

Abstract
Liver fibrosis occurs as a wound-healing response to chronic hepatic injuries irrespective of the underlying etiology and may progress to life-threatening cirrhosis. Here we show that CCN1, a matricellular protein of the CCN (CYR61/CTGF/NOV) family, is accumulated in hepatocytes of human cirrhotic livers. CCN1 is not required for liver development or regeneration, since these processes are normal in mice with hepatocyte-specific Ccn1 deletion. However, Ccn1 expression is upregulated upon liver injuries and functions to inhibit liver fibrogenesis induced by either carbon tetrachloride intoxication or bile duct ligation and promote fibrosis regression. CCN1 acts by triggering cellular senescence in activated hepatic stellate cells and portal fibroblasts by engaging integrin α6β1 to induce reactive oxygen species accumulation through the RAC1-NADPH oxidase 1 enzyme complex, whereupon the senescent cells express an antifibrosis genetic program. Mice with hepatocyte-specific Ccn1 deletion suffer exacerbated fibrosis with a concomitant deficit in cellular senescence, whereas overexpression of hepatic Ccn1 reduces liver fibrosis with enhanced senescence. Furthermore, tail vein delivery of purified CCN1 protein accelerates fibrosis regression in mice with established fibrosis. These findings reveal a novel integrin-dependent mechanism of fibrosis resolution in chronic liver injury and identify the CCN1 signaling pathway as a potential target for therapeutic intervention.
INTRODUCTION
Liver fibrosis, or the accumulation of excessive extracellular matrix (ECM) in the liver, is a pathology that develops as a wound-healing response to chronic hepatic injuries inflicted by diverse etiologies, including viral infection, alcoholism, biliary obstruction, and nonalcoholic steatohepatitis (1, 2). These conditions may lead to fibrosis that often develops insidiously over years, resulting in cirrhosis that can potentiate liver failure, portal hypertension, and hepatocellular carcinoma, thus imposing a significant burden on public health. Currently, there are no FDA-approved drugs for the treatment of liver fibrosis, and a better understanding of how this pathology can be resolved is of paramount importance. Injury repair in the liver, as in many organs and tissues, is associated with an inflammatory response and the deposition of a provisional ECM, which aids wound healing by enhancing the integrity of the injured tissue and promoting the proliferation of parenchymal cells (3). This provisional matrix is resolved and remodeled at later stages of repair to form the matured tissue. Chronic injuries, however, result in sustained inflammation and relentless stimulation of ECM production without adequate opportunity for matrix resolution and remodeling, leading to fibrosis as excess ECM distorts tissue architecture and replaces parenchyma.
The perisinusoidal hepatic stellate cells (HSCs) are the primary precursor cells that transdifferentiate into proliferative, ECM-producing myofibroblast-like cells when activated upon liver injury, whereas portal fibroblasts (PFs) also undergo myofibroblastic differentiation and play significant roles in fibrosis resulting from biliary obstruction (2, 4). Bone marrow-derived cells and epithelial cells may also differentiate into myofibroblasts (5, 6), although the extents of their contribution to liver fibrosis have been controversial (7, 8). Once thought to be irreversible, recent evidence showed that liver fibrosis caused by diverse etiologies both in animal models and in human patients gradually regresses if the underlying cause of disease is eliminated (1, 2). These results indicate the existence of an endogenous mechanism for liver fibrosis resolution. For example, ECM-producing myofibroblasts may be removed by apoptosis (2), and more recent findings show that these cells can also revert to an inactive, nonfibrogenic phenotype (9, 10) or enter a senescence state (11), leading to the cessation of ECM production. Whereas both myofibroblast apoptosis and reversion to an inactive phenotype prevent further ECM production, senescent myofibroblasts may actively contribute to the regression of fibrosis by expressing an antifibrosis genetic program as part of the senescence-associated secretory phenotype (SASP) characteristic of senescent cells, which includes upregulation of matrix-degrading enzymes and downregulation of ECM proteins such as collagen (12, 13). Moreover, senescent cells can be cleared by natural killer cells (11). Thus, the entry of myofibroblasts into senescence can prevent further proliferation of these ECM-producing cells, promote ECM degradation, and accelerate clearance of myofibroblasts from the site of injury. However, the involvement of cellular senescence in limiting hepatic fibrosis is an emerging concept that has been shown only in the context of CCl4-induced injury, and the molecular triggers by which myofibroblast senescence is induced are unknown.
Here we show that the matricellular protein CCN1 (also named CYR61) (14, 15) is highly accumulated in livers of human patients with cirrhosis, although it is not essential for liver development and function. Further, expression of CCN1 is induced upon hepatic injury in murine models, and it acts as a key regulator that triggers cellular senescence through the accumulation of reactive oxygen species (ROS) in activated HSCs and PFs, thereby limiting fibrogenesis and promoting regression of liver fibrosis induced by diverse injuries. Moreover, tail vein delivery of purified CCN1 protein in mice with established fibrosis accelerates fibrosis regression. These findings uncover a critical mechanism of fibrosis regression that functions through CCN1-mediated integrin signaling and identify potential targets for therapeutic intervention.
MATERIALS AND METHODS
Animals and liver fibrosis induction.
All mice used in liver fibrosis studies were 2- to 4-month-old males kept in barrier facilities with veterinary care. Ccn1dm/dm mice were generated in an svJ129-C57BL/6 mixed background and backcrossed >10 times into the C57BL/6 background (16, 17). To produce Ccn1flox/flox and Ccn1ΔHep mice, svJ129 mouse embryonic stem cells were transfected with the Ccn1-targeting construct by electroporation and selected for neomycin resistance. Transfected clones with double-recombination events incorporating the targeting construct in the Ccn1 genomic locus (see Fig. 2A) were selected and injected into blastocysts to produce targeted chimeric mice. Injection was done at the University of Illinois Transgenic Production Service facility. Mice were bred with EIIa-Cre deleters, and pups with the desired loxP2 to loxP3 deletion removing the neomycin cassette were selected by genomic DNA analysis using positional primers. Selected mice were backcrossed four times with C57BL/6 mice to obtain Ccn1flox/+ heterozygous animals and bred to obtain Ccn1flox/flox mice. Ccn1ΔHep mice were generated by crossing Ccn1flox/flox mice with Alb-Cre mice (18) (Jackson Laboratory) expressing Cre recombinase under the control of the albumin promoter. Deletion of Ccn1 was confirmed by PCR using liver genomic DNA (see Fig. 2B) and primers (see Fig. 2A). Transgenic mice overexpressing Ccn1 in hepatocytes were generated by microinjecting into C57BL/6 single-cell zygotes a construct in which full-length Ccn1 cDNA was cloned under the control of a minimal promoter linked to the albumin enhancer element in the pAlb.GH vector (19). To facilitate transgene detection, a V5 epitope sequence was added in frame 3′ to the Ccn1 cDNA. Transgene protein expression was detected by Western blotting of liver lysates using a monoclonal antibody against the V5 epitope (clone V5-10; Sigma).
Fig 2.

