

Abstract
p19ARF plays an essential role in the senescence of mouse cells, and its expression is lost by methylation or deletion of the ARF locus; otherwise, p53 is inactivated to bypass senescence. ARF expression is tightly regulated, but little is known about its posttranscriptional regulation. Here, we show that an RNA-binding protein, HuR (human antigen R), represses ARF mRNA translation, thereby maintaining the replicative life span of mouse embryonic fibroblasts (MEFs). Loss of HuR results in premature senescence, with concomitant increases in p19ARF but not p16Ink4a levels, and this senescence is not observed in ARF-null MEFs that retain an intact Ink4a locus. HuR depletion does not alter ARF transcription or stability but enhances ribosome association with ARF mRNA. Under these conditions, ARF mRNA accumulates in nucleoli, where it associates with nucleolin. Furthermore, adipose-specific deletion of the HuR gene results in increased p19ARF expression in aged animals, which is accompanied by decreased insulin sensitivity. Together, our findings demonstrate that p19ARF is also regulated at the translational level, and this translational regulation restrains the cellular life span and tissue functions in vivo.
INTRODUCTION
Most mammalian somatic cells have a limited replicative life span when cultured in vitro and eventually undergo irreversible growth arrest, called cellular senescence (1). Senescence is caused by excessive extracellular or intracellular stress, and senescent cells are observed in tissues of aged animals and in tissues that experience prolonged inflammation (2, 3). Two major tumor suppressor pathways, the p19ARF (p14ARF in humans)-p53 and p16Ink4a-retinoblastoma (Rb) pathways, play critical roles in inducing and maintaining permanent cell cycle arrest during cellular senescence (4, 5), and inactivation of these proteins bypasses cellular senescence, allowing damaged cells to survive and proliferate. Thus, senescence prevents the spread of damaged cells, eliminating potential malignant transformation, and acts as a potent tumor-suppressive mechanism in mammals (6, 7).
Human antigen R (HuR) is a ubiquitously expressed member of the ELAV/Hu protein family and is involved in diverse biological processes (8, 9). Loss of HuR causes midgestational embryonic lethality due to placental defects (10). Animals rescued from this defect can develop to later stages but mostly die by embryonic day 19.5 and exhibit prominent defects in skeletal and splenic development. HuR encodes an RNA-binding protein that controls the stability, translation, splicing, and intracellular localization of its target mRNA (11, 12). Canonically, HuR directly binds to AU-rich elements (ARE) in the 3′ untranslated region (3′UTR) of its target mRNA, and the biological consequence of HuR association varies depending on the mRNA to which it binds (13, 14). In most cases, HuR stabilizes the mRNA associated with it; for instance, HuR has been shown to stabilize VEGF, β-actin, DNMT3b, and TNF-α mRNA (15–18). The mechanisms by which HuR regulates mRNA stability are not fully understood, but competition with other ARE-binding proteins is likely to be involved (19). Additionally, HuR is involved in microRNA (miRNA) recruitment to target mRNA. In this case, binding of HuR adjacent to the let-7 binding site on c-Myc mRNA facilitates the recruitment of let-7-loaded miRNA-induced silencing complexes (RISC) (20). Conversely, HuR has been shown to compete with miR-494 on Nucleolin mRNA (21).
It has become evident that HuR controls replicative senescence in human diploid fibroblasts (HDFs) (22). HuR levels decline during senescence in HDFs and are low in aged human tissues, and miR-519 is responsible for the downregulation of HuR in senescent HDFs (23, 24). HuR destabilizes Ink4a mRNA, the encoded protein of which, p16Ink4a, plays an important role in the cellular senescence of HDFs. HuR recruits RISC to Ink4a mRNA, and this process does not require miRNA but is mediated by the direct interaction of RISC with HuR proteins on mRNA (25). Additionally, HuR controls the mRNA metabolism of other senescence-related genes, including p53, p21, and cyclin D1, in response to cellular stress, such as UV (19, 26, 27). Therefore, HuR likely participates in cellular senescence by organizing the expression of multiple genes.
