

Abstract
Hepatic scavenger receptor class B type I (SR-BI) plays an important role in selective high-density lipoprotein cholesterol (HDL-C) uptake, which is a pivotal step of reverse cholesterol transport. In this study, the potential involvement of microRNAs (miRNAs) in posttranscriptional regulation of hepatic SR-BI and selective HDL-C uptake was investigated. The level of SR-BI expression was repressed by miRNA 185 (miR-185), miR-96, and miR-223, while the uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)-HDL was decreased by 31.9% (P < 0.001), 23.9% (P < 0.05), and 15.4% (P < 0.05), respectively, in HepG2 cells. The inhibition of these miRNAs by their anti-miRNAs had opposite effects in these hepatic cells. The critical effect of miR-185 was further validated by the loss of regulation in constructs with mutated miR-185 target sites. In addition, these miRNAs directly targeted the 3′ untranslated region (UTR) of SR-BI with a coordinated effect. Interestingly, the decrease of miR-96 and miR-185 coincided with the increase of SR-BI in the livers of ApoE KO mice on a high-fat diet. These data suggest that miR-185, miR-96, and miR-223 may repress selective HDL-C uptake through the inhibition of SR-BI in human hepatic cells, implying a novel mode of regulation of hepatic SR-BI and an important role of miRNAs in modulating cholesterol metabolism.
INTRODUCTION
Atherosclerotic cardiovascular disease is one of the leading causes of morbidity and mortality in industrialized and developing nations. Numerous long-term clinical trials have demonstrated an inverse relationship between plasma high-density lipoprotein cholesterol (HDL-C) levels and atherosclerosis independent of low-density lipoprotein cholesterol (LDL-C); the latter is a well-established major risk factor for atherosclerosis (1). The protective effect of HDL may mainly be attributed to its role in reverse cholesterol transport (RCT), whereby peripheral (extrahepatic) cholesterol is transported back to the liver for excretion into the bile and ultimately the feces (2, 3). Lipoprotein receptors play critical roles in cholesterol homeostasis both physiologically and pathologically, as exemplified by endocytosis of LDL-C via the LDL receptor (4). The selective uptake of HDL-C into the liver, a pivotal step of RCT, is mediated by scavenger receptor class B type I (SR-BI, or SCARB1) (5, 6).
SR-BI is a single-chain plasma membrane glycoprotein structurally related to CD36; the human homologue of SR-BI (hSR-BI) is also known as CLA-1 (CD36 and lysosomal integral membrane protein II analogous 1), which is mapped to chromosome 12 (7). As a high-affinity HDL receptor, SR-BI is most highly expressed in the liver and steroidogenic tissues, where it delivers HDL-C for excretion and steroid hormone synthesis (5). Abundant data from SR-BI gene-manipulated mice have definitely established that hepatic SR-BI is a critical regulator of overall HDL-C metabolism and that it presents antiatherogenic activity in vivo (8). The SR-BI expression level in the liver directly modulates HDL metabolism, which is delicately regulated at the transcriptional level. For example, SR-BI is under the control of different nuclear receptors and transcription factors, including peroxisome proliferator-activated receptors (PPAR) (9), liver X receptor (LXR) (10), liver receptor homologue 1 (LRH-1) (11), and sterol regulatory element-binding proteins (SREBPs) (12). Some posttranscriptional regulation mechanisms have also been reported, best exemplified by the alternative splicing of SR-BI pre-mRNA (13) and the interaction of SR-BI with its tissue-specific adaptor PDZK1 (14).
MicroRNAs (miRNAs) are a class of highly conserved, single-stranded, noncoding small RNAs (approximately 22 nucleotides in length) that regulate gene expression on the posttranscriptional level by promoting the degradation or inhibiting the translation of its target mRNA (15). These short, noncoding RNAs can be transcribed from their own promoter or included in the introns of other genes (15, 16). Regardless, most original primary miRNA (pri-miRNA) is processed by the RNase Drosha cotranscriptionally, resulting in a pre-miRNA, which is then exported to the cytoplasm for further cleavage by RNase Dicer to generate the mature miRNA (15, 16). In this study, a significant increase of SR-BI expression was observed by silencing the miRNA-processing enzymes Drosha and Dicer with small interfering RNA (siRNA), implying that miRNAs are involved in a complex mechanism to ensure the appropriate SR-BI gene expression regulation. Therefore, we further investigated the contribution of miRNAs to the posttranscriptional regulation of SR-BI and their potential involvement in the modulation of HDL-mediated selective uptake. Three miRNAs (miR-185, miR-96, and miR-223) were identified as regulators to effectively repress hSR-BI expression and HDL-mediated cholesterol selective uptake in hepatic cells by directly targeting the binding sites within the hSR-BI 3′ untranslated region (UTR), suggesting the importance of the miRNAs in the modulation of cholesterol metabolism.
MATERIALS AND METHODS
Cell culture.
Human hepatoma HepG2 cells were cultured in Eagle minimal essential medium (MEM) (Invitrogen, Carlsbad, CA) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Gibco, Grand Island, NY), sodium pyruvate, and nonessential amino acids (Invitrogen). Human hepatocellular carcinoma Bel-7402 cells, a human normal hepatic immortal cell line (HL-7702), and acute monocytic leukemia THP-1 cells were grown in RPMI 1640 medium (Invitrogen) containing 10% (vol/vol) FBS. For differentiation to macrophages, THP-1 cells were cultured at a density of 200,000 cells per cm2 and stimulated with 160 nM phorbol-12-myristate-13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO) for 24 h. Chinese hamster ovary (CHO) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (high glucose; Invitrogen)–F-12K (1:1) containing 10% (vol/vol) FBS. The cells were maintained at 37°C in a humidified 5% CO2 incubator.
Animals.
Experiments were performed in ApoE knockout (KO) mice purchased from the Department of Laboratory Animal Science of the Beijing University Health Science Center (Beijing, China). All experimental procedures involving animals were approved by the Institutional Laboratory Animal Care and Use Committee of Beijing University Health Science Center (Beijing, China). Eight-week-old male ApoE KO mice were fed a high-fat diet (HFD) containing 0.15% cholesterol and 20% lard or a regular rodent chow as a control. After 8 weeks of feeding, the animals were sacrificed; cholesterol was then measured, and the tissues were collected for further analysis.
3′ UTR luciferase reporter assays.