Hepatocyte-specific deletion of Ccn1 does not impair liver development or regeneration. (A) Schematic diagrams showing the targeted Ccn1 genomic locus with the Ccn1flox-neo allele, the Ccn1flox allele with the neomycin resistance cassette deleted by recombination performed with loxP sites 2 and 3 (loxP2/3), and the Ccn1ΔHep locus following Cre-mediated excision. Left-pointing triangles represent loxP sites. The primer R sequence occurs only in the third loxP site. Cre-mediated recombination between loxP sites 2 and 3 leads to the deletion of PGK-neo (gray box) to yield the desired Ccn1flox allele. (B) The Ccn1flox allele was detected as a 391-bp DNA band when primers F2 and R were used in PCRs. Ccn1flox/flox mice were crossed with Alb-Cre mice to produce Ccn1ΔHep mice. The ΔHep allele was confirmed by the presence of a 119-bp DNA band using primers F1 and R. (C) Total liver RNA isolated from 6-week-old Ccn1flox/flox and Ccn1ΔHep mice was analyzed for Ccn1 expression by RT-PCR. Ccn1 mRNA band intensity was normalized against β-actin by Image J software analysis to correct for equal levels of sample loading. The low level of Ccn1 mRNA may have resulted from nonparenchymal cells. (D to F) Liver regeneration was determined by the percentage of remnant-liver weight/whole-liver weight on the indicated days after a 2/3 partial hepatectomy (PHx); data are expressed as means ± SD (n = 5 each). (D) Eight-week-old Ccn1flox/flox (Flox) and Ccn1ΔHep (ΔHep) mice were sacrificed on the indicated days after PHx, and liver weight was measured. (E) Mice of various ages were subjected to PHx and assessed 7 days later. (F) After PHx, remnant liver tissue was collected on the days indicated and analyzed for cell proliferation by immunohistochemical staining with antibodies against Ki67. Percentages of Ki67-positive cells relative to total number of cells were counted in 5 randomly chosen high-power fields (n = 3).
To induce liver fibrosis, male mice were injected intraperitoneally (i.p.) with CCl4 (1 ml/kg of body weight diluted 1:10 in olive oil) twice weekly for 4 to 6 weeks (20). Bile duct ligation (BDL) was performed as described previously (21) to induce chronic cholestasis, and sham operations were performed as controls. For CCN1 treatment, 40 μg of purified recombinant CCN1 protein (22) was diluted in 1 ml saline solution and injected through the tail vein at 6 ml/min. using a hydrodynamic delivery method (23). Saline solution alone was injected as a control. All procedures were approved by the University of Illinois Animal Care Committee.
Partial hepatectomy.
A 2/3 partial hepatectomy was performed on normal male mice according to published protocol (24). Mice were weighed on indicated days and sacrificed; thereafter, the liver was removed, weighed, and cryopreserved for cell proliferation analysis.
Isolation of hepatic cells.
Hepatocytes were isolated by a two-step collagenase perfusion method as previously described (25). After washing, low-speed centrifugation was performed at 50 × g and repeated four times. Hepatocytes and nonparencymal cells were harvested from the pellet and the supernatant, respectively. HSCs were isolated from normal adult male mice as described previously (26). HSCs, which store vitamin A and retinoids, were confirmed by staining with Oil Red O and immunocytochemical analysis using antibodies against glial fibrillary acidic protein (GFAP) (Abcam), desmin (clone DE-U-10; Sigma), and alpha smooth muscle actin (α-SMA). PFs were isolated from the hepatic hila according to a published protocol (27) and were confirmed by immunocytochemical analysis for α-SMA and elastin (Abcam). Human activated HSCs were purchased from ScienCell and grown in medium provided by the manufacturer.
Histology, immunohistochemistry, human liver tissue array, SA-β-Gal, and TUNEL assays.
To examine liver fibrosis, formalin-fixed, paraffin-embedded tissue sections (7 μm thick) were stained with Picrosirius Red solution (American MasterTech Scientific) following the manufacturer's protocol. Fibrotic lesions are stained red; microphotographs of six randomly selected areas (3.4 by 2.5 mm2) were taken from each animal and images analyzed using NIH Image J software to quantify the fibrotic area. For immunohistochemistry to detect CCN1 in a human liver tissue array (LV20812; US Biomax, Inc.) and mouse tissues, we immunized rabbits with a fusion protein of bacterial glutathione S-transferase (GST) linked to the mouse CCN1 von Willibrand factor type C repeat (vWC) domain, and we purified the resulting antisera by affinity column chromatography using the antigen as a ligand. The secondary antibody (GE Healthcare) was conjugated with horseradish peroxidase, and 3,3′-diaminobenzidine (DAB; Sigma) was used as a chromogen. Samples were counterstained with hematoxylin. Senescence-associated β-galactosidase (SA-β-Gal) staining for tissue cryosections (5 μm thick) was performed at pH 5.5 and for cultured cells at pH 6.0 as described previously (28). Six microphotographs of randomly selected areas (0.25 by 0.2 mm2) were taken from each section for every animal, and positive cells were counted by analyzing the digital images using the NIH Image J program. Liver cryosections were double stained with rabbit polyclonal anti-p16 antibodies (Santa Cruz) or a mouse monoclonal anti-SMA antibody (clone 1A4; Sigma). Secondary antibody recognizing rabbit IgG was conjugated with the red fluorescent dye Alexa Fluor 546 (Invitrogen), and secondary antibody against mouse IgG was labeled with the green fluorescent dye Alexa Fluor 488 (Invitrogen). Samples were counterstained with 4′,6′diamidino-2-phenylindole (DAPI) to reveal cell nuclei. Fluorescence microphotographs were acquired using a Zeiss Axiovert 200 M microscope. Cell proliferation in cryosections was assessed by immunohistochemistry using antibodies against Ki67 (Abcam). A terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed using an ApopTaq Red detection kit (Millipore) following the manufacturer's protocol. Eight randomly selected high-power microscopic fields of each sample were analyzed; numbers of Ki-67- or TUNEL-positive cells were counted and expressed as percentages of total cells.
Western blots.
Liver tissue was homogenized in Laemmli buffer and boiled, and equal amounts of protein were resolved by 12% SDS-PAGE followed by immunoblotting with enhanced chemiluminescence detection (GE Healthcare). Antibodies used included sheep anti-mouse CCN1 (R&D Systems), rabbit anti-α-SMA (Abcam), mouse monoclonal anti-V5 (clone V5-10), and anti-β-actin (mAbcam8226; Abcam).
RNA isolation and qRT-PCR.
Total RNA was purified from liver tissue and HSCs using an RNeasy minikit following the manufacturer's protocol (Qiagen). RNA (2 μg) was reverse transcribed using SuperScript Reverse Transcriptase III (Invitrogen). Quantitative reverse transcription-PCR (qRT-PCR) was performed by mixing cDNA and primers with the reaction buffer iQ SYBR green Supermix (Bio-Rad), and reactions were carried out using an iCycler thermal cycler (Bio-Rad). PCR specificity was confirmed by agarose gel electrophoresis and melting curve analysis. A housekeeping gene (Cyclophilin E) was used as an internal standard.
Measurement of tissue collagen content.
The amount of collagen in liver tissue was determined as the amount of hydroxyproline (μg) per mg dried liver tissue according to a previously published protocol (29). Pure hydroxyproline (Sigma) was used to generate standard curves.
ROS detection.