While the involvement of HuR in human cellular senescence has been documented, little is known about the function of HuR in the replicative senescence of mouse cells. Unlike in human cells, where the Ink4a-Rb pathway plays a pivotal role in senescence, in mouse cells, the ARF-p53 pathway is essential and p16Ink4a is dispensable for senescence (28, 29). We show here that HuR maintains a replicative life span by repressing expression of the p19ARF tumor suppressor in mouse embryonic fibroblasts (MEFs). RNA interference (RNAi)-mediated HuR silencing in MEFs prematurely induces cellular senescence by activating the ARF-p53 pathway. In HuR-depleted cells, p19ARF levels, but not p16Ink4a levels, are increased due to enhanced translation of ARF mRNA. HuR associates weakly with the 5′UTR of ARF mRNA in living cells. In the absence of HuR, ARF mRNA accumulates in the nucleolus, where it associates with nucleolin, and nucleolin is required for p19ARF induction in HuR knockdown cells. Translational regulation is also observed in vivo, and adipose-specific HuR knockouts revealed progressive insulin resistance, with concomitant increased expression of p19ARF. Thus, HuR translationally represses p19ARF expression under normal conditions, thereby inhibiting cellular senescence and maintaining tissue functions in vivo.
MATERIALS AND METHODS
Cells and culture conditions.
NIH 3T3 and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 100 U/ml penicillin-streptomycin. MEFs were cultured in medium supplemented with 0.1 mM nonessential amino acids, 55 μM 2-mercaptoethanol, and 10 μg/ml gentamicin instead of penicillin and streptomycin. To analyze mRNA or protein stability, cells were treated with 2 μg/ml of actinomycin D or 100 μg/ml of cycloheximide (CHX), respectively. To induce adipocytic differentiation, cells were kept confluent for 2 days and switched to differentiation medium (DMEM containing 5 μg/ml insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine [IBMX; Sigma Chemicals, St. Louis, MO]). For 5-ethynil-2′-deoxyuridine (EdU) staining, cells were pulse-labeled with 10 μM EdU for 45 min. Labeled cells were visualized using the Click-iT EdU Alexa Fluor imaging kit according to the manufacturer's instructions (Life Technologies, Carlsbad, CA).
Senescence-associated β-galactosidase and Oil Red O staining.
Cells were washed in phosphate-buffered saline (PBS), fixed for 3 to 5 min (room temperature) in 2% formaldehyde-0.2% glutaraldehyde, washed, and incubated at 37°C with β-galactosidase (β-Gal) stain solution containing 1 mg of 5-bromo-4-chloro-3-indolyl-β-d-galactoside, 40 mM citric acid-sodium phosphate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2. Staining was evident after 12 to 16 h (2). To stain adipocytes, cells were washed twice with PBS and then incubated with 60% filtered Oil Red O solution (3 mg/ml of 2-propanol) for 30 min at 37°C. Cells were washed with 60% 2-propanol briefly and then with water before visualization. Phase-contrast images were taken with a Plan FI 40×/0.60 lens (Olympus, Tokyo, Japan) at ambient temperature using an inverted microscope (model IX71; Olympus) equipped with a DP70 digital camera system (Olympus). Images were acquired using DP Controller software (Olympus).
Plasmids, transfection, retrovirus production, and infection.
Mouse ARF and Ink4a cDNAs were obtained by PCR using the following primers: for full-length ARF, 5′-AAGGATCCTCTCGAGGTGCCTCAACGCC-3′ (sense) and 5′-AACTCGAGGACATTTTAAAAAGTATC-3′ (antisense); for Δ3′UTR ARF, the full-length sense primer and 5′-AAACTCGAGCTATGCCCGTCGGTCTGGGC-3′ (antisense); for Δ5′UTR ARF, 5′-AAGGATCCATGGGTCGCAGGTTCTTGG-3′ (sense) and the full-length antisense primer; for full-length Ink4a, 5′-AAGGATCCACTGGTCACACGACTGGGC-3′ (sense) and 5′-AAGAATTCGACATTTTAAAAAGTATC-3′ (antisense); and for the Ink4a open reading frame (ORF), 5′-AAGGATCCATGGAGTCCGCTGCAGACAG-3′ (sense) and 5′-AAGAATTCTTAGCTCTGCTCTTGGG-3′ (antisense). PCR products were digested with BamHI and XhoI (ARF) or BamHI and EcoRI (Ink4a) and cloned into a pcDNA3 vector (Life Technologies). For ARF-MS2, ARF cDNA (full length or Δ5′UTR) was cloned into the BamHI/XbaI sites of pcDNA3.1 Hygro (Life Technologies). The plasmid was then digested with NotI and XbaI, and annealed oligonucleotides, including a 2×MS2 tag sequence (sense, GGCCGCAAACATGAGGATCACCCATGTCCATGGTCGACGAGCTCAAACATGAGGATCACCCATGTCT, and antisense, CTAGAGACATGGGTGATCCTCATGTTTGAGCTCGTCGACCATGGACATGGGTGATCCTCATGTTTGC), were ligated. MS2-enhanced green fluorescent protein (EGFP)-nuclear localization signal (NLS) cDNA (30) and GFP-L10a expression plasmids were provided by Takashi Funatsu (University of Tokyo) and Leo Tsuda (National Center for Geriatrics and Gerontology), respectively. GFP-L10 cDNA was cloned into a murine stem cell virus (MSCV) vector.