The firefly luciferase coding sequence was amplified from the pGL3-Basic vector (Promega, Madison, WI) and inserted upstream of the multiple cloning site in the pcDNA3.1 vector (Invitrogen) at the HindIII and XbaI sites, resulting in a pcDNA-luciferase reporter vector (pc-luc). The entire 3′ UTR of human SR-BI (U1) was generated by reverse transcription-PCR (RT-PCR) from total RNA extracted from HepG2 cells and fused downstream of the luciferase coding sequence in the pc-luc vector at the XbaI site. Using the U1 fragment as a template, other luciferase reporter constructs with a truncated SR-BI 3′ UTR, U2 to U6, were obtained by PCR. The deletions of the predicted seed regions of miR-185 and miR-96 within the human SR-BI 3′ UTR were generated by overlap extension PCR and then cloned into the pc-luc vector. All of the constructs were confirmed by sequencing. Each purified reporter plasmid (0.2 μg) was transfected into HepG2 cells at 70% confluence using Lipofectamine 2000 (Invitrogen) for 4 h before introduction of miRNA mimics, anti-miRNAs, or negative-control duplexes (ctl-miR) for another 48 h. The cells were cotransfected with 0.02 μg Renilla luciferase control reporter vector pRL-TK (Promega) to normalize the transfection efficiency. The luciferase activity was measured using the Dual-Glo luciferase assay system (Promega) and detected by a Victor X5 multilabel plate reader (PerkinElmer, Waltham, MA). Firefly luciferase activity was normalized to the corresponding Renilla luciferase activity and plotted as a percentage of the control miRNA (ctl-miR).
hSR-BI expression in CHO cells.
The full-length human SR-BI cDNA was amplified by RT-PCR from total RNA extracted from HepG2 cells and inserted into the multiple cloning sites of the pcDNA3.1 vector (Invitrogen) at HindIII and KpnI, generating the recombinant SR-BI expression plasmid pc-SR-BI. The entire 3′ UTR of human SR-BI (the U1 fragment) or with mutated miR-185 target sites (the DEL1 and DEL2 fragment) was then cloned downstream of the SR-BI coding sequence in pc-SR-BI at the XbaI site, generating pc-WT and pc-185m, respectively. These constructs were transfected into CHO cells using Lipofectamine 2000 (Invitrogen).
Transfection with small RNAs.
Transfection of miRNA mimics, anti-miRNAs, and siRNAs was carried out using the Lipofectamine RNAiMax reagent (Invitrogen) according to the manufacturer's instructions. The miRNA mimics (double-stranded RNA oligonucleotides), anti-miRNA (2′-O-methyl antisense oligonucleotides against the target miRNAs), and negative-control duplexes were obtained from Qiagen (Hilden, Germany) and applied at a final concentration of 100 nM. Predesigned siRNAs specifically targeting the mRNA of human Dicer, Drosha, and SR-BI and the scrambled negative-control siRNA were obtained from Invitrogen and used at a final concentration of 100 nM. After the incubation of the small RNAs for 24 to 72 h, the cells were harvested for further analysis.
Real-time RT-PCR.
HepG2, Bel-7402, or HL-7702 cells or PMA-induced THP-1 cells were plated in 24-well culture dishes (200,000 cells/well) and transfected with either miRNA mimics or anti-miRNAs, as well as their control miRNAs, for 48 or 72 h. For assessment of the mRNA level, total RNAs were isolated from the cultured cells with the SV total RNA isolation system (Promega) and quantified by Nanodrop spectrophotometry (Thermo Scientific, Wilmington, DE). RNA was reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen) following the manufacturer's instructions. Specific real-time PCR primers were used to quantify the mRNA levels. Real-time PCR was performed in the CFX96 real-time system (Bio-Rad, CA) using a SYBR green Master Kit (Roche, Mannheim, Germany) as described previously (17). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the normalization control. For the quantitative analyses of the miRNAs, an RNeasy minikit (Qiagen) was used to purify small RNAs from the cultured cells and tissue samples with the inclusion of a DNase I (Qiagen) treatment step. Reverse transcription was performed with the miScript II RT kit (Qiagen). miRNA expression levels in the sorted samples were assessed by real-time PCR using the miScript SYBR green PCR kit (Qiagen). U6 small nuclear RNA (RNU6) served as the endogenous control for the miRNA expression levels.
Western blotting.
Cells were treated as described above for transcript analysis. Whole-cell protein lysates were prepared using RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6) containing complete EDTA-free protease inhibitor cocktail (Roche). The protein extracts (50 μg) were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to a 0.45-μm polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). The membranes were probed with antibodies to hSR-BI (1:2,000; BD Biosciences, San Jose, CA) and β-actin (1:1,000; Cell Signaling Technology Inc., Beverly, MA). The proteins were visualized using the appropriate horseradish peroxidase-conjugated secondary antibodies (1:1,000; BD Biosciences) and chemiluminescence reagents (Thermo Scientific).
Analysis of cell surface expression by flow cytometry.
Cells were transfected with miRNA mimics, anti-miRNAs, or control miRNAs for 72 h, as described above. The cells were detached from the plates by trypsinization (0.25% trypsin with 0.02% EDTA) for 2 min before phosphate-buffered saline (PBS) containing 10% FBS was immediately added to stop the trypsin action. The cells were then washed with PBS and incubated with the primary mouse anti-human SR-BI monoclonal antibody (1:1,000; BD Biosciences) for 50 min at 4°C and, subsequently, with the secondary fluorescein isothiocyanate (FITC)-conjugated rabbit anti-mouse antibody (1:2,000; BD Biosciences) for 50 min at 4°C. For flow cytometry analysis, cells were collected by centrifugation (800 × g; 3 min; 4°C), and the pellet obtained was resuspended in PBS buffer. A total of 10,000 cells were counted, and FITC fluorescence was analyzed using an Accuri C6 flow cytometer (BD Biosciences).
Assays of the cellular uptake of DiI-HDL.
Different cells were transfected with either miRNA mimics, anti-miRNAs, or their control miRNAs as described above, followed by incubation with 2 μg/ml 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)-labeled HDL (Biomedical Technologies, Stoughton, MA) for 4 h at 37°C. After cell harvest and washes, fluorescence measurements were performed with the flow cytometer as described previously (18). For visualization, the cells were additionally stained with 4′,6-diamidino-2-phenylindole (DAPI) and photographed at 360 nm/460 nm (DAPI) and 480 nm/600 nm (DiI) with an IN Cell Analyzer 1000 (GE Healthcare, Piscataway, NJ) after washing with PBS twice.
Statistical analyses and bioinformatics.
The results presented are derived from at least three independent experiments. The data are presented as the mean and standard error of the mean (SEM), and statistical significance was evaluated by Student's t test. Multiple comparisons were made using analysis of variance (ANOVA) followed by a post hoc Newman-Keuls test. P values of <0.05 were considered statistically significant. The microRNA.org (http://www.microrna.org), TargetScan (http://www.targetscan.org), and miRBase (http://www.mirbase.org) databases were used for in silico computational target predictions and miRNA sequence analyses.
RESULTS
hSR-BI expression in hepatic cells is modulated by miRNAs.
miRNAs posttranscriptionally regulate gene expression by binding to the 3′ UTRs of their target mRNAs. The 3′ UTR of the human SR-BI gene is 959 bp long, which raises the possibility of its being regulated by miRNAs. RNA interference-mediated knockdown of the components of the miRNA biogenesis pathway was applied to detect the involvement of miRNAs in SR-BI gene regulation. The transfection of HepG2 cells with siRNAs directed against Drosha or Dicer (inhibitory efficiency, 64% or 62%, respectively), resulted in a marked increase of SR-BI mRNA and protein levels (Fig. 1A). Therefore, we hypothesized that the 3′ UTR of the SR-BI mRNA is susceptible to being targeted by miRNAs in either a direct or an indirect manner.
