

Abstract
We hypothesized that nucleophosmin (NPM), a nucleolar phosphoprotein, is critical for Bax-mediated cell death. To test this hypothesis, Bax activation was induced by metabolic stress. During stress, nucleolar NPM translocated into the cytosol, NPM-Bax complexes formed, and both NPM and Bax accumulated in mitochondria. Expression of a cytosol-restricted NPM mutant (NPM-ΔNLS), but not a nucleus-restricted NPM mutant, increased NPM-Bax complex formation, mitochondrial NPM and Bax accumulation, mitochondrial membrane injury, caspase 3 activation, and ischemia-induced cell death. Coexpression of NPM-ΔNLS with constitutively active Bax mutants caused nearly universal cell death in the absence of metabolic stress, whereas expression of active Bax or NPM-ΔNLS alone did not. A Bax peptide that disrupts NPM-Bax interaction significantly reduced cell death caused by exposure to metabolic inhibitors in vitro and preserved kidney function after ischemia in vivo. Thus, NPM-Bax interaction enhances mitochondrial Bax accumulation, organelle injury, and cell death. NPM-Bax complex formation is a novel target for preventing ischemic tissue injury.
INTRODUCTION
Although Bax toxicity is well characterized, the events that signal Bax translocation to mitochondria, a major target of Bax-mediated apoptosis, are unknown (1). Interestingly, more than 75% of Bax colocalizes with mitochondria during apoptogenic insults, whereas the remaining Bax translocates to nuclei and endoplasmic reticulum or exists unbound in the cytosol (2). Bax lacks a mitochondrial localizing sequence, and a mitochondrial Bax receptor has yet to be identified (3, 4), complicating efforts to dissect the molecular signals that drive intracellular Bax movement and toxicity. The possibility that a molecular chaperone facilitates mitochondrial Bax accumulation has been entertained recently (5, 6).
Several Bax chaperones have been proposed (7), including humanin, Ku-70, Bax interacting factor 1 (bif-1), clusterin, specific 14-3-3 isoforms, and nucleophosmin (NPM; also called B23). NPM is an abundant, ubiquitously expressed, 35-kDa phosphoprotein that localizes to the nucleolar region of resting cells (8). In healthy cells, NPM promotes RNA metabolism, as well as the packaging and transport of cell proteins, by reversibly shuttling between the cytosol and the nucleolar region (9, 10). NPM also migrates into the cytosol during cell division to promote ribosomal protein synthesis and cell proliferation before returning to the nucleolar region (5, 11, 12). These NPM functions are essential, since its elimination causes embryonic death in NPM knockout mice (11).
In cell-free systems, NPM interacts with Bax (6, 13). However, NPM only binds Bax that has undergone conformational change characterized by exposure of the amino-terminal 6A7 epitope (6, 14). The site responsible for binding NPM has been identified and localizes to the Bax carboxy-terminal domain (14). Furthermore, a synthetic, 21-amino-acid Bax peptide that includes this carboxy-terminal NPM binding site disrupts NPM-Bax interaction in a cell-free system (14). However, the role of NPM and NPM-Bax interaction during cell death is uncertain.
In order to interact with conformationally active Bax during stress, NPM must enter the cytosol. In healthy cells, well-characterized domains regulate NPM translocation from the nucleolar region to the cytosolic compartment. A nuclear export sequence (NES) controls NPM exit across the nuclear pore into the cytosol, whereas the nuclear localizing sequence (NLS) mediates nuclear uptake (15). Interestingly, NPM is highly susceptible to naturally occurring mutations that determine its location within cells. In humans with acute myelocytic leukemia, defects in the nuclear and cytosolic localizing sequences, as well as in NPM lysine residue 263, have been reported (16). In leukemic cells, NPM mutations that promote its cytosolic accumulation (e.g., deletion of the nuclear localizing sequence) enhance sensitivity to apoptogenic agents and improve patient prognosis. In contrast, mutations that restrict NPM to the nucleus (e.g., deletion of the nuclear export sequence or a single substitution mutation that precludes NPM sumoylation at lysine 263) promote resistance to radiation- or chemotherapy-induced apoptosis and worsen prognosis (15, 17, 18).
Based on these prior reports, we hypothesized that NPM, acting as a molecular chaperone, is critical for Bax-mediated mitochondrial injury and cell death after metabolic stress. This prediction was tested by interfering with several key steps that facilitate NPM-Bax interaction and potentially regulate cell death. In this report, we determined that stress promotes NPM translocation from the nucleus into the cytosol, where it binds to conformationally changed Bax. Neither expression of conformationally changed Bax nor cytosolic NPM accumulation alone is sufficient to cause cell death. In contrast, the combination of conformationally changed Bax and an NPM mutant that accumulates in the cytosol causes mitochondrial injury and cell death even in the absence of stress. During metabolic stress, diverse maneuvers that interfere with NPM-Bax complex formation improve cell survival or protect organ function. These findings strongly suggest that NPM-Bax interaction is a potential therapeutic target for ameliorating ischemic tissue ischemia, a pathological process that currently lacks an effective therapy.
MATERIALS AND METHODS
Animals.
All animals were maintained under the guidance and policies of the Boston University Animal Core facility, using NIH and IACUC guidelines. Male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were housed and bred at the Boston University Animal Core facility.
Cell culture.
Primary culture of murine proximal tubular cells was performed as previously described (19, 20). Briefly, 3-week-old mice were sacrificed, their kidneys were removed, and cortices were harvested. Individual cortical samples were minced and digested by collagenase IV (1 mg/ml) and then plated onto culture dishes in a renal epithelial cell growth medium (PromoCell, Heidelberg, Germany). Cultures of cells that exhibited proximal tubule characteristics were maintained for 5 to 7 days in 5% CO2 at 37°C (19, 20). HEK293NT lentiviral packaging cells (System Biosciences, Mountain View, CA) were maintained in Dulbecco's modified Eagle medium (DMEM) with high-glucose medium (Life Technologies BRL, Carlsbad, CA) containing 10% bovine serum and 10% penicillin-streptomycin at 37°C in a 5% CO2 incubator.
Metabolic stress.
Exposure to metabolic inhibitors is an established model of stress that reduces ATP content to <10% of baseline values until recovery is initiated (21–24). To achieve ATP depletion, cells were washed 3 times in glucose-free DMEM (containing 584 mg/liter l-glutamine, without pyruvate; Invitrogen, Carlsbad, CA), followed by incubation in glucose-free DMEM containing sodium cyanide (5 mM) and 2-deoxy-d-glucose (5 mM) for the indicated times. Recovery was initiated by replacing the ischemia medium described above with complete primary cell culture medium.
Male mice at an age of 4 weeks were subjected to bilateral renal pedicle clamping after being anesthetized with 2,2,2-tribromoethanol. A midline laparotomy and nontraumatic vascular clamp was placed on both renal pedicles for 25 min, an insult that produces severe, reversible acute kidney injury (25). The clamp was then removed and reperfusion was confirmed by visual inspection of the kidneys. Sham ischemia was performed by encircling each renal pedicle with a nonocclusive ligature.