Intracellular ROS levels were determined by fluorescence microscopy as previously described (30). Briefly, cells were loaded with 10 μM dihydrocalcein (DHC; Invitrogen) for 10 min, followed by treatment with CCN1 or other factors as previously described. Cells were counterstained with Hoechst 33342 (Invitrogen) (1 μg/ml) and immediately examined by fluorescence microscopy. Five randomly chosen high-power fields were photographed, and integrated DHC fluorescence intensities were determined with NIH Image J software.
RNA interference.
Cells were transfected with 10 nM small interfering RNA (siRNA) targeting Nox1, Nox4, Rac1, or a scrambled sequence control (Integrated DNA Technologies) using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol. Gene expression was analyzed by RT-PCR 48 h after transfection. The sequences (5′ to 3′) used for siRNA knockdown were as follows: for Nox1, CCAAGAATTTCTCTCGTGTAATAGTAC (siNox1#1) and GATGGGATTTAGCCAAGAATCTTCTTT (siNox1#2); for Nox4, AATTAGAAACTCTTATTGCGTAGGTAG; and for Rac1, GTGGTGTCGCACTTCAGGATACCACTT.
Statistical analysis.
All experiments were repeated at least three times with similar results. Student's t test was performed to determine the probability of statistically significant differences (P values), and the results are recorded in the figure legends. CCN1 immunohistochemical staining intensities in human liver tissue were classified as low, medium, and high levels, based on bright-field microscopy examination. Differences in CCN1 intensities between normal and cirrhotic liver tissues were analyzed by a Mann-Whitney U test to determine the probability of statistically significant difference.
RESULTS
CCN1 is highly induced in hepatocytes of human cirrhotic liver and in response to hepatic injury.
CCN1 is a matricellular protein involved in skin wound healing and bone fracture repair (31, 32); however, its role in normal liver development, function, or pathology is unknown. Examination of human liver specimens by immunohistochemistry revealed that the CCN1 protein level was low in normal livers but was greatly increased in cirrhotic livers (Fig. 1A). Approximately 75% of normal livers maintained low levels of CCN1, 25% exhibited medium level, and none showed a high level of accumulation. This distribution was dramatically shifted in cirrhotic livers, among which 40% displayed high levels of CCN1 and 44% and 16% showed medium and low levels, respectively (Fig. 1B). CCN1 was predominantly localized in the cytoplasm of hepatocytes, indicating that hepatocytes are the major source of CCN1 and suggesting that its secretion or internalization may be highly regulated.
Fig 1.

CCN1 is highly elevated in cirrhotic liver in humans and after CCl4 treatment in mice. (A) Representative images of CCN1 immunohistochemical staining (brown) in normal and cirrhotic human liver tissues in the human liver tissue array. Bar = 100 μm. (B) Staining intensity for CCN1 in the human liver tissue array was classified as low, medium, and high, and the difference between normal and cirrhotic tissues was analyzed by the Mann-Whitney U test. **, P = 4.8 × 10−6. (C) Immunohistochemistry of liver sections from untreated mice and mice injected with CCl4 twice weekly for 4 weeks probed with either control IgG or anti-CCN1 antibodies and counterstained with hematoxylin. Bar = 100 μm. (D) Liver lysates from untreated mice and mice injected with CCl4 twice weekly for 6 weeks were resolved on SDS-PAGE followed Western blotting using antibodies against CCN1 and β-actin (n = 3). (E) Ccn1 expression was analyzed by qRT-PCR in liver RNA isolated at the indicated times after CCl4 injection (n = 6). Data represent means ± standard deviations (SD) of the results determined in triplicate experiments. Rel., relative. (F) RNA was harvested from normal liver cells or from isolated hepatocytes (HC) and nonparenchymal cells (NP) from livers of untreated mice (normal) or mice 6 h after CCl4 treatment. Ccn1 expression was analyzed by qRT-PCR. Data represent means ± SD of the results determined in triplicate experiments. (G) Hepatocytes freshly isolated from normal mice were incubated with TNF-α (10 ng/ml), IL-6 (20 ng/ml), or bovine serum albumin (BSA) as a control at 37°C overnight. The Ccn1 mRNA level was analyzed by qRT-PCR (n = 3). Data represent means ± SD of the results determined in triplicate experiments. *, P < 0.004.
To examine the role of CCN1 in hepatic injury repair and fibrosis, we have used a murine model of liver fibrosis. The hepatotoxin CCl4 is metabolized by the cytochrome P450 system expressed mainly around the central veins, generating free radical intermediates that cause acute cell death and inflammation leading to fibrosis if injury is sustained. CCN1 was localized in hepatocytes in normal mouse liver and was accumulated to a high level around the central veins after repeated CCl4 insults (Fig. 1C). Furthermore, CCN1 staining was lost in livers of mice with hepatocyte-specific Ccn1 deletion (ΔHep) (see below), confirming that hepatocytes are the major cell type expressing CCN1 (Fig. 1C). Likewise, immunoblotting showed that CCN1 protein was detectable at a low level in normal liver but was greatly accumulated after CCl4-induced injury (Fig. 1D). The CCN1 level was nearly abolished in liver of mice with hepatocyte-specific Ccn1 deletion but was modestly increased after CCl4 treatment, most likely in nonparenchymal cells (Fig. 1D). Acute response to a single dose of CCl4 treatment rapidly induced the Ccn1 mRNA level in the liver by >80-fold within 6 h (Fig. 1E). The mRNA level gradually decreased but remained elevated compared to untreated control levels even after repeated CCl4 treatments to establish fibrosis. Primary hepatocytes freshly isolated from normal or CCl4-treated mice expressed levels of Ccn1 mRNA that were 4- or 5-fold higher than those seen with nonparenchymal cells from the liver, respectively (Fig. 1F), further indicating that hepatocytes are the major source of CCN1. Moreover, treatment of primary hepatocytes with the inflammatory cytokine tumor necrosis factor alpha (TNF-α), but not interleukin-6 (IL-6), induced the expression of Ccn1 (Fig. 1G). Together, these results indicate that Ccn1 is highly induced in hepatocytes in response to liver injury and may be regulated by inflammatory cytokines such as TNF-α that are rapidly elevated in liver injuries.
Since Ccn1 is essential for cardiovascular development, germ line Ccn1 knockout mice are embryonic lethal (33, 34). To examine the role of Ccn1 in liver development and function, we generated mice with hepatocyte-specific deletion of Ccn1 (Ccn1ΔHep) using a Cre-loxP strategy (Fig. 2A). Ccn1flox/flox mice were constructed in which the Ccn1 genomic locus is replaced by a Ccn1 allele with LoxP sites flanking the first two exons, deletion of which generates a true null allele (33). These mice were mated with mice expressing the Cre recombinase in hepatocytes under the control of the albumin promoter. Hepatocyte-specific Ccn1 deletion, confirmed by PCR analysis with position-specific primers (Fig. 2B), resulted in a >90% reduction in Ccn1 mRNA in the liver of Ccn1ΔHep mice compared to Ccn1flox/flox control mice (Fig. 2C) as well as virtual obliteration of the CCN1 protein staining in the liver by immunohistochemistry (Fig. 1C). Ccn1ΔHep mice are viable and fertile, show normal liver histology, and present no apparent behavioral or morphological abnormality, indicating that CCN1 is not essential for liver development or function. Furthermore, liver regeneration after 2/3 partial hepatectomy occurred at the same rate in Ccn1ΔHep mice as in Ccn1flox/flox control mice (Fig. 2D). The age of mice subjected to surgery did not change the outcome (Fig. 2E), and both Ccn1ΔHep and Ccn1flox/flox mice reached the same liver weight as wild-type (WT) mice 7 days after surgery (data not shown). Levels of hepatocyte proliferation, as detected by immunohistochemical analysis of Ki67 expression, were also indistinguishable (Fig. 2F). Together, these results show that Ccn1 is not required for normal liver development or hepatocyte proliferation. Rather, its induction in cirrhosis and in acute and chronic liver damage suggests a role in the liver injury response.