Fig 1.

miR-185, miR-96, and miR-223 are involved in SR-BI modulation in HepG2 cells. (A) Transfection of siRNAs against Drosha and Dicer results in increased expression of SR-BI in HepG2 cells. *, P < 0.05, and **, P < 0.01 versus scr-si. (B to D) SR-BI mRNA levels were detected by real-time RT-PCR (B), protein levels were analyzed by Western blotting (C), and cell surface SR-BI levels were detected by flow cytometry (D) in HepG2 cells transfected with either miRNA mimics (100 nM) or anti-miRNAs (100 nM), as well as ctl-miR (100 nM), for 72 h. *, P < 0.05, and **, P < 0.01 versus ctl-miR of miRNA mimics; #, P < 0.05 versus ctl-miR of anti-miRNAs. All of the data are presented as the mean values and SEM of 3 independent experiments. A representative result from 3 independent experiments is shown in panel C. scr-si, scrambled siRNA; Dicer-si, Dicer siRNA; Drosha-si, Drosha siRNA.
To identify the potential miRNA candidates that target the SR-BI 3′ UTR, both TargetScan (http://www.targetscan.org) and miRanda (http://www.microrna.org), two bioinformatics tools, were applied for miRNA target prediction. The former tool revealed 45 predicted miRNAs that target the SR-BI 3′ UTR, whereas the latter predicted 20 conserved miRNAs with good miRNA support vector regression (mirSVR) scores. To assess whether these potential miRNAs regulated SR-BI expression, the mRNA level of SR-BI was detected by RT-PCR after transfection with some of the predicted miRNAs in HepG2 cells. Interestingly, several effective miRNAs were found, and among them, miR-185, miR-96, and miR-223 had the most pronounced inhibitory effects on SR-BI mRNA levels.
miR-185, miR-96, and miR-223 repress hSR-BI expression in hepatic cells by targeting the 3′ UTR.
The transfection of HepG2 cells with 100 nM miR-185, miR-96, or miR-223 significantly decreased SR-BI at both the mRNA (Fig. 1B) and protein (Fig. 1C) levels. Similar results were also observed when the cell surface SR-BI protein level was measured by flow cytometry (Fig. 1D; see Fig. S1D in the supplemental material). To further determine whether these miRNAs are specifically involved in regulating SR-BI expression, antisense oligonucleotides directed against miR-185, miR-96, and miR-223 were applied. The results showed that inhibition of endogenous miRNAs by anti-miR-185, anti-miR-96, or anti-miR-223 significantly increased the expression of SR-BI in HepG2 cells at 72 h (Fig. 1B to D), further confirming that miR-185, miR-96, and miR-223 may be involved in regulating SR-BI expression. The results were similar in two other human hepatic cell lines, Bel-7402 and HL-7702 (see Fig. S1A, B, and C in the supplemental material), indicating that the modulating effects of these miRNAs are not due to cell line specificity.
The predicted binding sites for the 3 miRNAs are located in different sections of the hSR-BI mRNA 3′ UTR according to the bioinformatic miRNA target prediction (Fig. 2A), and there are 2 computationally predicted miR-185 binding sites (site 1 and site 2) (Fig. 2A). Sequence alignment revealed that the 4 sites for the 3 miRNAs are conserved among different mammals (Fig. 2B). As for rodents, the predicted target site for miR-96 is conserved in the mouse SR-BI 3′ UTR, while there is no predicted target site for miR-223 in the SR-BI 3′ UTR of mouse, rat, and guinea pig. For miR-185, site 2, but not site 1, is conserved in guinea pig. In addition, within a stretch of phylogenetically conserved sequence of the mouse and rat SR-BI 3′ UTRs, there is a G-U wobble at the second nucleotide of miR-185 target site 1, which is a thermodynamically favorable base pair in the RNA secondary structure. To assess the effects of the 3 miRNAs on the SR-BI mRNA 3′ UTR, a luciferase reporter assay was conducted. The results showed that miR-185, miR-96, and miR-223 markedly reduced the activity of the full-length SR-BI mRNA 3′ UTR reporter construct (U1) (Fig. 2C). According to the 4 predicted target binding sites for these 3 miRNAs, luciferase reporter constructs with different 5′-end-truncated SR-BI 3′ UTRs were constructed (U2 to U6) and applied to evaluate the effects of the 3 miRNAs on their target sites in HepG2 cells. The lack of a binding site for miR-185 (site 1, U2 versus U1; site 2, U3 versus U2), miR-96 (U4 versus U3), or miR-223 (U6 versus U5) markedly diminished each miRNA-induced repression of SR-BI mRNA 3′ UTR activity (Fig. 2C), implying that all 3 miRNAs are involved in SR-BI posttranscriptional regulation. It was further confirmed that direct removal of the seed region of the binding sites for miR-185 (DEL1, 84 bp to 89 bp, and/or DEL2, 274 bp to 281 bp) or miR-96 (461 bp to 468 bp) abolished miR-185- or miR-96-induced downregulation and anti-miR-185- or anti-miR-96-induced upregulation (Fig. 2D; see Fig. S2 in the supplemental material). Taken together, these results demonstrate that miR-185, miR-96, and miR-223 may regulate SR-BI posttranscriptionally by binding to their target sites located in the SR-BI 3′ UTR.
Fig 2.

miR-185, miR-96, and miR-223 modulate SR-BI at the posttranscriptional level through the 3′ UTR. (A) Human SR-BI harbors two putative miR-185 target sites, a miR-96 site, and a miR-223 site within its 3′ UTR. The relative positions of their binding sites are indicated by different colors. Position 1 represents the first nucleotide following the termination codon. The sequence alignment of the human hsa-miR-185, hsa-miR-96, and hsa-miR-223 with the binding sites of the human SR-BI 3′ UTR is shown below. (B) Sequence alignment of the seed regions of the binding sites for miR-185, miR-96, and miR-223 within the indicated species. The conserved sequences are shown in red. (C) Effects of the 3 miRNAs on the activities of the luciferase reporter constructs with different truncated 3′ UTRs of hSR-BI (U2 to U6) compared with the luciferase reporter construct containing the full-length 3′ UTR of hSR-BI (U1). The effect of control miRNA on the luciferase activities of different constructs (U1 to U6) was defined as 1. *, P < 0.05, and **, P < 0.01 versus ctl-miR; #, P < 0.05, and ##, P < 0.01; n = 3. (D) The SR-BI 3′ UTR with deletion of the indicated seed region of the miR-185 binding sites (DEL1 and DEL2) or the miR-96 binding site (DEL3). HepG2 cells were transfected with different constructs for 5 h, followed by transient transfection with miR-185/anti-miR-185 (100 nM) or miR-96/anti-miR-96 (100 nM) for 48 h. Firefly luciferase activity was detected and normalized with Renilla luciferase. The effect of the control miRNA on the luciferase activities of the different constructs was defined as 1. The data are shown as mean values and SEM and were obtained from three independent experiments performed in triplicate. *, P < 0.05, and **, P < 0.01 versus ctl-miR; #, P < 0.05, and ##, P < 0.01 versus the wild-type (WT) construct. Hsa, H. sapiens; Ptr, Pan troglodytes; Mml, Macaca mulatta; Rno, Rattus norvegicus; Cpo, Cavia porcellus; Mmu, Mus musculus.
miR-185, miR-96, and miR-223 regulate selective HDL-C uptake in hepatic cells.