For knockdown of nucleolin, 1 million MEFs were plated in a 10-cm-diameter dish and cultured in a medium without antibiotics. Synthetic small interfering RNA (siRNA) (Thermo Fisher Scientific, Lafayette, CO) was transfected using DhamaFect1 (Thermo Fisher Scientific) according to the manufacturer's instructions. Cells were collected at 48 h posttransfection for subsequent analysis.
For retrovirus production, 293T cells were transfected with retroviral expression plasmids together with helper plasmids, as described previously (31). Culture supernatants were harvested 24 to 60 h after transfection, pooled, and stored on ice. Exponentially growing cells in 10-cm-diameter culture dishes were infected with 3 ml of a fresh virus-containing supernatant in complete medium containing 8 μg/ml Polybrene. Infection was confirmed either by GFP expression or by selection for drug resistance.
RNA analyses.
RNAs were prepared from cells or immune complexes using TriPure isolation reagent (Roche, Indianapolis, IN), reverse transcribed using a PrimeScript reverse transcriptase (RT) reagent kit with the genomic DNA (gDNA) Eraser (TaKaRa, Shiga, Japan), and subjected to PCR using the following primers: for PAI-1, 5′-TCAGAGCAACAAGTTCAACTACACTGAG-3′ (sense) and 5′-CCCACTGTCAAGGCTCCATCACTTGCCCCA-3′ (antisense); for HuR, 5′-TTGGGCTACGGTTTTGTGAAC-3′ (sense) and 5′-CCCACTGATGTATAAGTTGGCAT-3′ (antisense); for ARF, 5′-GCCGCACCGGAATCCT-3′ (sense) and 5′-TTGAGCAGAAGAGCTGCTACGT-3′ (antisense); for Ink4a, 5′-CCCAACGCCCCGAACT-3′ (sense) and 5′-GCAGAAGAGCTGCTACGTGAA-3′ (antisense); for c-myc, 5′-TCTATTTGGGGACAGTGTTC-3′ (sense) and 5′-GGTCATAGTTCCTGTTGGTG-3′ (antisense); for p53, 5′-TGGAGAGTATTTCACCCTCAAGA-3′ (sense) and 5′-CTCCTCTGTAGCATGGGCATC-3′ (antisense); for β-actin, 5′-CTAAGGCCAACCGTGAAAAG-3′ (sense) and 5′-ACCAGAGGCATACAGGGACA-3′ (antisense); for 18S rRNA 5′-AGTCCCTGCCCTTTGTACACA-3′ (sense) and 5′-GATCCGAGGGCCTCACTAAAC-3′ (antisense); for AUF-1, 5′-TTTCTCCAGACACACCTGAAGA-3′ (sense) and 5′-CTGTTCCTTTGACATGGCTACTT-3′ (antisense); and for GFP, 5′-TCTGCACCACCGGCAAGCTG-3′ (sense) and 5′-TGCGCTCCTGGACGTAGCCT-3′ (antisense). Real-time PCR analysis was carried out on a Chromo4 real-time PCR system (Bio-Rad, Hercules, CA).
Immunoblotting and preparation of cytoplasmic and nuclear fractions.
