jejunum, duodenum), colonic lymph nodes, transverse colon, mesenteric lymph nodes, eye, nasal turbinate (right side), and the upper left lung lobe were removed, dissected on sterile trays, and placed in formalin-buffered saline for histology. All other lung lobes were collected for the assessment of immune responses.
Pathology and histology.
Representative sections from all tissues described above were processed to paraffin wax, cut at 5 μm, and stained with hematoxylin and eosin (H&E) for examination by microscopy.
Statistical analyses.
To compare the immune responses measured by ex vivo ELISpot assay in animals according to vaccination group, the area under the curve (AUC) for each response was calculated using SigmaPlot version 10 (Systat Software Inc., Hounslow, United Kingdom) for each animal. The AUCs calculated for the animals in each test group were compared to those for the animals in the other test group with a Mann-Whitney test using Minitab version 15 (Minitab Ltd., Coventry, United Kingdom).
To compare T-cell functional profiles between vaccination groups as measured by polyfunctional flow cytometry, the frequency of each functional subset (triple, dual, and monofunctional cytokine-producing CD4 or CD8 T cells) was compared using a Mann-Whitney test at each analysis time point.
Similarly, vaccine-induced changes in the T-cell functional profile, within each vaccination group, were assessed by comparing the frequency of functional subsets at each analysis time point with mean baseline values. Zero values were replaced by a small number that was lower than the minimum value detected.
RESULTS
In vitro studies to optimize delivery of MVA85A through the mesh nebulizer.
Initial studies demonstrated that the aerosolization process did not adversely affect the viability of MVA85A and showed that the level of viable MVA85A collected postaerosolization was not significantly different from that determined in the vaccine preparations prior to aerosolization (see Fig. S1A in the supplemental material). Nebulization rates were found to be highly reproducible between consecutive runs and between four different mesh caps and different devices (see Fig. S1B in the supplemental material). To ensure optimal delivery of the aerosol, a range of pediatric masks were tested for use with rhesus macaques. The mask with the best fit type and make was enhanced using a spacer to fill the gap under the chin.
Based on the results from the optimization studies, the strategy adopted for the vaccination of rhesus macaques in the immunogenicity study used separate, preprepared mesh caps and the same device for all vaccinations.
Safety of aerosol-delivered MVA85A.
Vaccination did not lead to perturbations beyond the normal ranges for body weight, temperature, peripheral lymph node size, red cell hemoglobin concentration, or erythrocyte sedimentation rate at any point during the study.
Chest radiographs collected through the study demonstrated normal pulmonary structure that remained unchanged postvaccination. Intradermal immunization with BCG and MVA85A led to mild induration and erythema at the vaccination sites. Seven of the eight animals developed local skin reactions at the site of BCG vaccination at 2 and 4 weeks postvaccination, with reactions still visible in 3 animals at the end of the study. Intradermal vaccination with MVA85A induced local skin reactions at the vaccination site 1 week postimmunization in all 4 animals, which resolved 3 weeks later. At the end of the study, 9 weeks after vaccination with MVA85A, gross pathology and histological assessments did not identify any adverse effects induced by vaccination. Overall, aerosol vaccination was well tolerated by all animals and caused no adverse effects.
Distribution of MVA in tissues.
No viable virus was detected in any of the NHP tissue samples provided, whether administered by i.d. or aerosol.
Evaluation of the T-cell response induced by each route of vaccination in peripheral blood.
The systemic profiles of immune responses that were induced following immunization with BCG and MVA85A by aerosol and intradermal injection were characterized. An increased frequency of PPD-specific IFN-γ-secreting cells was detected by ex vivo ELISpot assay in all 8 animals following vaccination with BCG. After intradermal boost with MVA85A, an increased response was seen in one animal, but not in any of the animals after aerosol MVA85A administration (Fig. 1A and B). Responses to the Ag85A peptides were not detectable after BCG vaccination. One week after vaccination with MVA85A, high responses were detected in two of the four animals of each immunization group, together with modest responses above prevaccination levels in one of the other two animals in each group. These responses contracted over the subsequent 3 weeks, as expected (Fig. 1C and D). Systemic immune responses in the group of animals that received intradermal MVA85A were higher than responses in the aerosol-administered group. Neither AUC analysis of the response during the 8-week period after vaccination (P = 0.665) nor comparison of the peak response with a Mann-Whitney test (P = 0.665) reached statistical significance. Multiparameter flow cytometry was performed to evaluate the production of the cytokines IFN-γ, TNF-α, and IL-2 in stimulated PBMCs isolated throughout the in vivo time course (Fig. 2). After intradermal MVA85A vaccination, there was an increase in the double-positive (TNF-α+ IFN-γ+) and triple-positive (TNF-α+ IFN-γ+ IL-2+) populations of antigen-specific CD4+ T cells, which was not seen in the aerosol-administered group. This population contracted, but was still detectable, 4 weeks after vaccination. Differences between the groups were not statistically significant. Interestingly, at 8 weeks postvaccination with MVA85A, there was a population of double-positive antigen-specific CD4+ T cells in both aerosol- and intradermally vaccinated animals (Fig. 2B and D).