CCN1 limits liver fibrosis due to diverse etiologies by inducing myofibroblast senescence.
To study CCN1 function in chronic liver injury, mice were injected with CCl4 i.p. twice weekly for 6 weeks to induce liver fibrosis (20). Remarkably, CCl4-treated Ccn1ΔHep mice exhibited prominent central-to-central bridging fibrosis and ∼4-fold more fibrotic areas of collagen deposition than Ccn1flox/flox control mice (Fig. 3A). The Ccn1ΔHep mice also accumulated ∼3-fold more hydroxyproline, a major modified amino acid in collagen, than control mice (Fig. 3B). Further, Ccn1ΔHep mice expressed a higher level of the myofibroblast marker α-SMA, indicating a more extensive activation of precursor cells to express the myofibroblastic phenotype (Fig. 3C). Expression of Mmp9 and Mmp13, which encode gelatinase B and collagenase 3, respectively, was substantially lower in Ccn1ΔHep mice, whereas expression of Col1a1 and the matrix metalloproteinase inhibitor Timp1 was significantly higher (Fig. 3D). However, Ccn1ΔHep and Ccn1flox/flox mice suffered the same level of liver damage in response to CCl4, since their levels of serum alanine aminotransferase (ALT) and extents of cell proliferation and apoptosis were indistinguishable (Fig. 3E to G). Thus, the loss of hepatic Ccn1 leads to exacerbated liver fibrosis after CCl4-induced injury, suggesting a role for CCN1 in limiting fibrosis following liver injury.
Fig 3.

CCN1 is antifibrotic in CCl4-induced liver fibrosis. (A to C) Ccn1flox/flox (Flox) and Ccn1ΔHep (ΔHep) mice were injected i.p. with either vehicle or CCl4 twice a week for 6 weeks to induce fibrosis (n = 7 each). (A) Liver sections were stained with Sirius Red to reveal collagen deposition. The percentages of fibrotic areas were assessed by Image J analysis of microphotographs of six randomly selected areas. Data represent means ± SD. CV, central vein; PT, portal triad. Bar = 100 μm. (B) Liver hydroxyproline content was determined. (C) Liver lysates were resolved on SDS-PAGE followed by Western blotting using antibodies against α-SMA and β-actin (n = 3). (D) Liver mRNA of the indicated genes was analyzed by qRT-PCR; data are shown as means ± SD. *, P < 0.005. (E to G) At the indicated times after a single CCl4 injection or after repeated CCl4 insults for 6 weeks (42 days) to induce fibrosis, serum ALT levels were measured (E), and livers were analyzed by immunohistochemistry using antibody against Ki-67 (F) or processed for a TUNEL assay (G). (H) qRT-PCR-quantified Ccn2 gene expression using total liver RNA isolated from chronic CCl4- or vehicle-treated WT, Ccn1dm/dm (Dm), Ccn1flox/flox (Flox), and Ccn1ΔHep (ΔHep) mice (n = 3 each).
CCN2, or connective tissue growth factor (CTGF), is a close homolog of CCN1 that has been shown to promote liver fibrosis and is thought to act in synergy with transforming growth factor β (TGF-β) (35, 36). Interestingly, although Ccn2 is induced in WT or Ccn1flox/flox mice upon CCl4-induced fibrosis, no induction was observed in mice where Ccn1 is defective, either by gene deletion (Ccn1ΔHep mice) or knock-in of a mutant allele (Ccn1dm/dm; see below). These results suggest that CCN1 upregulates Ccn2 expression upon chronic liver damage. Both Ccn1ΔHep and Ccn1dm/dm mice suffer enhanced fibrosis without Ccn2 overexpression (Fig. 3; see also Fig. 8), indicating that exuberant liver fibrosis can be dissociated from elevated Ccn2 expression.
Fig 8.
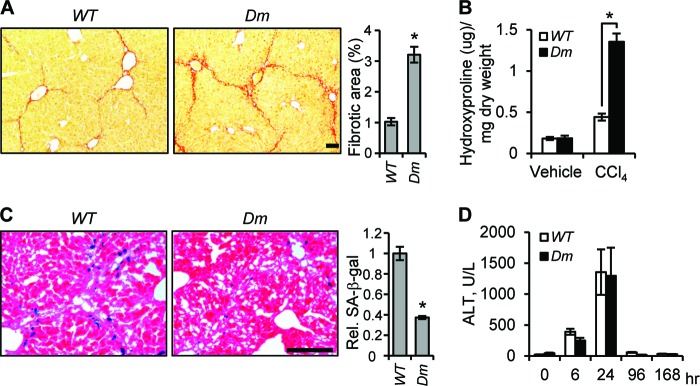
CCN1 acts through integrin α6β1 binding sites in vivo. Ccn1dm/dm (Dm) and WT mice (n = 6 each) were treated i.p. with CCl4 twice weekly for 6 weeks. (A) Tissue sections were stained with Sirius Red and fibrotic areas measured. (B) Liver hydroxyproline content was determined. (C) Frozen tissue sections were stained for SA-β-Gal activity. (D) Serum ALT levels were measured at the indicated time points after a single dose of CCl4 by injection. Bars = 100 μm.
The termination of fibrogenesis in liver injury may occur in part when ECM-producing myofibroblasts are purged by undergoing apoptosis or senescence (11, 37), and thus defects in these processes may result in enhanced fibrosis in Ccn1ΔHep mice. Numerous senescent cells were found surrounding the central veins in CCl4-treated Ccn1flox/flox mice as judged by the expression of senescence-associated β-galactosidase (SA-β-Gal) as a marker, but their numbers were reduced by >60% in Ccn1ΔHep mice (Fig. 4A), indicating that CCN1 is critical for senescence. The senescent cells, which also expressed the CDK inhibitor p16Ink4A (another senescence marker), were largely (>80%) α-SMA-expressing myofibroblasts (Fig. 4B). In contrast, Ccn1ΔHep and Ccn1flox/flox mice showed similar numbers of apoptotic cells, many of which (45%) were also α-SMA-expressing myofibroblasts (Fig. 4A and B). These results suggest that deficits in myofibroblast senescence, rather than impaired myofibroblast apoptosis, may underlie the exacerbated fibrosis in Ccn1ΔHep mice. To evaluate the role of CCN1 further, we generated transgenic mice that overexpressed Ccn1 in hepatocytes (OE) (Fig. 4C to E). Overexpression of Ccn1 resulted in a >50% lower level of fibrotic lesions and a 40% lower level of hydroxyproline due to CCl4 injury compared to wild-type (WT) mice (Fig. 4F and G). The number of senescent cells was concomitantly increased by 2.5-fold in OE mice (Fig. 4H), further indicating that CCN1 triggers myofibroblast senescence to inhibit fibrosis in response to liver injury.