Plasmids, cDNA, and cellular infections.
pCDH and pCDF lentivector systems (System Biosciences, Mountain View, CA) were used to generate wild-type NPM and Bax or their mutant constructs using mouse NPM and human Bax cDNA (cDNA plasmid; Addgene, Cambridge, MA) as the template. Mutagenic primers for generating these mutants were commercially synthesized (Table 1; also see Fig. 3A). PCR-based mutagenesis was used to generate both WT and mutant proteins. Flag tag was fused to the 3′ open reading frame (ORF) for each NPM construct, and a V5 tag was fused to the 5′ ORF for each Bax construct. NPM PCR products were cloned into pCDH vector BamHI/EcoRI sites, whereas Bax PCR products were cloned into pCDF vector XbaI/EcoRI sites. A series of the NPM mutants was created that included strains ΔNLS, ΔNES, ΔNuls K263R Mut, A mut, and C mut (Table 1) (15–18). A series of nonphosphorylatable and phosphomimetic Bax mutants was either created at a single site (i.e., S163 or S184) or combined and included S163A, S184A, S163E, S184E, S163E/S184A (EA), S163A/S184E (AE), S163A/S184A (AA), and S163E/S184E (EE). All constructs were confirmed by DNA sequencing prior to cotransfection with pPACKG1 and pPACKF1 lentivector packaging plasmids into HEK293NT cells to generate lenti-pseudoviral particles. The resultant pseudoviruses were harvested and purified, and titers were determined according to the manufacturer's instructions. Cells were plated for 24 h prior to each experiment and before exposure to virus (either empty vector, wild type, or mutant; multiplicities of infection [MOI] of 5 to 9) diluted in Opti-MEM (Invitrogen, Carlsbad, CA). After 6 h of incubation with virus-infected medium, a complete culture medium was substituted for an additional 16-h incubation period.
Table 1.
PCR primers for the NPM and Bax mutantsa
| Primer name | Sequence (5′–3′) |
|---|---|
| NPM (BamHI-EcoRI) | |
| 5′ Flag WT Fw | GAGCTCGGATCCGCCACCATGGACTACAAGGATGACGAGAAGATTCGATGGACATGGA |
| 3′ Rev WT | GGATATCTGCAGAATTCTTAAAGAGACTTCCT |
| ΔNLS Fw | GCGCCACCAGTCTTAAGCTTGCGAGATACTCCAGCCAAAAAT |
| ΔNLS Rev | ATCTCGCAAGCTTAAGACTGGTGGCGCTTTTTCTTCAGCTTC |
| ΔNES Fw | GAAAAAGCTTTTAGCGTTGAAGTGTGGTTCAGGGCC |
| ΔNES Rev | CGCTAAAAGCTTTTTCTGTGGTATTTCAAAGCCCCCAAGG |
| K263R Mut | TGCCAGAGATCTTGAATAGCCTCTTGGTCAGTCATCCGGAAGCAATTCTTCACATAATTGATGAATTTGGCTTCCACTCTGGGAAGAGAACCAC |
| Bax (XbaI-BamHI-EcoRI) | |
| V5 tag Fw | GAAGATTCTAGAGCCACCATGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGAAGCTTATGGACGGGTCCGGGGAGCA |
| 163A Fw | CGCGCCGGATCCAAGACCAGGGTGGTTGGGACGGCCTCCTCGCCTACTTTGGGACGCCCA |
| 163E Fw | CGCGCCGGATCCAAGACCAGGGTGGTTGGGACGGCCTCCTCGAATACTTTGGGACGCCCA |
| 184A Rev | GAGCTCGAATTCAGCCCATCTTCTTCCAGATGGTGAGTTCGGCGGTGAGCACTCCCGCCA |
| 184E Rev | GAGCTCGAATTCAGCCCATCTTCTTCCAGATGGTGAGGGCGGCGGTGAGCACTCCCGCCA |
| WT Rev | GAGCTCGAATTCAGCCCATCTTCTTCCAGATGGT |
Shown is a list of forward (Fw) and reverse (Rev) primers used for generating wild-type (WT) NPM and Bax mutants using standard PCR (see Materials and Methods), including cytosol-restricted NPM mutant (NPM-ΔNLS) and nucleus-restricted NPM mutants NPM-ΔNES and NPM-K263R. Bax Ser163 is the GSK3β-regulated site. Bax Ser184 is the Akt-regulated site. V5 is an epitope tag. A and E refer to point mutations that render Bax either constitutively active or inactive, respectively, at specific serine sites.
Antibodies.
Antibodies directed against NPM, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Sigma-Aldrich, St. Louis, MO), conformationally changed (i.e., 6A7 epitope-exposed) Bax and total Bax (Trevigen, Gaithersburg, MD), F1F0 ATPase (complex V β subunit; Invitrogen, Carlsbad, CA), and cytochrome c and caspase 3 (Clontech, Mountain View, CA) were purchased from commercial sources. Immunofluorescence was performed with secondary antibodies conjugated to Alexa Fluor 488 (Invitrogen), and immunoblot analyses were performed with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad).
RNA interference.
Small interfering RNA (siRNA) kits were used to deplete Bax (Cell Signaling Technology, Danvers, MA) and NPM (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer's protocol.
NPM-Bax blocking peptide.
For in vitro and in vivo experiments, peptides containing a cell-penetrating sequence as well as the region containing the Bax domain responsible for binding NPM or a nonspecific sequence (control) were synthesized (Biomatik, Wilmington, DE). The amino acid sequence of Bax-ΔNLS was Ac-KKKRKV-(βA)-TVTIFVAGVLTASLTIWKKMG-COOH (where β-A represents β-alanine). For peptide exposure during metabolic stress in vitro, MPT cells were plated in 6-well dishes. Cells were treated with peptide (1 mg/well) 30 min before and immediately after ATP depletion. For peptide administration during ischemia in vivo, 3-week-old mice weighing 12 to 15 g were used to perform 25-min ischemia reperfusion studies under standardized conditions as recently described (26). An NPM-Bax blocking or control peptide (100 μg/g of body weight) was administered intraperitoneally to mice 30 min before renal ischemia and again after 30 min of reperfusion. The serum blood urea nitrogen (BUN) level, a reliable marker of kidney function in this model (26), was serially measured in tail vein blood samples for up to 4 days after injury using a QuantiChrom BUN assay kit (BioAssay Systems, Hayward, CA).
Cell viability.
In this injury model, exposure to metabolic inhibitors activates Bax and causes apoptotic cell death with minimal necrosis (26–30). Cell viability, a surrogate for apoptosis in this model (31), was assayed using a modified colorimetric technique based on the ability of viable cells to convert 3-(4,5 dimethylthiazol)-2,5-diphenyl tetrazolium bromide (MTT) into purple formazan crystals (26, 29). The MTT assay positively correlates with the severity of apoptosis in cells exposed to metabolic inhibitors (31). The number of surviving cells is expressed as a percentage of viable control cells detected at baseline.
Protein isolation and immunoblot analysis.