Fig 1.
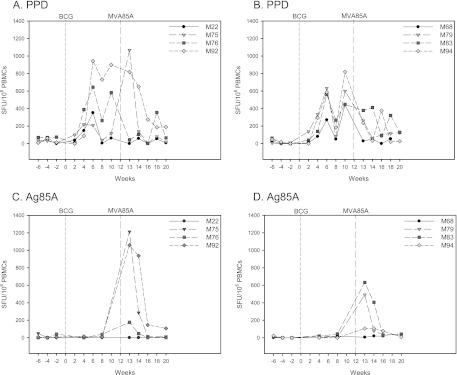
Immune response to vaccination in the peripheral blood. The frequency of vaccine-specific IFN-γ-secreting cells measured by ELISpot following BCG prime with MVA85A intradermal boost in peripheral blood mononuclear cells is shown in the panels on the left, and the panels on the right shows the frequencies following BCG prime with MVA85A aerosol boost. (A and B) Profiles of PPD-specific IFN-γ-secreting cells. (C and D) Profiles of Ag85A-specific IFN-γ-secreting cells. Vaccination with BCG at week 0 is indicated by the dotted line and vaccination with MVA85A indicated by the dashed line at week 12.
Fig 2.

Polyfunctional CD4+ and CD8+ T-cell response to vaccination in the peripheral blood. Polyfunctional CD4+ T-cell responses induced by BCG prime with MVA85A aerosol boost (A and B) and CD8+ T-cell responses (E and F) are shown, as are CD4+ T-cell responses induced by BCG prime/MVA intradermal boost (C and D) and CD8+ T-cell responses (G and H). (A, C, E, and G) Frequencies of T-cell populations responding to stimulation with PPD isolated from peripheral blood. (B, D, F and H) Functional profiles of T cells responding to PPD stimulation in cells isolated from the peripheral blood. The asterisk represents responses significantly higher than prevaccination levels (P < 0.05).
Double- and triple-positive CD8 T-cell populations were not induced following BCG or MVA85A vaccination, and single CD8+ T-cell populations were only detected sporadically and at low frequencies (Fig. 2F and H).
Evaluation of the T-cell response in bronchoalveolar lavage (BAL) samples induced by vaccination.
BCG vaccination induced increases in both the proportion of PPD-specific CD4+ T cells in the BAL samples capable of producing more than one cytokine and the frequency of specific triple-, double-, and single-positive CD4+ T cells from 4 weeks after BCG vaccination (Fig. 3) compared to prevaccination levels. Similarly, the frequency of PPD-specific CD4 T-cell populations peaked 1 week after vaccination with MVA85A (week 13) and then declined. Vaccination induced a number of differences in population frequency and proportion, although these were not significantly different between the two vaccination groups; however, these were significantly elevated compared to baseline responses in both groups. An increase in the proportion of double-positive (TNF-α+ IFN-γ+) and triple-positive (TNF-α+ IFN-γ+ IL-2+) PPD-specific cells compared to prevaccination levels was detected 4 weeks after BCG vaccination and was maintained throughout the study. One week after MVA85A vaccination, a higher frequency of PPD-specific single-positive IFN-γ-producing CD4 T cells (P = < 0.05), as well as a larger proportion of double-positive (TNF-α+ IFN-γ+) (P = < 0.05) and triple-positive (TNF-α+ IFN-γ+ IL-2+) PPD-specific cells (P = < 0.05), was seen in the group that received MVA85A by aerosol (Fig. 3B) compared to the intradermally vaccinated group (Fig. 3D). An increase in the frequency of single-positive IFN-γ-producing CD8+ T cells was detected 8 weeks postvaccination with BCG that further increased 1 week postvaccination with MVA85A (week 13 in both aerosol- and intradermally vaccinated animals). Vaccination did not alter the proportion of triple-, double-, or single-positive PPD-specific CD8+ T cells (Fig. 3). A small double-positive (IFN-γ+ TNF-α+) population was seen in the aerosol-vaccinated group at weeks 8 and 13 that was not detectable in the intradermally vaccinated group.