Fig 4.

CCN1 inhibits hepatic fibrosis by inducing myofibroblast senescence. (A) Adjacent serial liver cryosections from Ccn1flox/flox and Ccn1ΔHep mice treated with CCl4 to induce fibrosis were stained for SA-β-Gal (upper panels, blue; n = 7) and TUNEL (lower panels, red; n = 3) analysis and counterstained with eosin and DAPI, respectively. Boxed areas are enlarged and shown in inserts. Percentages of SA-β-Gal- and TUNEL-positive cells are shown as means ± SD. (B) Ccn1flox/flox liver sections prepared as described above were doubly stained for p16INK4a (red) and α-SMA (green) and counterstained with DAPI or doubly stained with TUNEL (red) and α-SMA (green) and counterstained with DAPI. Percentages of p16INK4a- and TUNEL-positive cells that were α-SMA positive or negative are shown. (C) A diagram of the DNA construct for Ccn1-overexpressing (OE) transgenic mice, showing Ccn1 cDNA driven by a minimal promoter fused to the murine albumin gene enhancer (PAlb). V5 epitope coding sequence was added in frame to the Ccn1 3′ end. pA, polyadenylation signal. (D) Liver lysates from OE and WT mice were resolved on SDS-PAGE and analyzed by Western blotting using antibodies against V5 epitope and β-actin. (E) Liver tissue sections from OE and WT mice were stained with anti-CCN1 antibodies (brown) and counterstained with hemotoxylin. (F) OE and WT mice were treated with CCl4 for 6 weeks (n = 6), and liver tissue sections were stained with Sirius Red; fibrotic areas were quantified. (G) Liver hydroxyproline content was determined. (H) Liver tissue sections were stained for SA-β-Gal to assess the number of senescent cells. White bar = 10 μm; black bars = 100 μm. *, P < 0.02.
The pattern of fibrotic lesions is dependent upon the etiology involved (2). Whereas chronic CCl4 intoxication causes hepatic fibrosis stemming from the central vein region, patients with viral hepatitis or biliary obstruction sustain fibrotic pathologies around the portal triad (2). Thus, we tested the role of CCN1 after bile duct ligation (BDL), a model of cholestasis that induces fibrosis around the portal tract (21). Ccn1ΔHep mice suffered ∼3-fold more fibrotic area and hydroxyproline content than control mice 3 weeks after BDL (Fig. 5A and B), with a 60% decrease in the level of SA-β-Gal-positive senescent cells (Fig. 5C). These mice showed enhanced expression of profibrogenic genes (Col1a1, Timp1, and Tgfb1) and downregulation of genes encoding ECM protein-degrading enzymes (Mmp2 and Mmp9) (Fig. 5D), consistent with inhibition of the SASP. Thus, inactivation of Ccn1 led to less senescence and more fibrosis after BDL, although Ccn1ΔHep and Ccn1flox/flox control mice suffered similar extents of liver injury as reflected by serum levels of ALT (Fig. 5E). In contrast, overexpression of Ccn1 in OE mice resulted in reduced fibrotic areas and hydroxyproline content (Fig. 5F and G), with a concomitant increase in the level of senescent cells (Fig. 5H). These results show that CCN1 induces cellular senescence to control liver fibrosis due to diverse etiologies and illustrate for the first time that senescence limits fibrosis that affects the portal triad after BDL.
Fig 5.

CCN1 induces cellular senescence to inhibit fibrosis due to chronic cholestasis. (A to E) Ccn1ΔHep and Ccn1flox/flox control mice (n = 4 each) were subjected to bile duct ligation (BDL) and sacrificed for analysis 3 weeks later. (A and B) Liver sections were stained with Sirius Red (A), and liver hydroxyproline content was determined (B). (C) Liver sections were stained with SA-β-Gal. The relative levels of fibrosis and senescence are expressed as means ± SD. (D) Expression of fibrosis-related genes was analyzed by qRT-PCR. (E) Serum levels of ALT were analyzed at various days after BDL as indicated. Data presented are means ± SD; n = 4. (F to H) WT and OE mice (n = 4 each) were subjected to BDL. (F and H) Liver sections are stained with Sirius Red (F) and SA-β-Gal (H) as described above to reveal fibrosis and senescence. (G) Liver hydroxyproline content was determined. CV, central vein; PT, portal triad. Bars = 100 μm. *, P < 0.02.
CCN1 induces cellular senescence in activated HSCs and PFs.
Upon liver injury, activated HSCs and PFs differentiate into myofibroblast-like cells and play critical roles in fibrogenesis (2). Freshly isolated HSCs stained positively with Oil Red O and expressed glial fibrillary acidic protein (GFAP) and desmin but not elastin; these cells became activated and differentiated into α-SMA-expressing myofibroblast-like cells within days in culture (data not shown). When isolated activated HSCs were treated with purified CCN1 protein, they entered senescence as they became flattened and enlarged, ceased proliferation, and expressed SA-β-Gal (Fig. 6A and B). CCN1 also enhanced expression of Mmp9, Mmp13, and IL-6 but decreased expression of profibrotic genes (Col1a1, Timp1, and Tgfb1), consistent with expression of the SASP (Fig. 6C). Several lines of evidence show that integrin α6β1, the major CCN1 receptor in fibroblastic cells (38), mediates CCN1-induced senescence in activated HSCs. First, the CCN1-DM mutant protein (39), which is disrupted in its α6β1 binding sites, was defective for the senescence-inducing activity since it did not suppress cell proliferation or induce SA-β-Gal expression (Fig. 6B and D). Second, coincubation of CCN1 with a function-blocking monoclonal antibody against integrin α6 (GoH3), but not control IgG, inhibited CCN1-induced senescence (Fig. 6D). Finally, an α6β1-binding peptide (T1) (40) that competes with ligand binding to α6β1 abrogated CCN1-induced senescence, whereas a mutated T1 peptide that does not bind α6β1 had no effect (Fig. 6E). Moreover, CCN1 also induced cellular senescence in an α6β1-dependent manner in activated human HSCs (Fig. 6I and J). We also isolated PFs, which expressed elastin but not desmin (27) (data not shown), and found that CCN1 triggered cellular senescence in activated PFs as it suppressed cell proliferation and induced the expression of SA-β-Gal and SASP (Fig. 6F to H).
Fig 6.