As the HDL receptor, the main function of SR-BI is to mediate selective HDL-C uptake in the liver, the final phase of RCT, which is critical to the antiatherosclerotic role of HDL. To assess the inhibitory effects of the 3 miRNAs on the selective uptake of lipids from HDL, fluorescence-labeled DiI-HDL uptake by HepG2 cells was measured. HepG2 cells were treated with miR-185, miR-96, or miR-223 mimics or their anti-miRNAs for 72 h, followed by an additional 4 h of incubation with DiI-HDL. All 3 miRNAs markedly attenuated DiI-HDL uptake into HepG2 cells (Fig. 3A), in accordance with the decreased cellular and cell surface SR-BI levels (Fig. 1C and D). Notably, antagonism of endogenous miR-185 or miR-96 significantly increased the uptake of DiI-HDL into HepG2 cells (Fig. 3A), along with the increased expression of SR-BI (Fig. 1C and D). Similar results were obtained in Bel-7402 and HL-7702 cells with miR-185/96 and anti-miR-185/96 treatment (see Fig. S1B and C in the supplemental material). Fluorescence visualization with the IN Cell Analyzer 1000 (GE Healthcare) detected lipid accumulation in HepG2 cells after incubation of 2 μg/ml DiI-HDL for 4 h (Fig. 3B, panel g versus panel c). Compared with ctl-miR-transfected cells, fluorescence was clearly decreased in miR-185-, miR-96-, or miR-223-transfected cells (Fig. 3B, panels k, o, and s versus panel g).
Fig 3.
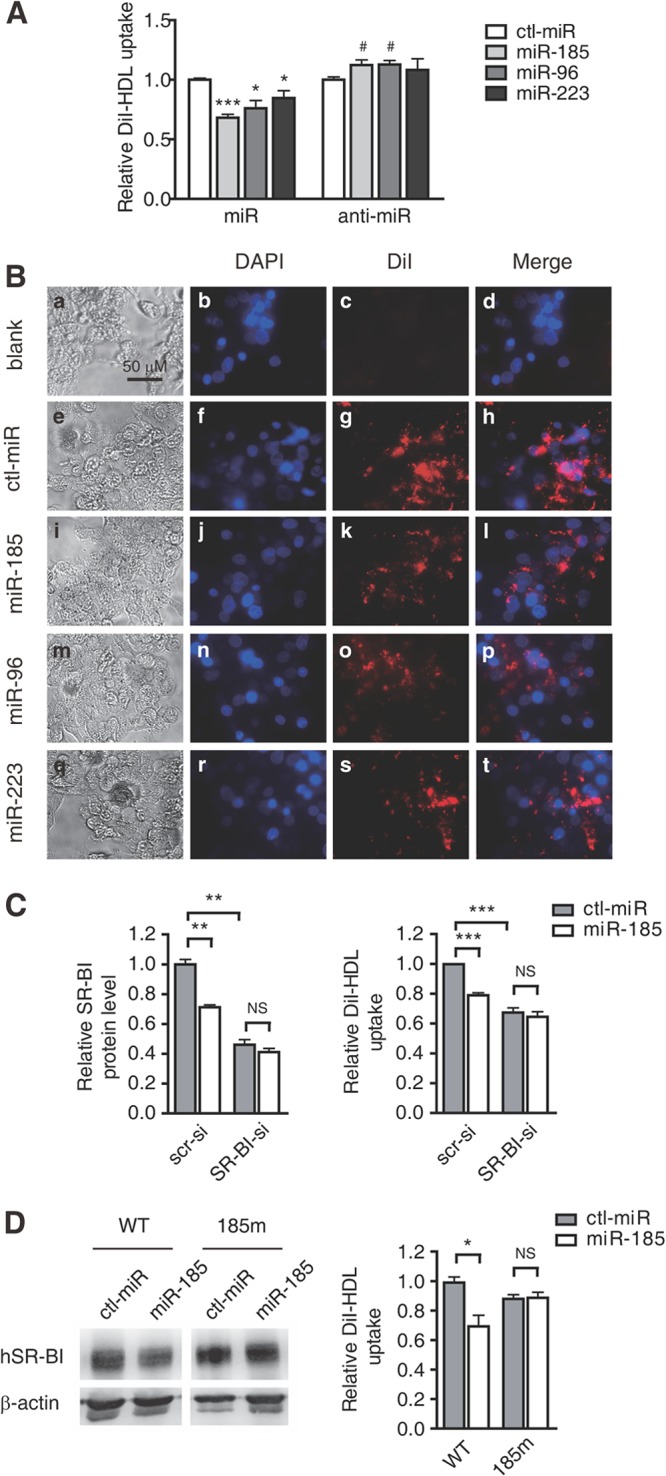
miR-185, miR-96, and miR-223 regulate selective HDL-C uptake into hepatic cells depending on SR-BI. (A) Flow cytometry analysis to determine the effects of miR-185, miR-96, and miR-223 on lipid uptake in HepG2 cells. HepG2 cells were incubated with 2 μg/ml DiI-HDL for 4 h after treatment with the miRNA mimics (100 nM) or the anti-miRNAs (100 nM) for 72 h. The data are shown as mean values and SEM of 3 independent experiments in triplicate. *, P < 0.05, and ***, P < 0.001 versus ctl-miR of miRNA mimics; #, P < 0.05 versus ctl-miR of anti-miRNA. (B) HepG2 cells were incubated with DiI-HDL after treatment with ctl-miR or the 3 miRNAs for 72 h and were visualized and photographed using the IN Cell Analyzer 1000 with a 20× objective. “Blank” indicates cells that were not incubated with DiI-HDL. The experiment was repeated 3 times, and the representative results are presented. (C) Effects of miR-185 on cell surface SR-BI protein levels and DiI-HDL selective uptake by HepG2 cells with SR-BI siRNA as analyzed using flow cytometry. (D) Effects of miR-185 on cellular SR-BI protein levels and DiI-HDL selective uptake in pc-WT- or pc-185m-transfected CHO cells. CHO cells were transfected with pc-WT or pc-185m for 6 h and subjected to miR-185 (100 nM) for 48 h. The cellular SR-BI protein levels were analyzed using Western blotting. The DiI-HDL uptake was analyzed by incubating the cells with 2 μg/ml DiI-HDL for another 4 h and subjecting them to flow cytometry. The experiment was repeated 3 times. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant. SR-BI-si, SR-BI siRNA.