Soluble cell protein was extracted with NP-40 buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.4% NP-40) containing a protease inhibitor cocktail (Set I; Calbiochem, San Diego, CA). Mitochondrial fractions were obtained and their purity confirmed as previously described (32). Briefly, cells were harvested in isotonic mitochondrial buffer containing protease inhibitor and homogenized by strokes in a dounce homogenizer. Lysates were centrifuged at 500 × g for 5 min to remove unbroken cells and nuclei. The membrane fraction pellets were solubilized in radioimmunoprecipitation assay (RIPA)-EDTA buffer containing protease inhibitor cocktail. The mitochondrial fraction was extracted by freezing and thawing on ice. Kidney cortical proteins were extracted from tissue homogenates in NP-40 buffer using an Ultra-Turrax disperser (IKA, Wilmington, NC) followed by sonication and centrifugation of samples as described above. Apoptosis-inducing factor (AIF) and cytochrome c, present in the cytosol and nucleus, were extracted from intact cells by permeabilizing the plasma and nuclear membranes with high-dose digitonin (Sigma) containing buffer (50 μg/ml digitonin, 145 mM NaCl, 11 mM KCl, 75 mM Tris-HCl, pH 7.4) to which a protease inhibitor cocktail was added (27), allowing proteins in the nonmitochondrial compartment to be released into the buffer to be quantified. NPM accumulation was measured in cytosolic samples extracted with low-dose digitonin (8 μg/ml). Cytosolic proteins were extracted from renal cortical samples using a commercially available tissue protein extraction kit (Biochain Institute, Newark, CA) by following the manufacturer's instructions. Briefly, 50 to 100 mg of kidney cortex tissue was minced on ice and spin washed with 1 ml ice-cold wash buffer prior to adding 100 to 200 μl of buffer C. The mixture was rotated at 4°C for 20 min and then centrifuged at 18,000 × g at 4°C for 20 min before being harvested. Protein levels were measured using the bicinchoninic acid (BCA) assay (Thermo Scientific, Rockford, IL). Total protein expression was determined using standard Western blotting.
Coimmunoprecipitation.
Cells were grown to confluence in 60-mm dishes prior to ATP depletion and recovery. The cells were lysed in 0.5 ml of PD buffer (40 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.1% Nonidet P-40, 6 mM EGTA) containing a protease inhibitor cocktail. The supernatant was immunoprecipitated overnight with specific antibodies against mouse Bax in the presence of protein A/G-agarose beads at 4°C. The immunoprecipitated complex was washed three times with PD buffer. The samples were fractionated on 15% SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and subjected to immunoblotting with antibodies specific for Bax or NPM.
Immunohistochemistry.
Cells were cultured on slides (MatTek Corp, Ashland, MA), stained with MitoTracker Green FM (200 nM; Invitrogen, Carlsbad, CA) for 1 h, and then fixed with 4% paraformaldehyde. Immunohistochemistry to detect NPM was performed according to the manufacturer's protocol (Sigma-Aldrich, St. Louis, MO).
Densitometry.
After digitizing each immunoblot image (Desk Scan II; Hewlett-Packard), selected band densities were quantified using NIH Image J software. Data are expressed as the means ± standard errors (SE).
Statistical analysis.
Data are expressed as means ± SE. Differences between groups were determined by a 2-tailed Student's t test. Comparisons involving more than two groups were determined by a 2-way analysis of variance (ANOVA), followed by a Holm-Sidak post hoc test for nonparametric data. P < 0.05 was considered significant.
RESULTS
Exposure to metabolic inhibitors caused dramatic NPM redistribution in primary renal epithelial cells, a major target of ischemic stress (33, 34). In resting cells, NPM was detected only in the nucleolar region (Fig. 1A, left). In contrast, stress caused marked NPM redistribution in many cells: nucleolar content decreased, and NPM became more widely distributed within nuclei and accumulated in the cytosolic compartment (Fig. 1A, middle). During recovery, NPM content decreased in both the nuclear and cytosolic compartments and returned to nucleoli, suggesting that NPM translocation is regulated. To quantify NPM redistribution into the cytosol, NPM was assessed by immunoblot analysis. As noted for intact cells by immunohistochemistry, NPM dramatically accumulated in cytosolic fractions harvested from cells subjected to metabolic stress in vitro, although total cell NPM content remained constant (Fig. 1B). A 25-fold increase in cytosolic NPM content was detected after in vitro stress (Fig. 1C). After 60 min of recovery, cytosolic NPM content remained more than 5-fold greater than baseline levels (i.e., prior to stress). At this same time point, NPM was no longer detected in the cytosol of intact cells by immunohistochemistry, suggesting that immunoblot analysis is more sensitive for quantifying stress-induced NPM translocation into the cytosol. In addition to causing cytosolic NPM accumulation, metabolic stress markedly increased NPM-Bax interaction, detected by immunoprecipitation, that persisted for at least 1 h thereafter (Fig. 2).
Fig 1.

(A to C) Effect of metabolic stress on intracellular NPM distribution. (A) At baseline, intact cells stained with NPM antibody show prominent nucleolar staining (open arrow). In many cells, stress (ATP Deplete) induces NPM redistribution into the cytosol (closed arrow), whereas in other cells, nucleolar to nuclear NPM redistribution is detected (open arrow). During recovery, NPM reaccumulates in nucleoli (open arrow). (B) NPM redistribution into the cytosolic fraction in cells permeabilized with low-dose digitonin (upper) (see Materials and Methods) and total NPM (lower), measured by immunoblot analysis. GAPDH and β-actin were used as loading controls. (C) Densitometric analysis of four separate immunoblot studies. *, P < 0.05 versus the baseline; n = 5 separate studies. CTL, control.
Fig 2.
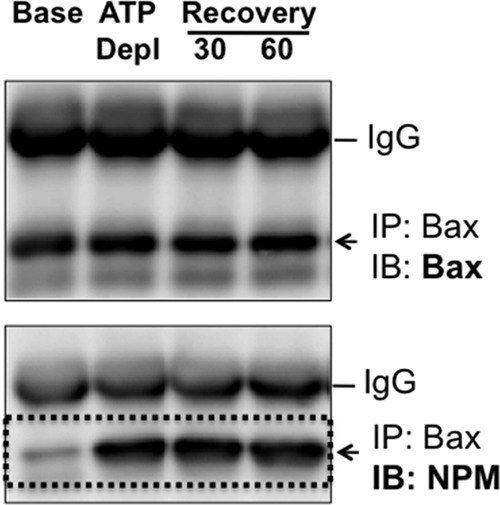
Effect of metabolic stress on NPM-Bax interaction. Immunoprecipitates (IP) were harvested from cells using an antibody directed against Bax at baseline (Base), immediately after 60 min of ATP depletion (ATP Depl), and after 30 or 60 min of recovery (Recovery) and then probed for Bax (upper) or NPM (lower) content. Immunoglobulin G (IgG) is shown as a control. Studies were performed on 400-μg cell lysate samples. IB, immunoblot.