CCN1 induces senescence in activated HSCs and PFs. (A) Isolated mouse HSCs were activated in culture and treated with purified recombinant CCN1 protein (2.5 μg/ml) or BSA for 6 days. Cell morphology (upper panel), SA-β-Gal assay (lower panel), and quantitation results are shown. (B) Cell numbers were counted at the indicated times after culturing with added CCN1, DM mutant protein, or BSA (2.5 μg/ml each). (C) Cells were treated with BSA or CCN1 for 3 days, and qRT-PCR was performed to quantify the expression of the indicated genes. (D) Cells were treated with BSA, CCN1, or DM or preincubated with function-blocking antibodies against integrin α6 (GoH3) or control IgG (50 μg/ml) before addition of CCN1 and assayed for SA-β-Gal after 3 days. (E) Cells were treated with BSA or CCN1 for 3 days with either the α6β1 integrin-binding T1 peptide or the nonbinding mutant (mut-T1; 0.5 mM each) as a competitor before assay for SA-β-Gal. (F) Activated PFs were treated with CCN1 (2.5 μg/ml) or BSA for 6 days and assayed for SA-β-Gal activity. SA-β-Gal assay and quantitation results are shown. (G) Numbers of cells grown in the presence of BSA, CCN1, or DM for the indicated days were counted. (H) PFs were treated with BSA or CCN1 for 3 days, and expression of the indicated genes was assessed by qRT-PCR. (I and J) Activated human HSCs were cultured with BSA or CCN1 for 3 days, and total cell numbers were counted (I) or cells were assayed for SA-β-Gal (J). Where indicated, cells were pretreated with T1 and mut-T1 peptide before addition of CCN1. Data are presented as means ± SD of triplicate determinations. *, P < 0.02. Bars = 10 μm.
The engagement of α6β1 by CCN1 in human skin fibroblasts activates the small G-protein RAC1 (41), which can serve multiple functions, including acting as an activating subunit of the NOX complex to induce the generation of ROS (42). CCN1 induces ROS in both activated HSCs and PFs (Fig. 7A and H), and coincubation of CCN1 with the ROS scavenger N-acetylcysteine inhibited senescence, indicating the requirement of ROS (Fig. 7B and I). Since the NOX inhibitor apocynin also effectively reduced senescence (Fig. 7B and I), we hypothesized that CCN1 generates ROS through a RAC-NOX-dependent mechanism. There are four NOX isoforms in mouse cells in addition to the more distantly related DUOX1 and DUOX2 (43). CCN1 treatment specifically elevated Nox1 and Rac1 expression, but not expression of other Nox or Rac isoforms or of the Nox2 cofactor p47phox (Fig. 7C). Knockdown of either Nox1 or Rac1 by specific siRNAs inhibited CCN1-induced ROS (Fig. 7E) and senescence as judged by cell proliferation and SA-β-Gal staining (Fig. 7F and G). Knockdown of Nox4, which is also expressed in fibroblasts, had no effect (Fig. 7E to G). Likewise, in activated PFs CCN1 induced ROS accumulation and senescence, which also required NOX-dependent ROS generation (Fig. 7H and I). These results show that CCN1 binds to integrin α6β1 and induces cellular senescence through the RAC1-NOX1-ROS axis, leading to the expression of antifibrosis genes associated with the SASP to inhibit fibrosis.
Fig 7.

CCN1-induced senescence occurs through the RAC1-Nox1-ROS axis. (A) Activated mouse HSCs were treated with CCN1 or BSA for 2 h and loaded with DHC for detection of ROS by fluorescence microscopy, with quantitation shown. (B) HSCs were pretreated with NAC (2.5 mM) or apocynin (10 μM) for 1 h before the addition of CCN1. NAC or apocynin was replenished daily, and SA-β-Gal activity was quantified after 3 days. (C) HSCs treated with CCN1 for 3 days were assayed for expression of the indicated genes by qRT-PCR. *, P < 0.003. (D) siRNA-mediated knockdown of Nox1 (siNox1#1 and siNox1#2), Nox4 (siNox4), and Rac1 (siRac1) was confirmed by RT-PCR. Scrambled-sequence siRNA was used as a control. (E to G) Knockdown cells were treated with CCN1 (2.5 μg/ml) for 2 h and loaded with DHC for fluorescence microscopy (E) and examined after 3 days of CCN1 treatment for relative cell numbers (F) and percentages of SA-β-Gal-positive cells (G). (H) Activated PFs were treated with CCN1 or BSA for 2 h and loaded with DHC for fluorescence microscopy. (I) PFs were pretreated with NAC (2.5 mM) and apocynin (10 μM) for 1 h followed by the addition of CCN1. NAC and apocynin were replenished daily, and SA-β-Gal activity was quantified after 3 days. Data are presented as means ± SD. Bars = 10 μm.
To test whether CCN1 induces senescence and limits fibrosis by acting through binding α6β1 in vivo, we utilized Ccn1dm/dm knock-in mice in which the Ccn1 genomic locus has been replaced by the Dm allele, which encodes a CCN1 mutant disrupted in the α6β1 binding sites (17, 38). Ccn1dm/dm mice subjected to chronic CCl4 treatment phenocopied Ccn1ΔHep mice and sustained exacerbated fibrosis as judged by Sirius Red staining and hydroxyproline content concomitant with a 60% reduction in SA-β-gal-positive senescent cells compared to WT mice (Fig. 8A to C), even though the two genotypes experienced the same level of liver damage (Fig. 8D). These results show that CCN1 acts through its α6β1 binding sites in vivo to induce senescence and limit liver fibrosis.
Tail vein delivery of CCN1 accelerates resolution of hepatic fibrosis.
Since CCN1 is an endogenous antifibrosis factor, we tested whether administration of purified CCN1 protein can promote fibrosis resolution in animals with established fibrosis. Ccn1dm/dm mice were injected with CCl4 for 4 weeks to induce liver fibrosis, and CCN1 protein or vehicle was delivered via tail vein injection every 4 days for a total of 4 times, beginning 4 days after the final CCl4 injection (Fig. 9A). Staining of liver sections 2 days after the final CCN1 injection showed that CCN1 protein treatment reduced the fibrotic area ∼50% more than the level seen with vehicle-treated controls (Fig. 9B) concomitant with ∼40% further reduction in hydroxyproline (Fig. 9C). CCN1 treatment also significantly reduced Col1a1 and Timp1 mRNA levels and increased Mmp9 expression by 3-fold, consistent with the SASP results (Fig. 9D). These data show that delivery of CCN1 protein accelerates fibrosis regression, suggesting potential therapeutic value in activating the CCN1-induced senescence pathway for the treatment of liver fibrosis.
Fig 9.

Tail vein delivery of CCN1 protein accelerates fibrosis resolution. (A) A schematic schedule of CCl4 i.p. injection into Ccn1dm/dm mice and CCN1 tail vein delivery. Arrowheads indicate points at which CCN1 was injected at 40 μg/mouse. (B) Livers from mice injected with CCN1 or vehicle during the resolution period were collected 2 days after the final injection and stained with Sirius Red; n = 7. Bar = 100 μm. (C) Hydroxyproline content was quantified in livers (n = 3; *, P < 0.02). Open bars, mice treated with vehicle or CCl4 for 4 weeks to induce fibrosis; gray bars, mice treated with CCl4 for 4 weeks, followed by 18 days of fibrosis resolution with tail vein injection of CCN1 or vehicle. (D) Livers of mice after tail vein injection of CCN1 or vehicle were analyzed for expression of the indicated genes by qRT-PCR. Data are presented as means ± SD; n = 3.
DISCUSSION
The most important findings of this study are the demonstration that liver fibrogenesis induced by disparate forms of hepatic injuries from CCl4 intoxication to bile duct ligation is effectively restrained as fibrogenic myofibroblasts enter senescence and that the maticellular protein CCN1, which is highly accumulated in human cirrhotic livers, plays a pivotal role in triggering myofibroblast senescence to combat fibrosis in these diverse etiologies. CCN1 induces senescence of liver myofibroblasts through the engagement of integrin α6β1 to activate ROS accumulation by activating the RAC1-NOX1 pathway. Thus, although NOX1-induced ROS have been shown to promote HSC activation and stimulate liver fibrogenesis (44, 45), they may also be critical for the regression of fibrosis. Test-of-principle experiments show that administration of CCN1 can accelerate fibrosis regression even in animals that have already established fibrosis, suggesting that activation of the CCN1-induced senescence pathway may hold therapeutic promise.