Fig 3.

Polyfunctional CD4+ and CD8 T-cell responses to vaccination in the lungs. Polyfunctional CD4+ T-cell responses induced by BCG prime with MVA aerosol boost (A and B) and CD8+ T-cell responses (E and F) are shown, as are CD4+ T-cell responses induced by BCG prime with MVA intradermal boost (C and D) and CD8+ T-cell responses (G and H). (A, C, E, and G) Frequencies of T-cell populations responding to stimulation with PPD in BAL. (B, D, F, and H) Functional profiles of T cells responding to PPD stimulation in cells in BAL. The asterisk represents responses significantly higher than prevaccination levels (P < 0.05).
Evaluation of the T-cell response induced by each route of vaccination in lymphoid tissues.
The frequencies of PPD- and Ag85A-specific IFN-γ-secreting cells were measured by ELISpot assay in a range of lymphoid tissues (spleen, bone marrow, lung, hilar, axillary, and inguinal lymph nodes) collected at necropsy. Responses detected in the tissues from animals that received MVA85A by aerosol were comparable to those for animals vaccinated intradermally (data not shown). However, there was a nonsignificant trend for the Ag85A-specific responses in the lungs from the aerosol group to be higher than those detected in the lungs from the intradermal group (P = 0.6625) (Fig. 4). In contrast, the response in the peripheral blood of the animals vaccinated intradermally was higher than that in the lungs.
Fig 4.
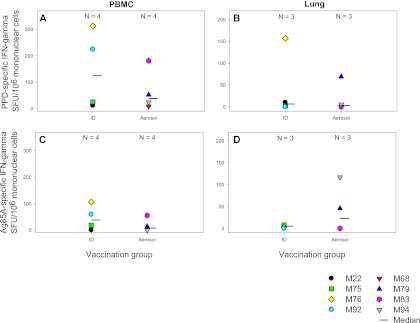
Antigen-specific immune responses measured in PBMCs and monocytes isolated from lung tissue at necropsy. Box plots display vaccination group medians and interquartile ranges of antigen-specific IFN-γ-secreting cells measured by ELISpot assay in mononuclear cells isolated from peripheral blood (A and C) and lung tissue samples collected at necropsy (B and D). Panels A and B show the profiles of PPD-specific IFN-γ-secreting cells, and panels C and D show the profiles of Ag85A-specific IFN-γ-secreting cells.
Evaluation of the humoral response to the vaccinia virus vaccine vector induced by each route of vaccination in peripheral blood.
Vector-specific IgG responses were detected in the serum collected from animals after intradermal vaccination with MVA85A. Responses were first detected 2 weeks after vaccination, with the peak increase in the response from prevaccination levels detected 4 weeks postvaccination (Fig. 5). In contrast, an anti-vaccinia virus antibody response was not detected in the serum samples from animals after aerosol vaccination with MVA85A.
Fig 5.

Vector-specific IgG response induced by vaccination with MVA85A measured by ELISA. The plot shows the median change from prevaccination level for the BCG prime with MVA85A intradermal boost group with the closed circles and for the BCG prime with MVA85A aerosol boost group with open triangles. Error bars show the standard deviations from the median. Vaccination with BCG at week 0 is indicated by the dotted line, and vaccination with MVA85A is indicated by the dashed line at week 12.
DISCUSSION
To be most effective, it would be advantageous for a vaccine to induce immune responses at the site where the pathogen is first encountered to allow a faster local response at the site. Importantly, in this study, the MVA85A was well tolerated by all the animals, with no adverse effects on any of the clinical parameters tested, and thus, aerosol vaccination with MVA85A was shown to be similar to that reported previously for other MVA- and (vaccinia virus strain) NYVAC-vectored vaccines (30).