To assess the role of SR-BI in the miRNA-mediated repression of selective HDL-C uptake, HepG2 cells transfected with small interfering RNA against SR-BI were used. The results showed that the inhibitory effect of miR-185 on cell surface SR-BI levels and DiI-HDL uptake by HepG2 cells was significantly attenuated after SR-BI silencing (Fig. 3C), suggesting that miR-185 regulates DiI-HDL uptake via SR-BI. To further confirm this, an SR-BI expression vector containing its entire 3′ UTR (pc-WT) or with mutated miR-185 target sites (pc-185m) was constructed and introduced into CHO cells, which exhibited a relatively low background of DiI-HDL uptake (data not shown). When the CHO cells transfected with the expression plasmids were subjected to miRNA-185 mimics, the hSR-BI protein level and DiI-HDL uptake were inhibited in pc-WT-transfected CHO cells but not in pc-185m-transfected CHO cells (Fig. 3D). Taken together, it was demonstrated that miR-185 blocks selective cholesterol uptake by repressing HDL receptor SR-BI expression.
Next, we evaluated whether the 3 miRNAs might function in a coordinated manner. The results showed that miR-185, miR-96, and miR-223 mimics decreased luciferase activity in HepG2 cells in a dose-dependent manner and reached maximal effect at 200 nM (Fig. 4A). Thus, coordinate effects of miR-185, miR-96, and miR-223 were detected at 200 nM in the full-length SR-BI 3′ UTR luciferase reporter assay. The results showed that the combination of two or three miRNAs was much more effective than either alone (Fig. 4B), implying their coordination with each other in SR-BI gene regulation. The coordinated effect of these miRNAs was further confirmed by the selective HDL-C uptake in HepG2 cells (Fig. 4C).
Fig 4.

miR-185, miR-96, and miR-223 repress luciferase activity and selective HDL-C uptake in a coordinated manner. (A) Activity of the luciferase reporter construct containing the 3′ UTR of hSR-BI (U1) transfected in HepG2 cells. Different concentrations (50, 100, 200, and 250 nM) of the 3 miRNAs were transfected for 48 h. The effect of control miRNA on luciferase activity was defined as 1 for each concentration. The data are shown as mean values and SEM and were obtained from 3 to 5 independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Luciferase reporter analysis of a coordinated effect of miR-185, miR-96, and miR-223. miRNA mimics (200 nM) were transfected individually or together for 48 h before detection. (C) The selective uptake of DiI-HDL by HepG2 cells transfected with the 3 miRNAs individually or together for 72 h was detected using flow cytometry. All of the data shown are significant compared to ctl-miR (a); #, P < 0.05, and $, P < 0.01 compared to miR-185 plus miR-96 plus miR-223 (b); n = 3. **, P < 0.01.
miRNA expression and regulation by cholesterol in vivo.
The functional significance of miR-96 was addressed by determining its in vivo expression in various ApoE KO mouse tissues for its predicted binding sites within the mouse SR-BI 3′ UTR. As shown in Fig. 5A, miR-96 is broadly expressed in different tissues and is particularly abundant in the kidney, liver, and intestine compared with other tissues. The expression of miR-185 was also detected in the different tissues (Fig. 5A), although there is a G-U wobble in the seed region of the target site for miR-185 in the mouse SR-BI 3′ UTR. To check whether these miRNAs are modulated under cholesterol-loaded physiological conditions in vivo, their expression in the livers of ApoE KO mice fed either a normal chow diet or HFD for 8 weeks was measured. As expected, the HFD-treated ApoE KO mice showed a notable increase in body weight and plasma total cholesterol and LDL cholesterol levels compared with the normally fed ApoE KO mice (Fig. 5B). Interestingly, hepatic levels of miR-96 were significantly decreased in the HFD-treated group, whereas the expression of SR-BI showed an inverse effect (Fig. 5C), suggesting the possible involvement of miR-96 in the regulation of SR-BI functioning in the delivery of excess cholesterol to the liver. The hepatic miR-185 was also decreased in the HFD-treated group (Fig. 5C), implying that miR-185 may be involved in mouse SR-BI regulation in a direct or an indirect manner.
Fig 5.

Distribution and dietary regulation of miR-96 and miR-185 in mice. (A) Expression profile of miR-96 and miR-185 in selected mouse tissues by quantitative RT-PCR. (B) Measurement of body weight, total cholesterol plasma levels, and LDL cholesterol levels of ApoE KO mice fed a chow diet (chow) or HFD for 8 weeks. (C) Real-time RT-PCR analysis of miRNA levels and SR-BI expression in the livers of ApoE KO mice fed a chow diet or HFD for 8 weeks. miRNA levels were normalized to U6. *, P < 0.05; **, P < 0.01; n = 6 (chow) and n = 5 (HFD). U6, U6 small nuclear RNA.
miR-185, miR-96, and miR-223 regulate SR-BI expression in macrophages.
In addition to its pivotal role in RCT in the liver, human SR-BI is expressed in cultured macrophages and atherosclerotic lesions (19). Therefore, we further evaluated the expression levels of the miRNAs identified above in PMA-stimulated THP-1 cells, which display macrophage-like differentiation. The levels of the 3 miRNAs in the PMA-stimulated THP-1 cells was about 1 order of magnitude higher than that in the untreated THP-1 cells (Fig. 6A). The expression levels of the 3 miRNAs in peritoneal macrophages from mice, as well as in mouse RAW 264.7 macrophages, were also detected. The results showed that the miRNA levels were much lower than those from PMA-induced THP-1 cells, which might be attributed to the species difference (see Fig. S3 in the supplemental material). The inhibitory effects of these miRNAs on SR-BI expression were detected in PMA-induced THP-1 cells. As shown in Fig. 6B, the transfection of miR-185 and miR-96 mimics markedly decreased SR-BI mRNA levels, while miR-223 exerted little effect. Further analysis showed that miR-185 significantly repressed DiI-HDL uptake, as expected (Fig. 6C), indicating that miR-185 regulated DiI-HDL uptake by targeting SR-BI in PMA-induced THP-1 cells. Surprisingly, miR-96 increased DiI-HDL uptake (Fig. 6C), hinting that miR-96 might also regulate other pathways involved in cholesterol delivery in macrophages.
Fig 6.

miR-185 and miR-96 regulate macrophage SR-BI expression and HDL-C uptake. (A) Abundance of miR-185, miR-96, and miR-223 in PMA-induced or noninduced THP-1 cells by quantitative RT-PCR. miRNA levels were normalized by U6. (B) SR-BI mRNA levels in PMA-induced THP-1 cells. THP-1 cells were seeded and stimulated with 100 ng/ml PMA for 24 h before transfection with either the miRNA mimics (100 nM) or the ctl-miR (100 nM) for another 48 h. Next, the SR-BI mRNA was analyzed by real-time RT-PCR. (C) Flow cytometry analysis was performed to determine the effects of miR-185, miR-96, and miR-223 on lipid uptake by macrophages. PMA-induced THP-1 cells were incubated with 2 μg/ml DiI-HDL for 4 h prior to detection. The data are presented as mean values and SEM of 3 independent experiments. **, P < 0.01 versus ctl-miR.