CCN1 is an angiogenic inducer involved in skin wound healing and bone fracture repair (32, 46). However, its role in liver development, function, and pathology is unknown. Here we show that, although hepatocytes are the major source of CCN1 in the liver, hepatocyte-specific ablation of Ccn1 has no apparent effect on liver development and function or role in hepatocyte proliferation in liver regeneration after partial hepatectomy. These results show for the first time that Ccn1 is not required for liver development or hepatocyte proliferation. Rather, Ccn1 is highly induced in response to both acute and chronic liver injury and CCN1 protein levels are substantially elevated in human patients with cirrhosis (Fig. 1). Although the major function of CCN1 is to inhibit fibrogenesis and promote fibrosis regression, chronic persistent liver injuries may overwhelm the antifibrosis activities of endogenously expressed Ccn1, and cirrhosis can develop despite elevated levels of CCN1 accumulation.
Cellular senescence is a form of irreversible cell cycle arrest that occurs in response to cellular stresses such as DNA damage, oxidative stress, oncogene activation, chromatin disruption, and telomere erosion (47, 48). Whereas senescence is a well-recognized mechanism of tumor suppression, emerging evidence indicates that it may play a role in aging, tissue repair, and even malignancy (12). Although a previous report (11) has suggested that senescence can limit fibrosis, this emerging concept has been tested only in CCl4-induced injury in the liver. In this study, we extended these findings to show that both CCl4- and BDL-induced liver injuries respond to CCN1-triggered senescence. Senescent myofibroblasts cease to proliferate and express an antifibrosis genetic program. Thus, by triggering cellular senescence in hepatic myofibroblasts, CCN1 blocks the proliferation of these ECM-producing cells and converts them into ECM-degrading cells, thereby accelerating fibrosis regression. Accordingly, senescence in response to CCl4- and BDL-induced liver injuries was greatly reduced in mice with hepatocyte deletion of Ccn1, resulting in exacerbated fibrosis (Fig. 4 and 5).
The observation that targeted cell type-specific overexpression and deletion of Ccn1 in hepatocytes elicit significant effects in fibrosis by regulating senescence of HSCs and PFs indicates that CCN1 acts in a paracrine manner (Fig. 3 to 5). This mode of action is consistent with CCN1 acting extracellularly by binding to its receptors on the target cell surface. In this regard, it is remarkable that the Ccn1dm/dm knock-in mice, which express mutant CCN1 defective for binding the integrin receptor α6β1, essentially phenocopied Ccn1ΔHep mice in the hepatic fibrosis response (Fig. 3 and 8), indicating that the effects of CCN1 are largely mediated through α6β1. These results strongly indicate the extracellular and paracrine action of CCN1. Thus, although cytoplasmic CCN1 can be observed by immunohistochemical staining of liver tissues, this may represent inefficient CCN1 secretion or reinternalization of CCN1.
Members of the CCN protein family may play contrasting roles in fibrosis (14, 49). CCN2/CTGF, a homolog of CCN1, has been shown to be a profibrotic regulator whose high expression is associated with fibrosis of many organs and tissues, including the liver (50). Consequently, monitoring CCN2 levels is being developed as a potential diagnostic tool for kidney, lung, and liver fibrosis (14). Here we show that exacerbated fibrosis occurs when CCN1 is defective without increased Ccn2 expression (Fig. 3H), indicating that exuberant fibrosis can be dissociated from elevated Ccn2 expression. Furthermore, the CCN2 level is not increased in Ccn1ΔHep and Ccn1dm/dm mice even upon chronic CCl4 injury, indicating that CCN1 may regulate Ccn2 expression in vivo.
CCN1 triggers cellular senescence in activated HSCs and PFs by engaging integrin α6β1, leading to the generation of ROS through a RAC-NOX1-dependent mechanism (Fig. 6 and 7). A high level of ROS may lead to DNA damage response and the activation of a p53- and p16INK4/pRb-dependent pathway of cellular senescence (16, 48). Thus, it is likely that CCN1 acts by inducing senescence through ROS-dependent activation of the p53- and p16INK4/pRb pathway in the liver. Support for this notion is found in the observation that CCl4-induced fibrosis is attenuated in p53−/−; p16INK4/ARF−/− mice, which are defective for this senescence pathway (11).
ROS are produced in all cell types and are critical for many cellular functions (51). The accumulation of ROS in HSCs contributes to their activation and fibrogenic response, and several clinical trials are currently testing the efficacy of antioxidants in reversing liver fibrogenesis (52). Both NOX1 and NOX2 expressed in HSCs have been found to play key roles in liver fibrosis (44, 45), and GKT137831, a NOX1/4 inhibitor, curbs the fibrogenic response in HSCs and attenuates liver fibrosis in mice (53, 54). Interestingly, CCN1 specifically induces the NOX1 isoform and its activator RAC1 in activated HSCs and mediates senescence through this enzyme (Fig. 7). Thus, although NOX1-mediated ROS generation is critical for both the fibrogenic activation of HSCs and liver fibrosis, it is paradoxically also essential for fibrosis regression through the entry of activated HSCs into senescence. These findings suggest that targeting ROS generation in HSCs as fibrosis therapy may be most effective while fibrogenesis is actively ongoing but may impede fibrosis regression through myofibroblast senescence during resolution.
A broad range of antifibrosis therapies, including various means of eliminating the causes of injury, reducing inflammation, inhibiting HSC activation and fibrogenesis, and promoting HSC apoptosis, are currently under development (55). The identification of the CCN1-induced senescence pathway as an effective endogenous mechanism of liver fibrosis resolution suggests a novel therapeutic paradigm through activating the CCN1 senescence pathway. Proof of concept for this approach is provided by the finding that administration of CCN1 protein through tail vein injection accelerates fibrotic regression in mice with established fibrosis (Fig. 9). Explorations of approaches that drive hepatic myofibroblasts into senescence, including the delivery of genetic information or chemical regulators targeting HSCs (56) and PFs, might prove informative in the development of novel therapeutic strategies for the treatment of liver fibrosis.
ACKNOWLEDGMENTS
We thank our colleagues for helpful discussions and Mark A. Magnuson for providing the pAlb.GH vector.
This work was supported by grants from the National Institutes of Health (GM78492 and AR61791).