This study has shown that MVA85A delivered by aerosol or by intradermal injection induced antigen-specific immune responses in all animals, either in the periphery or the lungs, or in both, with a trend towards the highest response when the compartment and route of delivery were matched. This is in line with the report from Corbett et al. (30), where aerosol delivery of a NYVAC-vectored HIV vaccine to rhesus macaques induced both systemic and mucosa-associated immune responses in the vaginal and rectal tissues that were comparable to those induced by intramuscular immunization. The ability of poxvirus-vectored vaccines delivered by the systemic route to induce responses in the mucosal immune compartment in macaques is in contrast to the observations made in mice, where the independent compartmentalization of the mucosal versus systemic immune systems has been clearly described (31) and where systemic immunization with vaccinia virus only induced cellular and antibody responses in the systemic lymphoid tissues. This difference in responses by the mouse and primate immune systems emphasizes the important role of the macaque in the evaluation of new vaccine approaches that are targeted for use in humans.
Live viruses, such as vaccinia virus and adenovirus, are widely used as delivery systems in the development of new vaccines; however, their use can be limited by preexisting immunity induced either by a prior natural infection (32, 33) or by previous vaccinations (34, 35). Approaches that might circumvent previous immunity would be extremely useful in the development of improved vaccine strategies. In this study, an antibody response to the MVA vector was detected in the serum samples of animals that received MVA85A by intradermal injection but not in the serum samples of animals that were vaccinated with MVA85A by aerosol. This parallels the report from Corbett et al. (30), where the systemic vector-specific IgG response detected after the first vaccination with the NYVAC-vectored vaccine was significantly higher in the animals that received the vaccine intramuscularly than in those that received the aerosol vaccination. This suggests that the delivery of vaccines to the lungs is an immunization strategy that limits the induction of systemic anti-vector immunity and provides the potential for multiple vaccinations with poxvirus-based strategies using mucosal, followed by systemic, delivery. This study demonstrated the feasibility of delivering a booster vaccination to the mucosal immune system by aerosol and, as mucosal administration of vaccinia virus-vectored vaccines has been shown previously to improve the immunogenicity of vaccinia virus-vectored vaccines (31), this also provides the potential to enhance the immunogenicity induced by a prime or boost vaccine regimen. Repeated vaccination with the same vaccine in multiple-boost regimens might in turn reduce the need for multiple formulations of the same vaccine antigens for prime or boost vaccination regimens; this might reduce the time and cost of their development. Use of the same vector system to deliver vaccines against different diseases might also become feasible without one vaccine limiting the immunity induced by the other. In addition, this approach might allow the use of commonly encountered vaccine vectors that induce immunity in large sectors of the population, such as human adenovirus 5 (AdHu5).
Aerosol vaccination offers many practical advantages over delivery via needles, and this study demonstrated that a single device can be used to vaccinate multiple subjects, which represents a highly cost-effective system for use in resource-poor settings. The feasibility of immunization by the inhalation of aerosolized vaccines for mass vaccination campaigns has been extensively evaluated for measles by the World Health Organization and was shown to be highly successful (36). The advantages associated with aerosol administration of TB vaccines are equally applicable to vaccines targeting not only other respiratory pathogens, such as influenza and pneumococcus, but also other pathogens, such as HIV, that enter via mucosal surfaces.
As the nebulizer used in the study is licensed for human use, the vaccination approach is therefore directly translatable to human vaccine delivery and these data support the evaluation of this promising route of vaccine delivery in a human clinical trial. Further studies are also required to evaluate the protective efficacy of this route of administration.
Supplementary Material
ACKNOWLEDGMENTS
This work was commissioned by the National Institute for Health Research. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
We thank the staff of the Biological Investigations Group. We also thank Graham Hall, Emma Rayner, and Geoff Pearson for histopathological analysis of tissues, Kim Hatch for histology support, Victoria Graham for assistance with the anti-vector serology, and Fergus Gleeson for analysis of the thoracic radiographs.
Footnotes
Published ahead of print 27 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00690-12.