DISCUSSION
SR-BI, the first molecularly defined and physiologically relevant HDL receptor, is abundantly expressed in the liver, the organ occupying a central position in the RCT pathway. Numerous studies have established that hepatic SR-BI is a major lipoprotein receptor mediating the selective uptake of periphery-derived cholesterol from HDL particles and that it possesses atheroprotective activity (8). A better understanding of the molecular mechanisms that regulate SR-BI expression in the liver may provide new insight into the global modulation of HDL metabolism. In addition to elaborate regulation at the transcriptional level, SR-BI regulation in the liver is also exerted at the posttranscriptional level, allowing fast and adaptable modifications in the amount of available SR-BI mRNA and/or protein (20). A wide variety of this type of regulation of SR-BI has been reported in the liver in previous studies involving the alternative splicing of pre-mRNA, protein stabilization, and subcellular translocation (13, 21, 22). However, the repression of mRNA transcripts by miRNAs, one centrally important mode of posttranscriptional regulation, has been poorly studied in hepatic SR-BI modulation.
Bioinformatic predictions generate dozens of potential miRNAs targeting the hSR-BI 3′ UTR, suggesting that miRNAs are involved in the complex mechanism to ensure appropriate SR-BI gene expression regulation. In this work, we first examined the potential involvement of miRNAs by knocking down Dicer and Drosha, which are two important enzymes required for miRNA maturation. The results provided primary evidence that miRNAs are associated with the regulation of the HDL receptor SR-BI in hepatic cells in either a direct or an indirect manner. Next, we presented the first experimental evidence of the involvement of miR-185, miR-96, and miR-223 in modulating cellular HDL cholesterol delivery by repressing the expression of SR-BI in different human liver cell lines. We also demonstrated that the miRNAs identified above regulated endogenous SR-BI expression and DiI-HDL uptake by treating the cells with antisense inhibitors. Furthermore, the inhibitory effect of miR-185 on the selective uptake of lipids from HDL was diminished when SR-BI was expressed in CHO cells with mutated miR-185 target sites in its 3′ UTR. Together with the application of SR-BI siRNA, it was indicated that SR-BI is the functional target of miR-185 in this process. However, it was found in some assays that the effect of the anti-miRNAs did not achieve statistical significance, while the miRNA mimics significantly repressed SR-BI expression and function. This phenomenon may be attributed to the low endogenous miRNA expression level in the cell lines. In addition, the transfection of miRNA mimics resulted in overexpression hundreds of times greater than the endogenous miRNA level, while the introduction of anti-miRNAs resulted in much less reduction (see Table S3 in the supplemental material), suggesting that the effect of the cell enrichment of the miRNA mimics is much greater than that of the anti-miRNAs. Interestingly, transfection with anti-miR-96 resulted in a significant increase in miR-185 levels (data not shown), which might have an inverse effect on SR-BI expression against anti-miR-96. All of these may contribute to the relatively nonapparent effects of some of the anti-miRNAs.
The direct targets within the hSR-BI 3′ UTR for miR-185, miR-96, and miR-223 were further confirmed by a detailed analysis of these sites in the luciferase reporter assay. Luciferase repression in HepG2 cells was relieved when the specific target site(s) was deleted, and the results of anti-miRNAs are consistent with the above-mentioned regulation of endogenous SR-BI expression. We also observed repression when two or three miRNAs were combined significantly greater than that conferred by each miRNA alone. It seems likely that these miRNAs simultaneously repress the hSR-BI transcript in human hepatic cells by targeting different regions of the 3′ UTR. Other studies have reported the coordinated regulation of a single mRNA transcript by multiple miRNAs under certain circumstances (23, 24).
There is a predicted target site for miR-96 in the mouse SR-BI 3′ UTR and a target site for miR-185 with a G-U wobble in the seed region located within a stretch of the phylogenetically conserved sequence. The G-U wobble is a thermodynamically favorable base pair in RNA secondary structure, and there are validated examples of miRNA-target interactions with wobbles within the seed region (25). Whether miR-185 is involved in the regulation of mouse SR-BI expression needs to be further investigated. Taken together, it will be interesting to know the changes in SR-BI and the expression levels of the miRNAs in rodents in vivo. The ApoE KO mice fed with an HFD showed repressed levels of miR-96 and miR-185 and a more than 2-fold-increased level of SR-BI in the liver. A similar increase in SR-BI expression was also observed in HFD-treated ApoE KO mice in other reports (26). The expression of SR-BI might be activated by numerous pathways independent of miRNAs, based on the large changes in the mouse metabolism (Fig. 5B). However, there is a possibility that the decreased expression of miR-96 and miR-185 is involved in the mechanisms to increase SR-B1 expression and to regulate the function of SR-BI in the delivery of excess cholesterol to the liver. The actual involvement of miRNAs in mouse SR-BI regulation may be further investigated by experiments using anti-miRNAs in vivo. As the target site of miR-223 is missing in the rodent SR-BI 3′ UTR, the presence of miR-223 in the human SR-BI 3′ UTR suggests species specificity and perhaps a more elaborated regulation of SR-BI by miRNAs in primates.
Interestingly, among the three miRNAs identified above, we found that miR-185 suppressed SR-BI expression and selective HDL-C uptake in PMA-induced THP-1 cells, which are cultured human monocyte-derived macrophages. Macrophages play a pivotal role in the deposition of excess cholesterol in atherogenesis. Studies have shown that SR-BI liver-specific conditional-knockout mice develop less atherosclerosis than SR-BI global-knockout mice, suggesting an atheroprotective role for SR-BI in peripheral tissues (27), although the mechanisms by which SR-BI modulates atherosclerosis are unclear. A recent study in cultured mouse bone marrow-derived macrophages found that SR-BI played a significant role in facilitating macrophage bidirectional cholesterol flux in the absence of lipid loading (28). Taken together, miR-185 may serve as a key regulator of lipid metabolism under normal physiological or pathological conditions by targeting SR-BI, not only in the liver, but also in macrophages. Surprisingly, miR-96 increased DiI-HDL uptake in PMA-induced THP-1 cells (Fig. 6C), suggesting that miR-96 might also be involved in other cholesterol delivery pathways in macrophages. Interestingly, there is a predicted target site of miR-96 in the 3′ UTR of the human ABCA1 gene, encoding an ATP-binding cassette transporter, which plays a pivotal role in the process of cholesterol efflux from macrophages (29).