Cell lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Proteins were detected with antibodies to γ-tubulin (GTU-88; Sigma Chemicals, St. Lois, MO), p19ARF, p16Ink4a, HuR, nucleolin, CDK4, lamin A/C, GFP, α-tubulin, peroxisome proliferator-activated receptor gamma (PPARγ) CCAAT/enhancer binding protein α (C/EBPα), C/EBPβ, C/EBPδ (5-C3-1, M-156, 3A2, MS-3, C-22, H-110, FL, B-7, E-8, 14AA, C-19, and C-22; all from Santa Cruz Biotechnology, Santa Cruz, CA), RPL10 (Atlas Antibodies, Stockholm, Sweden), RPL11 (3A4A7; Life Technologies), and RPS6 (54D2; Cell Signaling Technology, Danvers, MA). The intensities of the bands were determined using NIH's ImageJ. Cytoplasmic and nuclear fractions were generated using a Paris kit (Life Technologies).
Immunoprecipitation/RT-PCR.
To analyze ribosome-mRNA association, cells were incubated in the presence of 0.1 mM CHX for 5 min and then suspended in ice-cold extraction buffer containing 10 mM HEPES-KOH (pH 7.4), 150 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol (DTT), 100 μg/ml CHX, protease inhibitors, and 40 U recombinant RNase inhibitor (Toyobo, Osaka, Japan) with a homogenizer. Homogenates were centrifuged for 10 min at 2,000 × g to pellet large cell debris, and Nonidet P-40 (NP-40; Nacalai Tesque, Kyoto, Japan) was added to the supernatant at a final concentration of 1%. After incubation on ice for 5 min, clarified lysates were cleared by centrifugation for 10 min at 13,000 × g. Protein A magnetic beads (Millipore) and anti-GFP (mFx73; Wako, Osaka, Japan) were preincubated at room temperature for 30 min and added to the supernatant. The mixture was incubated at 4°C with end-over-end rotation for 3 h. Beads were subsequently collected on a magnetic rack, washed three times with high-salt wash buffer (10 mM HEPES-KOH [pH 7.4], 350 mM KCl, 5 mM MgCl2, 1% NP-40, 0.5 mM DTT, and 100 μg/ml CHX), and immediately placed in TriPure isolation reagent to extract bound RNAs from polysomes (32). RNA was subjected to real-time PCR analysis as described above.
For polysomal fractionation, cells were incubated in the presence of 0.1 mM cycloheximide for 5 min and lysed in a buffer containing 50 mM Tris-Cl (pH 7.5), 0.1 M NaCl, 10 mM MgCl2, 2 mM DTT, 200 U/ml RNase inhibitor (Toyobo, Osaka, Japan), 100 μg/ml cycloheximide, 200 μg/ml Heparan, 0.5% NP-40, and protease inhibitors. One milliliter of lysates cleared by centrifugation was loaded onto 15-to-40% sucrose gradients in a buffer (9 ml) containing 150 mM NaCl, 5 mM MgCl2, and 25 mM Tris-Cl (pH 7.5), centrifuged using an Sw41 rotor (Beckman Coulter, Fullerton, CA) (34,000 rpm, 140 min, 4 °C), and separated into 120 fractions. RNA and protein were recovered from each fraction and analyzed by real-time PCR and immunoblotting, respectively.
To analyze HuR-RNA and nucleolin-RNA complexes, cell lysates prepared using a buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM DTT, 0.2% Tween 20, 10% glycerol, and protease inhibitors were incubated at 4°C for 3 h together with protein A magnetic beads preincubated with antihemagglutinin (anti-HA) (3F10; Roche, Indianapolis, IN), anti-HuR (3A2; Santa Cruz Biotechnology), or antinucleolin (MS-3; Santa Cruz Biotechnology). Magnetic beads were washed three times with the buffer and suspended in TriPure isolation reagent to recover RNA associated with HA-tagged or endogenous HuR protein. RNAs were quantified using real-time PCR.