REFERENCES
- 1. World Health Organization 2012. Global tuberculosis report 2012. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, Mosteller F. 1994. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 271:698–702 [PubMed] [Google Scholar]
- 3. Hesseling AC, Marais BJ, Gie RP, Schaaf HS, Fine PE, Godfrey-Faussett P, Beyers N. 2007. The risk of disseminated Bacille Calmette-Guerin (BCG) disease in HIV-infected children. Vaccine 25:14–18 [DOI] [PubMed] [Google Scholar]
- 4. Mayr A, Hochstein-Mintzel V, Stickl H. 1975. Abstammung eigenschaften und verwendung des attenuierten vaccinia-stammes MVA. Infection 105:6–14 [Google Scholar]
- 5. Sutter G, Moss B. 1992. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. U. S. A. 89:10847–10851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carroll MW, Moss B. 1997. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198–211 [DOI] [PubMed] [Google Scholar]
- 7. Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss BA. 1994. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 12:1032–1040 [DOI] [PubMed] [Google Scholar]
- 8. Hirsch VM, Fuerst TR, Sutter G, Carroll MW, Yang LC, Goldstein S, Piatak M, Jr, Elkins WR, Alvord WG, Montefiori DC, Moss B, Lifson JD. 1996. Patterns of virus replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV vaccine in modified vaccinia Ankara. J. Virol. 70:3741–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McShane H, Brookes R, Gilbert SC, Hill AV. 2001. Enhanced immunogenicity of CD4+ T-cell responses and protective efficacy of a DNA-modified vaccinia virus Ankara prime-boost vaccination regimen for murine tuberculosis. Infect. Immun. 69:681–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Williams A, Hatch GJ, Clark SO, Gooch KE, Hatch KA, Hall GA, Huygen K, Ottenhoff TH, Franken KL, Andersen P, Doherty TM, Kaufmann SH, Grode L, Seiler P, Martin C, Gicquel B, Cole ST, Brodin P, Pym AS, Dalemans W, Cohen J, Lobet Y, Goonetilleke N, McShane H, Hill A, Parish T, Smith D, Stoker NG, Lowrie DB, Källenius G, Svenson S, Pawlowski A, Blake K, Marsh PD. 2005. Evaluation of vaccines in the EU TB Vaccine Cluster using a guinea pig aerosol infection model of tuberculosis. Tuberculosis (Edinb.) 85:29–38 [DOI] [PubMed] [Google Scholar]
- 11. Verreck FAW, Vervenne RAW, Kondova I, van Kralingen KW, Remarque EJ, Braskamp G, van der Werff NM, Kersbergen A, Ottenhoff THM, Heidt PJ, Gilbert SC, Gicquel B, Hill AVS, Martin C, McShane H, Thomas AW. 2009. MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PLoS One 4:e5264 doi:10.1371/journal.pone.0005264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vordermeier HM, Dean GS, Rosenkrands I, Agger EM, Andersen P, Kaveh DA, Hewinson RG, Hogarth PJ. 2009. Adjuvants induce distinct immunological phenotypes in a cattle tuberculosis vaccine model. Clin. Vaccine Immunol. 16:1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. 2003. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guérin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J. Immunol. 171:1602–1609 [DOI] [PubMed] [Google Scholar]
- 14. Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, Zganiacz A, Hitt M, Xing Z. 2004. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 173:6357–6365 [DOI] [PubMed] [Google Scholar]
- 15. Kaufmann SH. 1996. gamma/delta and other unconventional T lymphocytes: what do they see and what do they do? Proc. Natl. Acad. Sci. U. S. A. 93:2272–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flynn JL, Chan J. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129 [DOI] [PubMed] [Google Scholar]
- 17. Beveridge NE, Fletcher HA, Hughes J, Pathan AA, Scriba TJ, Minassian A, Sander CR, Whelan KT, Dockrell HM, Hill AV, Hanekom WA, McShane H. 2008. A comparison of IFNgamma detection methods used in tuberculosis vaccine trials. Tuberculosis (Edinb.) 88:631–640 [DOI] [PubMed] [Google Scholar]
- 18. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. 1993. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178:2243–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jouanguy E, Lamhamedi-Cherradi S, Altare F, Fondanèche MC, Tuerlinckx D, Blanche S, Emile JF, Gaillard JL, Schreiber R, Levin M, Fischer A, Hivroz C, Casanova JL. 1997. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guérin infection and a sibling with clinical tuberculosis. J. Clin. Invest. 100:2658–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonecini-Almeida MG, Chitale S, Boutsikakis I, Geng J, Doo H, He S, Ho JL. 1998. Induction of in vitro human macrophage anti-Mycobacterium tuberculosis activity: requirement for IFN-gamma and primed lymphocytes. J. Immunol. 160:4490–4499 [PubMed] [Google Scholar]