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Bataller R, Brenner DA. 2005. Liver fibrosis. J. Clin. Invest. 115:209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hernandez-Gea V, Friedman SL. 2011. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6:425–456 [DOI] [PubMed] [Google Scholar]
- 3. Gurtner GC, Werner S, Barrandon Y, Longaker MT. 2008. Wound repair and regeneration. Nature 453:314–321 [DOI] [PubMed] [Google Scholar]
- 4. Iwaisako K, Brenner DA, Kisseleva T. 2012. What's new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 27(Suppl 2):65–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baertschiger RM, Serre-Beinier V, Morel P, Bosco D, Peyrou M, Clement S, Sgroi A, Kaelin A, Buhler LH, Gonelle-Gispert C. 2009. Fibrogenic potential of human multipotent mesenchymal stromal cells in injured liver. PLoS One 4:e6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi SS, Diehl AM. 2009. Epithelial-to-mesenchymal transitions in the liver. Hepatology 50:2007–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Higashiyama R, Moro T, Nakao S, Mikami K, Fukumitsu H, Ueda Y, Ikeda K, Adachi E, Bou-Gharios G, Okazaki I, Inagaki Y. 2009. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 137:1459–1466 [DOI] [PubMed] [Google Scholar]
- 8. Taura K, Miura K, Iwaisako K, Osterreicher CH, Kodama Y, Penz-Osterreicher M, Brenner DA. 2010. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 51:1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, Evans SM, Dillmann W, Glass CK, Brenner DA. 2012. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. U. S. A. 109:9448–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, Pradere JP, Friedman RA, Schwabe RF. 2012. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143:1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134:657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodier F, Campisi J. 2011. Four faces of cellular senescence. J. Cell Biol. 192:547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5:99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jun JI, Lau LF. 2011. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 10:945–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lau LF. 2011. CCN1/CYR61: the very model of a modern matricellular protein. Cell. Mol. Life Sci. 68:3149–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jun J-I, Lau LF. 2010. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12:676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen C-C, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF. 2007. Cytotoxicity of TNFα is regulated by integrin-mediated matrix signaling. EMBO J. 26:1257–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274:305–315 [DOI] [PubMed] [Google Scholar]
- 19. Postic C, Magnuson MA. 2000. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26:149–150 [DOI] [PubMed] [Google Scholar]
- 20. Constandinou C, Henderson N, Iredale JP. 2005. Modeling liver fibrosis in rodents. Methods Mol. Med. 117:237–250 [DOI] [PubMed] [Google Scholar]
- 21. Kountouras J, Billing BH, Scheuer PJ. 1984. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br. J. Exp. Pathol. 65:305–311 [PMC free article] [PubMed] [Google Scholar]
- 22. Kireeva ML, Mo Yang F-EGP, Lau LF. 1996. Cyr61, product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell. Biol. 16:1326–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu F, Song Y, Liu D. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6:1258–1266 [DOI] [PubMed] [Google Scholar]
- 24. Mitchell C, Willenbring H. 2008. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 3:1167–1170 [DOI] [PubMed] [Google Scholar]
- 25. Mathijs K, Kienhuis AS, Brauers KJ, Jennen DG, Lahoz A, Kleinjans JC, van Delft JH. 2009. Assessing the metabolic competence of sandwich-cultured mouse primary hepatocytes. Drug Metab. Dispos. 37:1305–1311 [DOI] [PubMed] [Google Scholar]
- 26. Weiskirchen R, Gressner AM. 2005. Isolation and culture of hepatic stellate cells. Methods Mol. Med. 117:99–113 [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. 2007. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 46:1246–1256 [DOI] [PubMed] [Google Scholar]
- 28. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. 2009. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4:1798–1806 [DOI] [PubMed] [Google Scholar]
- 29. Edwards CA, O'Brien J. 1980. Modified assay for determination of hydroxyproline in tissue hydrolyzate. Clin. Chim. Acta 104:161–167 [DOI] [PubMed] [Google Scholar]
- 30. Juric V, Chen CC, Lau LF. 2009. Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol. Cell. Biol. 29:3266–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen C-C, Mo F-E, Lau LF. 2001. The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J. Biol. Chem. 276:47329–47337 [DOI] [PubMed] [Google Scholar]
- 32. Athanasopoulos AN, Schneider D, Keiper T, Alt V, Pendurthi UR, Liegibel UM, Sommer U, Nawroth PP, Kasperk C, Chavakis T. 2007. Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J. Biol. Chem. 282:26746–26753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. 2002. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell. Biol. 22:8709–8720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mo F-E, Lau LF. 2006. The matricellular protein CCN1 is essential for cardiac development. Circ. Res. 99:961–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gressner OA, Gressner AM. 2008. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 28:1065–1079 [DOI] [PubMed] [Google Scholar]
- 36. Huang G, Brigstock DR. 2012. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 17:2495–2507 [DOI] [PubMed] [Google Scholar]
- 37. Elsharkawy AM, Oakley F, Mann DA. 2005. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 10:927–939 [DOI] [PubMed] [Google Scholar]
- 38. Chen N, Chen CC, Lau LF. 2000. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275:24953–24961 [DOI] [PubMed] [Google Scholar]
- 39. Leu S-J, Chen N, Chen C-C, Todorovic V, Bai T, Juric V, Liu Y, Yan G, Lam SCT, Lau LF. 2004. Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J. Biol. Chem. 279:44177–44187 [DOI] [PubMed] [Google Scholar]
- 40. Leu S-J, Liu Y, Chen N, Chen CC, Lam SC, Lau LF. 2003. Identification of a novel integrin α6β1 binding site in the angiogenic inducer CCN1 (CYR61). J. Biol. Chem. 278:33801–33808 [DOI] [PubMed] [Google Scholar]
- 41. Chen C-C, Chen N, Lau LF. 2001. The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J. Biol. Chem. 276:10443–10452 [DOI] [PubMed] [Google Scholar]
- 42. Cheng G, Diebold BA, Hughes Y, Lambeth JD. 2006. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 281:17718–17726 [DOI] [PubMed] [Google Scholar]
- 43. Lambeth JD, Kawahara T, Diebold B. 2007. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 43:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, Kisseleva T, Brenner DA. 2011. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 53:1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cui W, Matsuno K, Iwata K, Ibi M, Matsumoto M, Zhang J, Zhu K, Katsuyama M, Torok NJ, Yabe-Nishimura C. 2011. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 54:949–958 [DOI] [PubMed] [Google Scholar]
- 46. Jun JI, Lau LF. 2010. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campisi J, d'Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8:729–740 [DOI] [PubMed] [Google Scholar]
- 48. Collado M, Blasco MA, Serrano M. 2007. Cellular senescence in cancer and aging. Cell 130:223–233 [DOI] [PubMed] [Google Scholar]
- 49. Yoon PO, Lee MA, Cha H, Jeong MH, Kim J, Jang SP, Choi BY, Jeong D, Yang DK, Hajjar RJ, Park WJ. 2010. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 49:294–303 [DOI] [PubMed] [Google Scholar]
- 50. Brigstock DR. 2009. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: lessons from transgenic animals. J. Cell Commun. Signal. 4:1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lambeth JD, Krause KH, Clark RA. 2008. NOX enzymes as novel targets for drug development. Semin. Immunopathol. 30:339–363 [DOI] [PubMed] [Google Scholar]
- 52. De MS, Brenner DA. 2008. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J. Gastroenterol. Hepatol. 23(Suppl 1):S98–S103 [DOI] [PubMed] [Google Scholar]
- 53. Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schroder K, Brandes RP, Devaraj S, Torok NJ. 2012. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 53:289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. 2012. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56:2316–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cohen-Naftaly M, Friedman SL. 2011. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap. Adv. Gastroenterol. 4:391–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, Takimoto R, Takada K, Miyanishi K, Matsunaga T, Takayama T, Niitsu Y. 2008. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 26:431–442 [DOI] [PubMed] [Google Scholar]