To determine whether stress-induced NPM translocation is regulated or due to disruption of the nuclear membrane, epitope-tagged NPM mutants observed in human with myelogenous leukemia (15, 17, 18, 35–37), with defects in the sequences known to regulate its movement, were constructed and introduced into cells using lentiviral vectors (Fig. 3A). Equivalent expression of each epitope-tagged mutant was confirmed by immunoblot analysis of cell lysates (Fig. 3B). Compared to the wild type, NPM mutants that lacked the export sequence (NPM-ΔNES) or were altered by point mutation at the sumoylation site required for nuclear export (NPM-K263R) (38) localized exclusively within nuclei of cells both at baseline and during stress (Fig. 4A). Thus, stress-induced NPM exit from the nucleus requires a functional export sequence and an intact sumoylation site at residue 263. Interestingly, only the cytosol-restricted NPM mutant lacking the nuclear localization sequence (NPM-ΔNLS) significantly increased cell death after stress (Fig. 4B). NPM-ΔNLS expression also markedly increased both mitochondrial accumulation of NPM and Bax (Fig. 5A) and leakage into the cytosol of apoptosis inducing factor (AIF) and cytochrome c (Fig. 5B and quantified in Fig. 5C), key mediators of apoptosis that normally reside in the mitochondrial intramembranous space. The increase in cytochrome c leakage in cells that expressed the cytosol-restricted NPM mutant was associated with a doubling in stress-induced caspase 3 activation (Fig. 5D and E). In these studies, death caused by stress did not significantly differ between nontransfected cells and cells transfected with wild-type NPM (data not shown).
Fig 3.
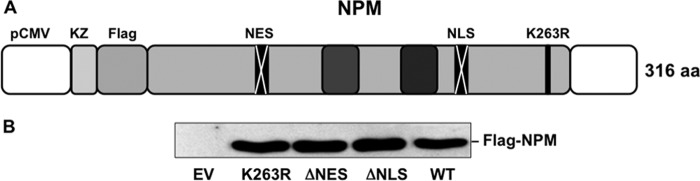
(A and B) NPM mutant expression. (A) Diagram of Flag-NPM constructs cloned into lentiviral vectors. The diagram indicates key sites that regulate intracellular NPM localization. Deletion of the NLS prevents nuclear NPM entry, and deletion of the NES prevents NPM from exiting nuclei. A single-amino-acid substitution at lysine 263 (K263R) prevents NPM sumoylation, which is required for nuclear NPM export. (B) Expression of wild-type NPM and mutants was confirmed by immunoblot analysis using an anti-Flag antibody.
Fig 4.
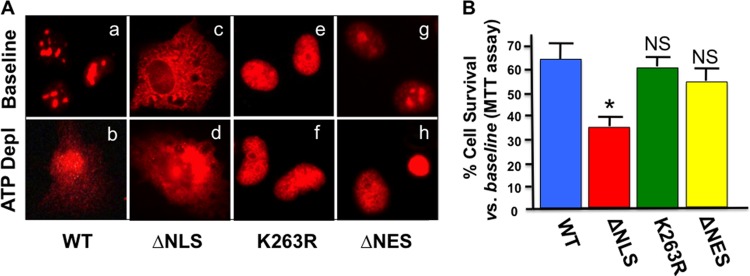
(A and B) Distribution of wild-type NPM and mutants in stressed and nonstressed cells. (A) NPM distribution in intact primary cells that express wild-type (WT) NPM or mutants lacking either the NLS (ΔNLS), the sumoylation site (K263R), or the NES (ΔNES) before (Baseline) or after (ATP Depl) stress; cells were stained with anti-Flag, antirhodamine secondary antibody. (B) Effect of wild-type NPM and mutant expression on the percentage of cell survival after metabolic stress compared to the baseline using the MTT assay (see Materials and Methods). n = 3 to 4 separate experiments; *, P < 0.05 compared to the wild-type baseline. NS, survival not significantly different from that of the wild type (P > 0.05).
Fig 5.

(A to E) Effect of wild-type NPM or NPM mutant expression on stress-induced mitochondrial NPM and Bax accumulation and outer mitochondrial membrane injury. (A) Immunoblot analysis of NPM (upper) and Bax (lower) accumulation in the mitochondrial fraction of cells expressing either empty vector (EV), NPM lacking the nuclear localizing sequence (NPM-ΔNLS), or wild-type NPM (NPM-WT) at baseline (Base), after 60 min of ATP depletion (ATP), or after 20 min of recovery (Rec). F1F0-ATPase, an intrinsic outer membrane protein, was used as the loading control. (B) Nonmitochondrial AIF and cytochrome c (Cyto-C) in cells before and after stress (assessed in the presence of high-dose digitonin; see Materials and Methods), with GAPDH as a loading control. (C) Densitometric analysis of 3 to 4 separate immunoblot studies performed as described for panel B. (D) Immunoblot analysis of active caspase 3 (17- and 19-kDa bands) in lysates harvested from cells that express either empty vector (EV), NPM lacking the nuclear localizing sequence (NPM-ΔNLS), or wild-type NPM (NPM-WT) at baseline (Base), immediately after 60 min of ATP depletion (ATP), and after 60 min of recovery (Rec). (E) Densitometric analysis of 3 to 4 separate immunoblot studies performed as described for panel D.
To establish a causal role for NPM in Bax-mediated cell death, several experimental maneuvers that interfere with the NPM-Bax complex formation were performed. First, exposure to Bax siRNA sufficient to decrease Bax content (Fig. 6B) also rescued cells from the toxicity caused by the cytosol-restricted NPM mutant (Fig. 6A), suggesting that cytosolic NPM facilitates Bax-mediated cell death. Second, partial NPM depletion by >60% with siRNA (Fig. 7A) significantly improved cell survival after metabolic stress compared to either nonspecific siRNA or vehicle alone (Fig. 7B).
Fig 6.
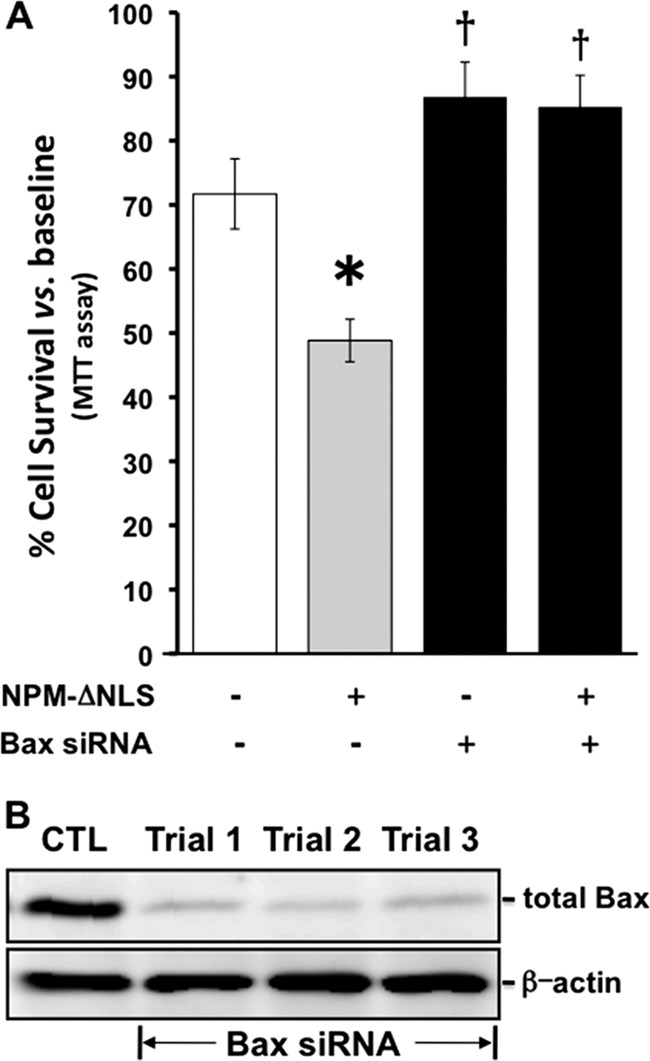
(A and B) Effect of Bax siRNA on Bax expression and survival after metabolic stress in cells that express NPM lacking the nuclear localizing sequence. (A) Percent survival after metabolic stress in cells that express the toxic NPM mutant (NPM-ΔNLS) in the presence and absence of specific (Bax siRNA +) or control (Bax siRNA −) siRNA. *, P < 0.05 versus the wild type; †, P < 0.05 versus NPM-ΔNLS. (B) Bax content in the lysates of cells exposed to Bax siRNA. Trials 1 to 3 represent three separate siRNA experiments.