- 21. Beveridge NE, Price DA, Casazza JP, Pathan AA, Sander CR, Asher TE, Ambrozak DR, Precopio ML, Scheinberg P, Alder NC, Roederer M, Koup RA, Douek DC, Hill AV, McShane H. 2007. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur. J. Immunol. 37:3089–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Capuano SV, III, Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, Bissel S, Fuhrman C, Klein E, Flynn JA. 2003. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 71:5831–5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McMurray DN. 2000. A nonhuman primate model for the preclinical testing of new tuberculosis vaccines. Clin. Infect. Dis. 30(Suppl 3):S210–S212 [DOI] [PubMed] [Google Scholar]
- 24. Barclay WR, Anacker RL, Brehmer W, Leif W, Ribi E. 1970. Aerosol-induced tuberculosis in sub-human primates and the course of the disease after intravenous BCG vaccination. Infect. Immun. 2:574–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barclay WR, Busey WM, Dalgard DW, Good RC, Janicki BW, Kasik JE, Ribi E, Ulrich CE, Wolinsky E. 1973. Protection of monkeys against airborne tuberculosis by aerosol vaccination with bacillus Calmette-Guerin. Am. Rev. Respir. Dis. 107:351–358 [DOI] [PubMed] [Google Scholar]
- 26. Chaparas SD, Good RC, Janicki BW. 1975. Tuberculin-induced lymphocyte transformation and skin reactivity in monkeys vaccinated or not vaccinated with Bacille Calmette-Guerin, then challenged with virulent Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 112:43–47 [DOI] [PubMed] [Google Scholar]
- 27. Good RC. 1968. Simian tuberculosis: immunologic aspects. Ann. N. Y. Acad. Sci. 154:200–213 [DOI] [PubMed] [Google Scholar]
- 28. Janicki BW, Good RC, Minden P, Affronti LF, Hymes WF. 1973. Immune responses in rhesus monkeys after bacille Calmette-Guerin vaccination and aerosol challenge with Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 107:359–366 [DOI] [PubMed] [Google Scholar]
- 29. Sharpe SA, McShane H, Dennis MJ, Basaraba RJ, Gleeson F, Hall G, McIntyre A, Clark S, Gooch K, Beveridge NE, Nuth E, White A, Marriott A, Dowall S, Hill AV, Williams A, Marsh PD. 2010. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clin. Vaccine Immunol. 17:1170–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corbett M, Bogers WM, Heeney JL, Gerber S, Genin C, Didierlaurent A, Oostermeijer H, Dubbes R, Braskamp G, Lerondel S, Gomez CE, Esteban M, Wagner R, Kondova I, Mooij P, Balla-Jhagjhoorsingh S, Beenhakker N, Koopman G, van der Burg S, Kraehenbuhl JP, Le Pape A. 2008. Aerosol immunization with NYVAC and MVA vectored vaccines is safe, simple, and immunogenic. Proc. Natl. Acad. Sci. U. S. A. 105:2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belyakov IM, Moss B, Strober W, Berzofsky JA. 1999. Mucosal vaccination overcomes the barrier to recombinant vaccinia immunization caused by preexisting poxvirus immunity. Proc. Natl. Acad. Sci. U. S. A. 96:4512–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lasaro MO, Ertl HC. 2009. New insights on adenovirus as vaccine vectors. Mol. Ther. 17:1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCoy K, Tatsis N, Korioth-Schmitz B, Lasaro MO, Hensley SE, Lin SW, Li Y, Giles-Davis W, Cun A, Zhou D, Xiang Z, Letvin NL, Ertl HC. 2007. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 81:6594–6604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, Huygen K, Fletcher HA, Hill AV. 2004. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat. Med. 10:1240–1244 [DOI] [PubMed] [Google Scholar]
- 35. Sharpe S, Polyanskaya N, Dennis M, Sutter G, Hanke T, Erfle V, Hirsch V, Cranage M. 2001. Induction of simian immunodeficiency virus (SIV)-specific CTL in rhesus macaques by vaccination with modified vaccinia virus Ankara expressing SIV transgenes: influence of pre-existing anti-vector immunity. J. Gen. Virol. 82:2215–2223 [DOI] [PubMed] [Google Scholar]
- 36. Low N, Kraemer S, Schneider M, Restrepo AM. 2008. Immunogenicity and safety of aerosolized measles vaccine: systematic review and meta-analysis. Vaccine 26:383–398 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.