For UV cross-linking and immunoprecipitation, cells in a 10-cm-diameter dish were washed twice with PBS and irradiated with UV (150 mJ/cm2) in the presence of 6 ml PBS. Irradiated cells were harvested and resuspended in a buffer containing 50 mM Tris (pH 7.4), 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 40 U/ml RNase inhibitor, and protease inhibitors. Lysates were treated with DNase I at 37°C for 3 min, cleared by centrifugation, and incubated with magnetic protein G beads pretreated with anti-HuR for 3 h at 4°C. Beads were washed twice with high-salt buffer (50 mM Tris [pH 7.4], 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, and 0.5% sodium deoxycholate) and then twice with wash buffer (20 mM Tris [pH 7.4], 10 mM MgCl2, and 0.2% Tween 20) and resuspended in PK buffer (100 mM Tris [pH 7.4], 50 mM NaCl, and 10 mM EDTA) containing proteinase K for 20 min at 37°C. An equal amount of PK buffer containing 7 M urea was added, the mixture was incubated for 20 min at 37°C, and supernatants were collected and subjected to phenol-chloroform extraction. RNAs were isolated from the aqueous phase by ethanol precipitation and subjected to real-time PCR analysis as described above.
Immunohistochemistry.
Immunohistochemistry was performed on frozen sections of adipose tissue from HuRfloxed/floxed (HuRfl/fl) or HuRfl/fl AP2-Cre mice. Sections were fixed with 4% paraformaldehyde in PBS for 15 min, rinsed, and then placed into a covered Coplin jar containing citrate buffer (1.8 mM citric acid, 8 mM sodium citrate, pH 6.0) that had been preheated to 100°C for 30 min. Sections were rinsed in PBS for 10 min, blocked with 10% FCS in 0.1% Triton X-100–PBS for 1 h, and incubated with 5 μg/ml of anti-p19ARF (5-C3-1) and anti-HuR (H-280; Santa Cruz Biotechnology) in Can Get Signal Solution A (Toyobo) at 4°C overnight. Proteins were visualized with Cy3- or Alexa Fluor 488-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA), and slides were mounted using Vectashield and DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA). Fluorescence images were taken with a Nikon CFI Plan Apo λ 40×/0.95-numerical-aperture lens at ambient temperature using an inverted microscope (Biorevo BZ-9000; Keyence, Osaka, Japan). Images were acquired using BZ-II Viewer software (Keyence).
Insulin and glucose tolerance tests.
Animals were fasted for 4 h (for the insulin test) or overnight (for the glucose test) and intraperitoneally injected with insulin (0.75 unit/kg of body weight) or glucose (1 g/kg). Following these injections, tail vein blood (approximately 5 μl) was collected, and glucose was measured using a glucose meter (LifeScan, Milpitas, CA).
RESULTS
HuR regulates the replicative senescence of murine fibroblasts.
HuR levels decline during senescence in human fibroblasts (22). We checked HuR levels in early- and late-passage mouse embryonic fibroblasts. As in human fibroblasts, HuR was downregulated in senescent MEFs where p19ARF and p16Ink4a levels were increased (Fig. 1A). To test if downregulation of HuR was sufficient to induce replicative senescence, MEFs were infected with retroviruses encoding short hairpin RNA (shRNA) that inhibits short hairpin HuR (sh-HuR) expression or control sh scramble (sh-SCR) (33). Infected cells were selected with puromycin and cultured according to the NIH 3T3 protocol (34). As shown in Fig. 1B, HuR knockdown cells had much shorter replicative life spans than control shRNA-expressing cells, and these cells showed the typical characteristics of cellular senescence, including a flattened shape and increased activity of senescence-associated β-galactosidase (SA-β-Gal) (Fig. 1C). In addition, another senescence marker, plasminogen activator inhibitor-1 (PAI-1), was increased in HuR-depleted cells (Fig. 1D) (35). These data suggest that loss of HuR results in acute cellular senescence in mouse fibroblasts.
Fig 1.