Fig 7.
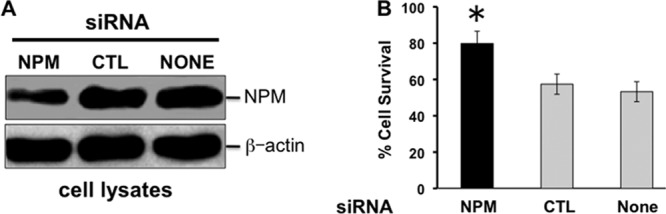
(A and B) Effect of NPM siRNA on NPM expression and cell survival after metabolic stress. (A) Effect of NPM or control (CTL) NPM siRNA versus no siRNA (None) on NPM expression in primary renal cells. β-Actin was used as a loading control. (B) Cell survival after metabolic stress. *, P < 0.05 versus CTL or no siRNA; n = 3 to 4 separate experiments.
To assess the role of Bax conformational changes on cell death, the wild type or one of eight epitope-tagged Bax mutants was expressed. These mutants have alterations at the Bax serine residues that are specifically acted on by Akt and/or glycogen synthase kinase 3β (GSK3β) (serines 164 and 183, respectively), rendering Bax either conformationally active or inactive (39, 40). In the absence of exogenous stress (i.e., at baseline), survival exceeded 80% in control cells that expressed an empty vector, wild-type Bax, or one of several Bax mutant proteins (Table 2). However, death was nearly universal (80 to 100%) in nonstressed cells that coexpressed the cytosol-restricted NPM mutant (NPM-ΔNLS) together with any of the constitutively active Bax Ser184 mutants that cannot be phosphorylated by Akt (i.e., Bax Ser184A mutants 3, 7, and 8) (Table 2). In contrast, coexpression of NPM-ΔNLS with Ser163A-mutated Bax, constitutively activated at the site regulated by GSK3β, caused minimal cell toxicity (Bax mutant 6) and failed to increase death when combined with the toxic Ser184A mutation (Bax mutant 8).
Table 2.
Effect of cytosol-restricted NPM and/or Bax mutants on cell survivala
| No. | Bax type | V5-Bax alone | V5-Bax + NPM NPM-ΔNLS | Bax Ser163 regulated by GSK3β | Bax Ser184 regulated by Akt |
|---|---|---|---|---|---|
| 1 | Control | + | + | NM | NM |
| 2 | Wild type | + | ++ | NM | NM |
| 3 | 184A | + | ++++ | NM | Inactive |
| 4 | 184E | −/+ | + | NM | Active |
| 5 | 163E | −/+ | + | Active | NM |
| 6 | 163A | −/+ | ++ | Inactive | NM |
| 7 | 163A_184A | +/++ | ++++/+++++ | Inactive | Inactive |
| 8 | 163E_184A | ++ | ++++/+++++ | Active | Inactive |
| 9 | 163A_184E | −/+ | + | Inactive | Active |
| 10 | 163E_184E | −/+ | + | Active | Active |
A cytosol-restricted NPM mutant (NPM-ΔNLS) and/or V5-tagged wild-type Bax, or mutants with alterations at specific serine residues regulated by Akt (Ser184) or GSK3β (Ser163), were coexpressed in cells in the absence of exogenous stress. Each plus sign represents 20% cell death as evaluated by a blinded observer; n = 3 separate experiments. The control was an empty vector. NM, not mutated.
To directly assess the pathogenic role of NPM-Bax interaction, cells were exposed to a novel synthetic peptide that contained a cell-permeable sequence (41) linked to the 20-amino-acid Bax C-terminal sequence that includes the NPM binding domain (14). Exposure to this NPM-Bax blocking peptide significantly reduced stress-induced cell death (Fig. 8A). Despite equivalent conformational Bax activation (as detected by a 6A7 epitope-specific Bax antibody), exposure to the NPM-Bax blocking peptide markedly reduced both cytochrome c leakage and caspase 3 activation during stress (Fig. 8B) and significantly improved cell survival (Fig. 8A).
Fig 8.
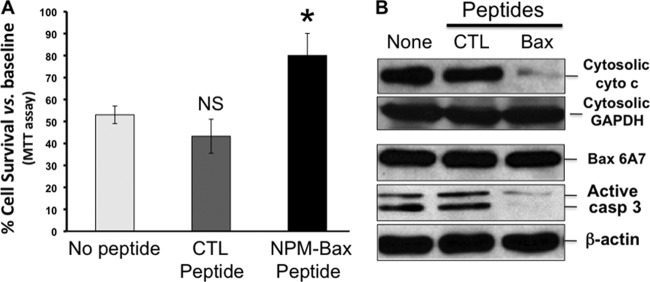
(A and B) Effect of an NPM-Bax blocking peptide on cell survival after metabolic stress. (A) Effect of a cell-permeable NPM-Bax peptide added to the medium 1 h before, during, or after subjecting proximal tubule cells to 60 min of ATP depletion on cell survival versus no peptide or a control (CTL) peptide. *, P = 0.08 versus CTL or no siRNA peptide; n = 3 to 4 separate experiments. (B) Immunoblot analyses of cytochrome c leakage (Cytosolic cyto c), active Bax content (Bax 6A7), and active caspase 3 (active Casp 3) after stress. GAPDH and β-actin were used as loading controls.
In order to evaluate the pathogenic role of NPM in ischemic tissue injury, mice were subjected to bilateral renal pedicle clamping, an insult associated with Bax conformational change, apoptosis, and organ failure (26). In vivo tissue ischemia caused a 4-fold increase in cytosolic NPM accumulation (Fig. 9A and B). Intraperitoneal injection of only 2 doses of the NPM-Bax blocking peptide into mice significantly protected kidney function on days 1 to 3 after ischemia (Fig. 10).
Fig 9.