Since the first two miRNAs, lin-4 and let-7, were discovered in the nematode Caenorhabditis elegans (30, 31), more than 1,000 human miRNAs have been identified. miRNAs have been found to be molecular “rheostats” or “switches” that modulate multiple facets of biological processes, including differentiation, development, proliferation, and apoptosis (32–34). In the previous study, some miRNAs have been reported to regulate lipid metabolism-related genes and pathways in the liver. miR-122 is most highly expressed in the liver, and silencing of miR-122 in mice altered the gene expression involved in cholesterol synthesis and fatty acid oxidation, resulting in the reduction of total plasma cholesterol and liver triglyceride content and an increase in the rate of fatty acid β-oxidation (35). Another well-known miRNA involved in lipid regulation is miR-33. It was independently reported by several groups that miR-33a/b represses multiple genes involved in cellular cholesterol trafficking, especially ABCA1, a key transporter in the generation of HDL and the removal of excess cholesterol from peripheral tissues, the first step of RCT (36, 37). Recently, miR-758 was discovered as another regulator directly targeting the ABCA1 mRNA 3′ UTR (38). Interestingly, HDL was reported to mediate miRNA transport (including miR-223) in plasma and delivery to recipient cells depending on SR-BI (39). As hSR-BI harbors a putative binding site for miR-223, the Renilla–SR-BI–3′ UTR luciferase reporters in SR-BI-expressing hamster kidney (BHK) cells were used to demonstrate that SR-BI-dependent HDL-miRNA delivery may result in the direct targeting of specific mRNAs. During the preparation of this article, there was a report on miRNA-125a-5p and miRNA-455 repressing SR-BI in steroidogenic cells (40). It was mentioned that miRNA-125a-5p inhibited the expression and function of SR-BI in Hepa 1-6 cells, a mouse hepatoma cell line. We examined whether miRNA-125a-5p regulated the expression level of hSR-BI in HepG2 cells; however, no effect was found on either the hSR-BI 3′ UTR reporter construct or the hSR-BI mRNA levels (see Fig. S4 in the supplemental material). These findings suggest that the effects of miR-125a-5p on SR-BI are species and/or cell specific.
In this work, we found that miR-185, miR-96, and miR-223 are involved in HDL cholesterol selective uptake as the posttranscriptional regulators of hepatic SR-BI. Gene analysis identified the sequences encoding miR-185 within the first intron of the unknown gene C22orf25 (Homo sapiens chromosome 22 open reading frame 25), which is located in the 22q11.21 chromosome region. miR-96 and miR-223 are located intergenically on chromosomes 7 and X, respectively. Previous studies have shown that miR-185 could regulate multiple cellular processes relevant to cancer initiation and progression, and some miR-185-targeted genes have been reported (41, 42). miR-96 was reported to be expressed in neurosensory cells and to be involved in the progression of cochlear hair cell differentiation in the mammalian auditory system (43). miR-223 was shown to be highly expressed in myeloid cells of the bone marrow, acting as “a fine tuner” of granulocytic differentiation and maturation (44). miR-223 has also been reported to be a potent regulator of some inflammatory responses (44, 45). Both miR-96 and miR-223, similar to miR-185, have been reported to regulate the genes involved in the proliferation of different tumor cells (46–49). Fortunately, miR-185, miR-96, and miR-223 did not influence the growth of the hepatic cancer cell lines used in this work (see Fig. S5 in the supplemental material). Until recently, the roles of miR-185, miR-96, and miR-223 in lipid metabolism had not been systematically explored.
In summary, our study demonstrated for the first time that the miR-185-, miR-96-, and miR-223-mediated repression of hepatic hSR-BI may be an important regulatory mechanism that modulates HDL cholesterol transport. Subsequent studies will further characterize the roles of these miRNAs in regulating lipid metabolism pathways and explore their in vivo effects on HDL delivery by applying the antagonism of endogenous miRNAs. These findings will provide new avenues to understand the complex genetic networks that regulate lipid homeostasis and may open new possibilities to develop potentially useful therapeutic strategies for the treatment of atherosclerosis and cardiovascular disease by enhancing SR-BI expression and HDL-C transport.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81102442 and 90813027) and National Mega-project for Innovative Drugs (2012ZX09301002-003, 2012ZX09301002-001, and 2010ZX09401-403).
We thank Jing Zhang for her help in the operation of the IN Cell Analyzer 1000. We thank Yi Bao for helpful discussions.
Footnotes
Published ahead of print 4 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01580-12.
REFERENCES
- 1. Boden WE. 2000. High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs High-Density Lipoprotein Intervention Trial. Am. J. Cardiol. 86:19L–22L [DOI] [PubMed] [Google Scholar]
- 2. Glomset JA, Norum KR. 1973. The metabolic role of lecithin: cholesterol acyltransferase: perspectives form pathology. Adv. Lipid Res. 11:1–65 [PubMed] [Google Scholar]
- 3. Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. 2012. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125:1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldstein JL, Brown MS. 1974. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 249:5153–5162 [PubMed] [Google Scholar]
- 5. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. 1996. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271:518–520 [DOI] [PubMed] [Google Scholar]
- 6. Krieger M. 1999. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu. Rev. Biochem. 68:523–558 [DOI] [PubMed] [Google Scholar]
- 7. Calvo D, Vega MA. 1993. Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem. 268:18929–18935 [PubMed] [Google Scholar]
- 8. Trigatti BL, Krieger M, Rigotti A. 2003. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 23:1732–1738 [DOI] [PubMed] [Google Scholar]
- 9. Lopez D, McLean MP. 2006. Activation of the rat scavenger receptor class B type I gene by PPARalpha. Mol. Cell. Endocrinol. 251:67–77 [DOI] [PubMed] [Google Scholar]
- 10. Malerod L, Juvet LK, Hanssen-Bauer A, Eskild W, Berg T. 2002. Oxysterol-activated LXRalpha/RXR induces hSR-BI-promoter activity in hepatoma cells and preadipocytes. Biochem. Biophys. Res. Commun. 299:916–923 [DOI] [PubMed] [Google Scholar]
- 11. Schoonjans K, Annicotte JS, Huby T, Botrugno OA, Fayard E, Ueda Y, Chapman J, Auwerx J. 2002. Liver receptor homolog 1 controls the expression of the scavenger receptor class B type I. EMBO Rep. 3:1181–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lopez D, McLean MP. 1999. Sterol regulatory element-binding protein-1a binds to cis elements in the promoter of the rat high density lipoprotein receptor SR-BI gene. Endocrinology 140:5669–5681 [DOI] [PubMed] [Google Scholar]
- 13. Zhang X, Moor AN, Merkler KA, Liu Q, McLean MP. 2007. Regulation of alternative splicing of liver scavenger receptor class B gene by estrogen and the involved regulatory splicing factors. Endocrinology 148:5295–5304 [DOI] [PubMed] [Google Scholar]