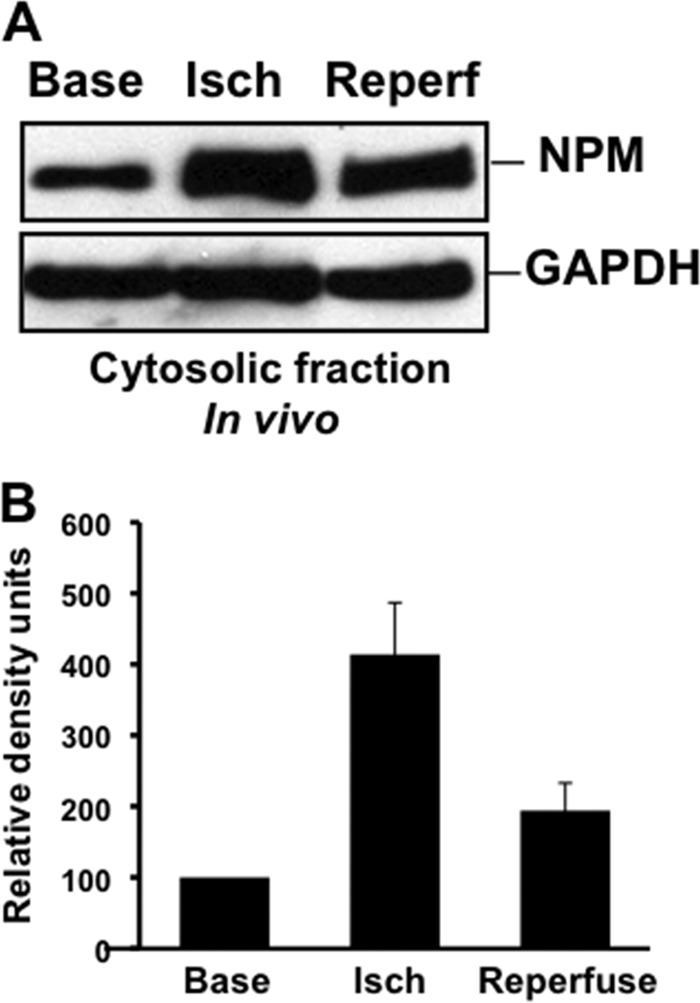
(A and B) Effect of renal ischemia in vivo on cytosolic NPM translocation. (A) NPM content in the cytosolic fraction of renal cortex at baseline (Base), immediately after 30 min of ischemia (Isch), and after 15 min of reperfusion (Reperf). GAPDH was used as a loading control. (B) Densitometric analysis of 3 to 4 separate immunoblot studies performed as described for panel A.
Fig 10.
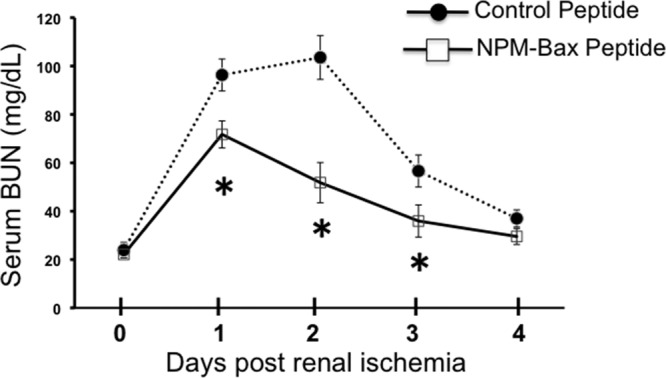
Effect of a Bax blocking peptide on kidney function after renal ischemia in vivo. Shown is the effect of a cell-permeable NPM-Bax blocking peptide (NPM-Bax peptide), versus a nonspecific (control) peptide, on kidney function (BUN) on days 1 to 4 after 25 min of ischemia; 100 μg/g peptide was administered 30 min before and again 30 min after ischemia; n = 6 mice per group; *, P < 0.05 versus control peptide.
DISCUSSION
If NPM acts as a key Bax chaperone, then several intracellular events are expected to occur: (i) nucleolar NPM will enter the cytosol during stress; (ii) cytosolic NPM will interact with conformationally active Bax; and (iii) maneuvers that limit NPM-Bax complex formation (e.g., depleting NPM or Bax, reducing cytosolic NPM accumulation, decreasing conformational Bax change, or preventing NPM-Bax complex formation) will ameliorate stress-induced mitochondrial membrane injury and cell death. In fact, data derived from current experiments strongly support each of these predictions, showing that NPM is critical for Bax-mediated cell death.
First, the present study shows that both in vitro (Fig. 1A) and in vivo (Fig. 9A), stress causes a fraction of nuclear NPM to enter the cytosol. Interestingly, cytosolic NPM accumulation alone is insufficient to cause cell death. This is not unexpected, since NPM promotes protein synthesis and cell proliferation by shuttling between the nucleus and the cytosol (9, 10) and accumulates in the cytosol during cell division, when the nuclear membrane is transiently disrupted (data not shown). Similarly, expression of a cytosol-restricted NPM is also harmless in the resting cells (Fig. 4). In contrast, both nucleus-restricted NPM mutants failed to translocate during stress (Fig. 4A), demonstrating that cytosolic NPM accumulation is not the result of nonselective disruption of the nuclear membrane. Similarly, NPM rapidly reenters the nucleolar region during recovery in surviving cells (Fig. 1A), a process regulated by the nuclear localizing sequence (Fig. 4A) (14, 15). How NPM is released from the nucleus during stress is still debated. Both conformationally active Bax (42) and RelA, a key component of the NF-κB complex (43), have been implicated in causing NPM translocation during apoptogenic insults. Regardless of the mechanism responsible for NPM translocation during stress, it is clear that cytosolic NPM accumulation per se is nontoxic.
On the other hand, if NPM localization is an important determinant of cell death, then expressing translocation-defective NPM mutants first characterized in leukemic patients (15, 17, 18, 35) would be expected to modify cell injury caused by metabolic stress. In fact, the cytosol-restricted NPM mutant (NPM-ΔNLS) significantly increased cell death after stress compared to wild-type NPM or either of two nucleus-restricted NPM mutants (NPM-ΔNES and NPM-K263R) (Fig. 4B). Compared to empty vector or wild-type NPM, NPM-ΔNLS significantly increased mitochondrial NPM and Bax accumulation by 3- to 4-fold (Fig. 5A), enhanced leakage of proapoptotic AIF and cytochrome c, apoptotic mediators normally restricted to the intramembranous mitochondrial space by 10-fold (Fig. 5B and C), and doubled caspase 3 activation (Fig. 5D and E). These observations implicate cytosolic NPM accumulation as a key determinant of mitochondrial Bax accumulation and cell death after stress. Furthermore, NPM translocation into the cytosol is a regulated process that requires both the NPM nuclear export sequence and site-specific NPM sumoylation.
Since cytosol-restricted NPM expression is not sufficient to injure resting cells but is highly toxic following stress, NPM likely requires a cofactor, such as Bax, to commit cells to apoptosis. After its arrival at the mitochondrial surface, Bax oligomerizes and inserts into the outer membrane, creating pores that leak apoptogens, such as apoptosis-inducing factor (AIF) and cytochrome c, into the cytosol (27, 44). In the present study, in vitro stress markedly increases the interaction between NPM and conformationally changed Bax (Fig. 2) and increases the mitochondrial accumulation of both NPM and Bax (Fig. 5A). If cytosolic NPM enhances Bax-mediated toxicity, then Bax depletion should limit death caused by expressing the cytosol-restricted NPM mutant. Indeed, partial Bax depletion rescued cells from toxicity caused by NPM-ΔNLS (Fig. 6A). Similarly, siRNA-mediated NPM depletion (Fig. 7A) significantly improved cell survival after metabolic stress (Fig. 7B). These results indicate that together, NPM and Bax cause death in cells that are highly susceptible to ischemic injury (33, 34). How NPM induces mitochondrial Bax accumulation is unknown, since NPM (like Bax) lacks a known mitochondrial localizing sequence.
Several laboratories have suggested that site-specific serine phosphorylation by stress kinases, including Akt and GSK3β, mediate conformational change and activation that precede Bax translocation to mitochondria (39, 40). Interestingly, activity of these stress kinases is altered during in vitro and in vivo ischemia in a manner expected to promote conformational Bax change (26, 45, 46). Specifically, either a decrease in Akt-mediated phosphorylation at Bax serine184 or a gain of phosphorylation at serine163 by GSK3β has been shown to be sufficient to change Bax conformation and cause cell death (26, 39, 40). To determine the extent to which conformational Bax activation contributes to cell toxicity caused by the cytosol-restricted NPM mutant (NPM-ΔNLS), Bax mutants with alterations at two key serine phosphorylation sites that regulate its activation were expressed and then introduced into cells. Only Bax mutants inactivated at the Akt-sensitive serine phosphorylation site (S184A) caused nearly universal cell death in cells that coexpressed NPM-ΔNLS (Table 2). In contrast, alterations at the Ser163 phosphorylation site alone were nontoxic and did not further increase cell death caused by the S184A mutation, which results in constitutive Bax activation at the Akt site. Our observation is consistent with a recent report showing that the Akt-specific Ser184 site is primarily responsible for Bax conformational change and cell death (47).
Finally, to directly implicate the NPM-Bax complex in stress-induced cell death, a permeable peptide linked to the Bax C-terminal region that binds NPM (14) was generated, and a total of two doses were introduced into cells before and after metabolic stress. Despite an equivalent increase in Bax conformational change, the NPM-Bax blocking peptide significantly decreased cytochrome c leakage and caspase 3 activation (Fig. 8B) and improved cell survival after metabolic stress (Fig. 8A). Thus, protection by the peptide occurs downstream of Bax conformational change, prompting us to investigate the role of NPM and NPM-Bax interaction during ischemic tissue injury in vivo.
Marked NPM translocation accompanied in vivo tissue injury in mice subjected to transient renal hypoperfusion (Fig. 9), an established model of ischemic tissue injury (25, 26). Furthermore, intraperitoneal administration of the NPM-Bax blocking peptide significantly reduced the level of BUN, a reliable marker of kidney function (25, 26, 28), on the first 3 days following the ischemic insult (Fig. 10), long after administration of the blocking peptide. Although peptides administered to cells or animals typically have a short half-life, measured in minutes, peptides bound to intracellular ligands exhibit half-lives as long as 12 h (48). Although the Bax peptide used in the present study interferes with NPM-Bax interaction (14), the cellular target(s) that mediates protection against renal ischemia requires further investigation. Taken together, our in vitro and in vivo observations demonstrate that NPM is a critical Bax chaperone that enhances mitochondrial membrane injury and cell death during metabolic stress. Since the NPM-Bax complex persists after the insult, its disruption may be an effective target for treating ischemic tissue injury.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DK-53387 (S.C.B.), DK-090143 (A.H.), and DK-090558-01 (R.B.).
Footnotes
Published ahead of print 4 March 2013
REFERENCES
- 1. Sharpe JC, Arnoult D, Youle RJ. 2004. Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta 1644:107–113 [DOI] [PubMed] [Google Scholar]
- 2. Valentijn AJ, Upton JP, Bates N, Gilmore AP. 2008. Bax targeting to mitochondria occurs via both tail anchor-dependent and -independent mechanisms. Cell Death Differ. 15:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghibelli L, Diederich M. 2010. Multistep and multitask Bax activation. Mitochondrion 10:604–613 [DOI] [PubMed] [Google Scholar]
- 4. Kroemer G, Galluzzi L, Brenner C. 2007. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87:99–163 [DOI] [PubMed] [Google Scholar]
- 5. Frehlick LJ, Eirin-Lopez JM, Ausio J. 2007. New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioessays 29:49–59 [DOI] [PubMed] [Google Scholar]
- 6. Kerr LE, Birse-Archbold JL, Short DM, McGregor AL, Heron I, Macdonald DC, Thompson J, Carlson GJ, Kelly JS, McCulloch J, Sharkey J. 2007. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 26:2554–2562 [DOI] [PubMed] [Google Scholar]
- 7. Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM. 2006. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim. Biophys. Acta 1757:1301–1311 [DOI] [PubMed] [Google Scholar]
- 8. Scaloni F, Gianni S, Federici L, Falini B, Brunori M. 2009. Folding mechanism of the C-terminal domain of nucleophosmin: residual structure in the denatured state and its pathophysiological significance. FASEB J. 23:2360–2365 [DOI] [PubMed] [Google Scholar]
- 9. Cha H, Hancock C, Dangi S, Maiguel D, Carrier F, Shapiro P. 2004. Phosphorylation regulates nucleophosmin targeting to the centrosome during mitosis as detected by cross-reactive phosphorylation-specific MKK1/MKK2 antibodies. Biochem. J. 378:857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mattern KA, Humbel BM, Muijsers AO, de Jong L, van Driel R. 1996. hnRNP proteins and B23 are the major proteins of the internal nuclear matrix of HeLa S3 cells. J. Cell. Biochem. 62:275–289 [DOI] [PubMed] [Google Scholar]
- 11. Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine JC, Helin K, Falini B, Pelicci PG. 2005. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol. Cell. Biol. 25:8874–8886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okuwaki M. 2008. The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 143:441–448 [DOI] [PubMed] [Google Scholar]
- 13. Lindenboim L, Borner C, Stein R. 2011. Nuclear proteins acting on mitochondria. Biochim. Biophys. Acta 1813:584–596 [DOI] [PubMed] [Google Scholar]