- 14. Yesilaltay A, Kocher O, Rigotti A, Krieger M. 2005. Regulation of SR-BI-mediated high-density lipoprotein metabolism by the tissue-specific adaptor protein PDZK1. Curr. Opin. Lipidol. 16:147–152 [DOI] [PubMed] [Google Scholar]
- 15. Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297 [DOI] [PubMed] [Google Scholar]
- 16. Olena AF, Patton JG. 2010. Genomic organization of microRNAs. J. Cell. Physiol. 222:540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bao Y, Wang L, Xu Y, Yang Y, Si S, Cho S, Hong B. 2012. Salvianolic acid B inhibits macrophage uptake of modified low density lipoprotein (mLDL) in a scavenger receptor CD36-dependent manner. Atherosclerosis 223:152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bao Y, Yang Y, Wang L, Gao L, Jiang W, Si S, Hong B. 2009. Identification of trichostatin A as a novel transcriptional up-regulator of scavenger receptor BI both in HepG2 and RAW 264.7 cells. Atherosclerosis 204:127–135 [DOI] [PubMed] [Google Scholar]
- 19. Chinetti G, Gbaguidi FG, Griglio S, Mallat Z, Antonucci M, Poulain P, Chapman J, Fruchart JC, Tedgui A, Najib-Fruchart J, Staels B. 2000. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 101:2411–2417 [DOI] [PubMed] [Google Scholar]
- 20. Leiva A, Verdejo H, Benitez ML, Martinez A, Busso D, Rigotti A. 2011. Mechanisms regulating hepatic SR-BI expression and their impact on HDL metabolism. Atherosclerosis 217:299–307 [DOI] [PubMed] [Google Scholar]
- 21. Hirano K, Ikegami C, Tsujii K, Zhang Z, Matsuura F, Nakagawa-Toyama Y, Koseki M, Masuda D, Maruyama T, Shimomura I, Ueda Y, Yamashita S. 2005. Probucol enhances the expression of human hepatic scavenger receptor class B type I, possibly through a species-specific mechanism. Arterioscler. Thromb. Vasc. Biol. 25:2422–2427 [DOI] [PubMed] [Google Scholar]
- 22. Shetty S, Eckhardt ER, Post SR, van der Westhuyzen DR. 2006. Phosphatidylinositol-3-kinase regulates scavenger receptor class B type I subcellular localization and selective lipid uptake in hepatocytes. Arterioscler. Thromb. Vasc. Biol. 26:2125–2131 [DOI] [PubMed] [Google Scholar]
- 23. Guttilla IK, White BA. 2009. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J. Biol. Chem. 284:23204–23216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. 2005. Combinatorial microRNA target predictions. Nat. Genet. 37:495–500 [DOI] [PubMed] [Google Scholar]
- 25. Thomas M, Lieberman J, Lal A. 2010. Desperately seeking microRNA targets. Nat. Struct. Mol. Biol. 17:1169–1174 [DOI] [PubMed] [Google Scholar]
- 26. Yesilaltay A, Daniels K, Pal R, Krieger M, Kocher O. 2009. Loss of PDZK1 causes coronary artery occlusion and myocardial infarction in Paigen diet-fed apolipoprotein E deficient mice. PLoS One 4:e8103 doi:10.1371/journal.pone.0008103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huby T, Doucet C, Dachet C, Ouzilleau B, Ueda Y, Afzal V, Rubin E, Chapman MJ, Lesnik P. 2006. Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J. Clin. Invest. 116:2767–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ji A, Meyer JM, Cai L, Akinmusire A, de Beer MC, Webb NR, van der Westhuyzen DR. 2011. Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis 217:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yvan-Charvet L, Wang N, Tall AR. 2010. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 30:139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854 [DOI] [PubMed] [Google Scholar]
- 31. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906 [DOI] [PubMed] [Google Scholar]
- 32. Ambros V. 2004. The functions of animal microRNAs. Nature 431:350–355 [DOI] [PubMed] [Google Scholar]
- 33. Belver L, Papavasiliou FN, Ramiro AR. 2011. MicroRNA control of lymphocyte differentiation and function. Curr. Opin. Immunol. 23:368–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu N, Olson EN. 2010. MicroRNA regulatory networks in cardiovascular development. Dev. Cell 18:510–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. 2006. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3:87–98 [DOI] [PubMed] [Google Scholar]
- 36. Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. 2010. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328:1566–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. 2010. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328:1570–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramirez CM, Davalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, Suarez Y, Fernandez-Hernando C. 2011. MicroRNA-758 regulates cholesterol efflux through posttranscriptional repression of ATP-binding cassette transporter A1. Arterioscler. Thromb. Vasc. Biol. 31:2707–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. 2011. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 13:423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu Z, Shen WJ, Kraemer FB, Azhar S. 2012. MicroRNAs-125a and 455 repress lipoprotein-supported steroidogenesis by targeting scavenger receptor class B, type I (SR-BI) in steroidogenic cells. Mol. Cell. Biol. 10.1128/MCB.01002-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Z, Tang H, Wang Z, Zhang B, Liu W, Lu H, Xiao L, Liu X, Wang R, Li X, Wu M, Li G. 2011. MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Mol. Cancer 10:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu M, Lang N, Chen X, Tang Q, Liu S, Huang J, Zheng Y, Bi F. 2011. miR-185 targets RhoA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 301:151–160 [DOI] [PubMed] [Google Scholar]
- 43. Kuhn S, Johnson SL, Furness DN, Chen J, Ingham N, Hilton JM, Steffes G, Lewis MA, Zampini V, Hackney CM, Masetto S, Holley MC, Steel KP, Marcotti W. 2011. miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc. Natl. Acad. Sci. U. S. A. 108:2355–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. 2008. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451:1125–1129 [DOI] [PubMed] [Google Scholar]
- 45. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, Li H, Wang G, Evans AR, Safe S, Wu C, Zhou B. 2012. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation 125:2892–2903 [DOI] [PubMed] [Google Scholar]
- 46. Xu XM, Qian JC, Deng ZL, Cai Z, Tang T, Wang P, Zhang KH, Cai JP. 2012. Expression of miR-21, miR-31, miR-96 and miR-135b is correlated with the clinical parameters of colorectal cancer. Oncol. Lett. 4:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin H, Dai T, Xiong H, Zhao X, Chen X, Yu C, Li J, Wang X, Song L. 2010. Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS One 5:e15797 doi:10.1371/journal.pone.0015797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li J, Guo Y, Liang X, Sun M, Wang G, De W, Wu W. 2012. MicroRNA-223 functions as an oncogene in human gastric cancer by targeting FBXW7/hCdc4. J. Cancer Res. Clin. Oncol. 138:763–774 [DOI] [PubMed] [Google Scholar]
- 49. Zeng X, Xiang J, Wu M, Xiong W, Tang H, Deng M, Li X, Liao Q, Su B, Luo Z, Zhou Y, Zhou M, Zeng Z, Shen S, Shuai C, Li G, Fang J, Peng S. 2012. Circulating miR-17, miR-20a, miR-29c, and miR-223 combined as non-invasive biomarkers in nasopharyngeal carcinoma. PLoS One 7:e46367 doi:10.1371/journal.pone.0046367 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.