- 14. Thompson J, Finlayson K, Salvo-Chirnside E, MacDonald D, McCulloch J, Kerr L, Sharkey J. 2008. Characterisation of the Bax-nucleophosmin interaction: the importance of the Bax C-terminus. Apoptosis 13:394–403 [DOI] [PubMed] [Google Scholar]
- 15. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. 2005. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352:254–266 [DOI] [PubMed] [Google Scholar]
- 16. Liu X, Liu Z, Jang SW, Ma Z, Shinmura K, Kang S, Dong S, Chen J, Fukasawa K, Ye K. 2007. Sumoylation of nucleophosmin/B23 regulates its subcellular localization, mediating cell proliferation and survival. Proc. Natl. Acad. Sci. U. S. A. 104:9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falini B, Martelli MP, Bolli N, Bonasso R, Ghia E, Pallotta MT, Diverio D, Nicoletti I, Pacini R, Tabarrini A, Galletti BV, Mannucci R, Roti G, Rosati R, Specchia G, Liso A, Tiacci E, Alcalay M, Luzi L, Volorio S, Bernard L, Guarini A, Amadori S, Mandelli F, Pane F, Lo-Coco F, Saglio G, Pelicci PG, Martelli MF, Mecucci C. 2006. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 108:1999–2005 [DOI] [PubMed] [Google Scholar]
- 18. Meani N, Alcalay M. 2009. Role of nucleophosmin in acute myeloid leukemia. Expert Rev. Anticancer Ther. 9:1283–1294 [DOI] [PubMed] [Google Scholar]
- 19. Borkan SC, Wang YH, Lieberthal W, Burke PR, Schwartz JH. 1997. Heat stress ameliorates ATP depletion-induced sublethal injury in mouse proximal tubule cells. Am. J. Physiol. 272:F347–F355 [DOI] [PubMed] [Google Scholar]
- 20. Lieberthal W, Menza SA, Levine JS. 1998. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. 274:F315–F327 [DOI] [PubMed] [Google Scholar]
- 21. Price VR, Reed CA, Lieberthal W, Schwartz JH. 2002. ATP depletion of tubular cells causes dissociation of the zonula adherens and nuclear translocation of beta-catenin and LEF-1. J. Am. Soc. Nephrol. 13:1152–1161 [DOI] [PubMed] [Google Scholar]
- 22. Schwartz JH, Shih T, Menza SA, Lieberthal W. 1999. ATP depletion increases tyrosine phosphorylation of beta-catenin and plakoglobin in renal tubular cells. J. Am. Soc. Nephrol. 10:2297–2305 [DOI] [PubMed] [Google Scholar]
- 23. Wang YH, Borkan SC. 1996. Prior heat stress enhances survival of renal epithelial cells after ATP depletion. Am. J. Physiol. 270:F1057–F1065 [DOI] [PubMed] [Google Scholar]
- 24. Wang YH, Knowlton AA, Li FH, Borkan SC. 2002. HSP 72 expression enhances survival in adenosine triphosphate-depleted renal epithelial cells. Cell Stress Chaperones 7:84–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z, Gall JM, Bonegio RG, Havasi A, Hunt CR, Sherman MY, Schwartz JH, Borkan SC. 2011. Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int. 79:861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, Schwartz JH, Borkan SC. 2010. GSK3beta promotes apoptosis after renal ischemic injury. J. Am. Soc. Nephrol. 21:284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li F, Mao HP, Ruchalski KL, Wang YH, Choy W, Schwartz JH, Borkan SC. 2002. Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am. J. Physiol. Cell Physiol. 283:C917–C926 [DOI] [PubMed] [Google Scholar]
- 28. Gall JM, Wang Z, Liesa M, Molina A, Havasi A, Schwartz JH, Shirihai O, Borkan SC, Bonegio RG. 2012. Role of mitofusin 2 in the renal stress response. PLoS One 7:e31074 doi:10.1371/journal.pone.0031074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gall JM, Wong V, Pimental DR, Havasi A, Wang Z, Pastorino JG, Bonegio RG, Schwartz JH, Borkan SC. 2011. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 79:1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruchalski K, Mao H, Singh SK, Wang Y, Mosser DD, Li F, Schwartz JH, Borkan SC. 2003. HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am. J. Physiol. Cell Physiol. 285:C1483–C1493 [DOI] [PubMed] [Google Scholar]
- 31. Wang Y, Knowlton AA, Christensen TG, Shih T, Borkan SC. 1999. Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int. 55:2224–2235 [DOI] [PubMed] [Google Scholar]
- 32. Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. 2003. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 22:4385–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Havasi A, Borkan SC. 2011. Apoptosis and acute kidney injury. Kidney Int. 80:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelly KJ. 2006. Acute renal failure: much more than a kidney disease. Semin. Nephrol. 26:105–113 [DOI] [PubMed] [Google Scholar]
- 35. Rau R, Brown P. 2009. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol. Oncol. 27:171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, Nicoletti I. 2009. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 23:1731–1743 [DOI] [PubMed] [Google Scholar]
- 37. Falini B, Lenze D, Hasserjian R, Coupland S, Jaehne D, Soupir C, Liso A, Martelli MP, Bolli N, Bacci F, Pettirossi V, Santucci A, Martelli MF, Pileri S, Stein H. 2007. Cytoplasmic mutated nucleophosmin (NPM) defines the molecular status of a significant fraction of myeloid sarcomas. Leukemia 21:1566–1570 [DOI] [PubMed] [Google Scholar]
- 38. Xiao J, Zhang Z, Chen GG, Zhang M, Ding Y, Fu J, Li M, Yun JP. 2009. Nucleophosmin/B23 interacts with p21WAF1/CIP1 and contributes to its stability. Cell Cycle 8:889–895 [DOI] [PubMed] [Google Scholar]
- 39. Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. 2004. Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J. Neurosci. 24:9993–10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM. 2004. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 279:21085–21095 [DOI] [PubMed] [Google Scholar]
- 41. McConnell KW, Muenzer JT, Chang KC, Davis CG, McDunn JE, Coopersmith CM, Hilliard CA, Hotchkiss RS, Grigsby PW, Hunt CR. 2007. Anti-apoptotic peptides protect against radiation-induced cell death. Biochem. Biophys. Res. Commun. 355:501–507 [DOI] [PubMed] [Google Scholar]
- 42. Lindenboim L, Blacher E, Borner C, Stein R. 2010. Regulation of stress-induced nuclear protein redistribution: a new function of Bax and Bak uncoupled from Bcl-x(L). Cell Death Differ. 17:346–359 [DOI] [PubMed] [Google Scholar]
- 43. Khandelwal N, Simpson J, Taylor G, Rafique S, Whitehouse A, Hiscox J, Stark LA. 2011. Nucleolar NF-kappaB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 18:1889–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruchalski K, Mao H, Li Z, Wang Z, Gillers S, Wang Y, Mosser DD, Gabai V, Schwartz JH, Borkan SC. 2006. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J. Biol. Chem. 281:7873–7880 [DOI] [PubMed] [Google Scholar]
- 45. Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. 2001. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335 [DOI] [PubMed] [Google Scholar]
- 46. Miao W, Luo Z, Kitsis RN, Walsh K. 2000. Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J. Mol. Cell. Cardiol. 32:2397–2402 [DOI] [PubMed] [Google Scholar]
- 47. Wang Q, Sun SY, Khuri F, Curran WJ, Deng X. 2010. Mono- or double-site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One 5:e13393 doi:10.1371/journal.pone.0013393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fotin-Mleczek M, Voss S, Brock R. 2007. The import mechanism of cationic cell-penetrating peptides and its implication for the delivery of peptide inhibitors of signal tranduction, p 161–182 In Langel U. (ed), Handbook of cell-penetrating peptides, 2nd ed. CRC Press, Boca Raton, FL [Google Scholar]