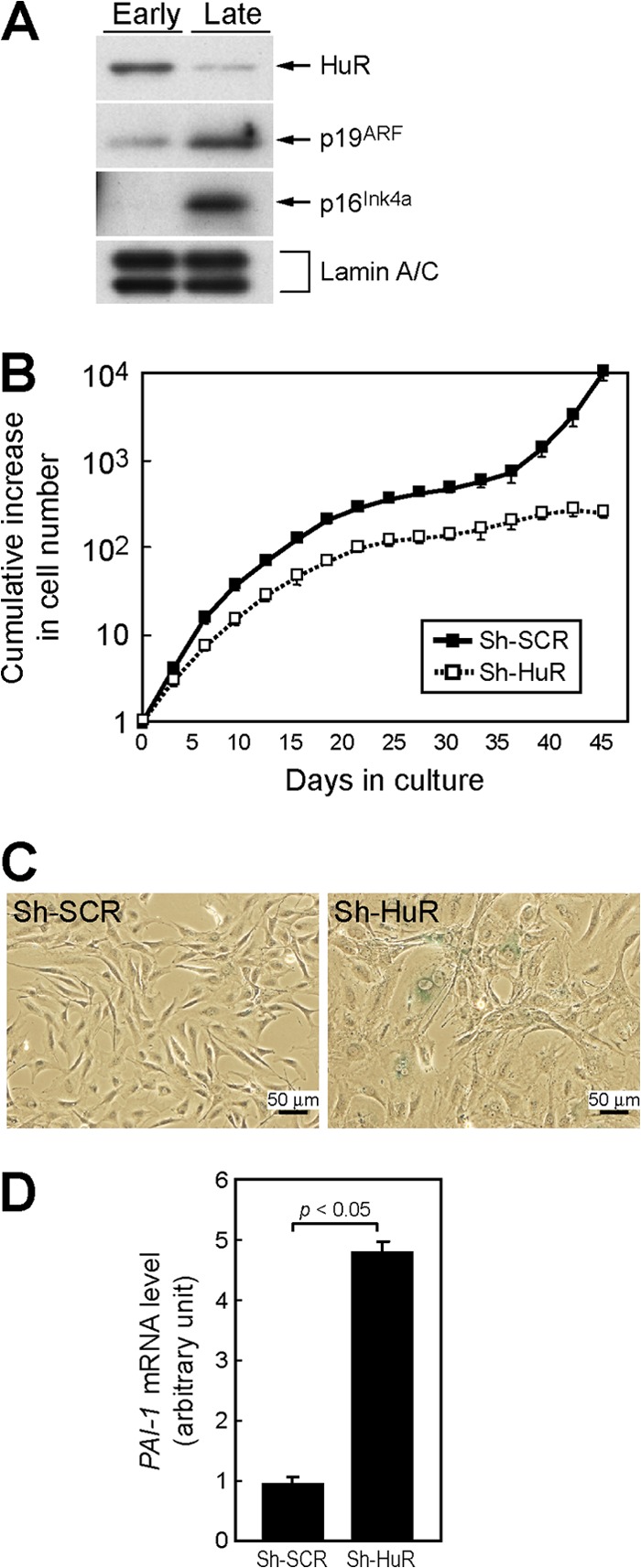
Loss of HuR leads to acute cellular senescence in MEFs. (A) Lysates from early-passage (passage 2 [P2]) and late-passage (P10) MEFs were analyzed for expression of the indicated proteins by immunoblotting. Lamin A/C was used as a loading control. (B) Wild-type MEFs infected with control (sh-SCR) or sh-HuR retroviruses were cultured by the NIH 3T3 protocol. Error bars represent standard errors of the means (SEM) of results from triplicate wells. (C) Cells (10 days postinfection) were stained with SA-β-Gal. (D) Expression of PAI-1 mRNA was analyzed by real-time PCR. Values were normalized to those for GAPDH in each sample. Data are representative of three independent experiments. Error bars represent SEM of results from triplicate samples.
The ARF-p53 pathway plays an essential role in mouse cellular senescence. ARF- or p53-null cells do not undergo cellular senescence upon serial passage or oncogenic activation (36–39). To clarify the roles of the ARF-p53 pathway in the senescence of HuR knockdown MEFs, cells derived from ARF−/− or p53−/− animals were infected with retroviruses encoding sh-SCR or sh-HuR. Unlike in wild-type MEFs, no SA-β-Gal staining was observed in ARF−/− and p53−/− cells (Fig. 2A). HuR depletion consistently caused significant cell growth arrest only in wild-type MEFs, and ARF- or p53-null cells continued to proliferate irrespective of HuR status (Fig. 2B). However, HuR knockdown had a weak growth-inhibitory effect in these cells, which is consistent with earlier reports showing that HuR targets several growth-related genes (40). These results suggest that cellular senescence induced by HuR depletion is strictly dependent on the ARF-p53 pathway, while other factors may also be involved in cell growth regulation by HuR.
Fig 2.
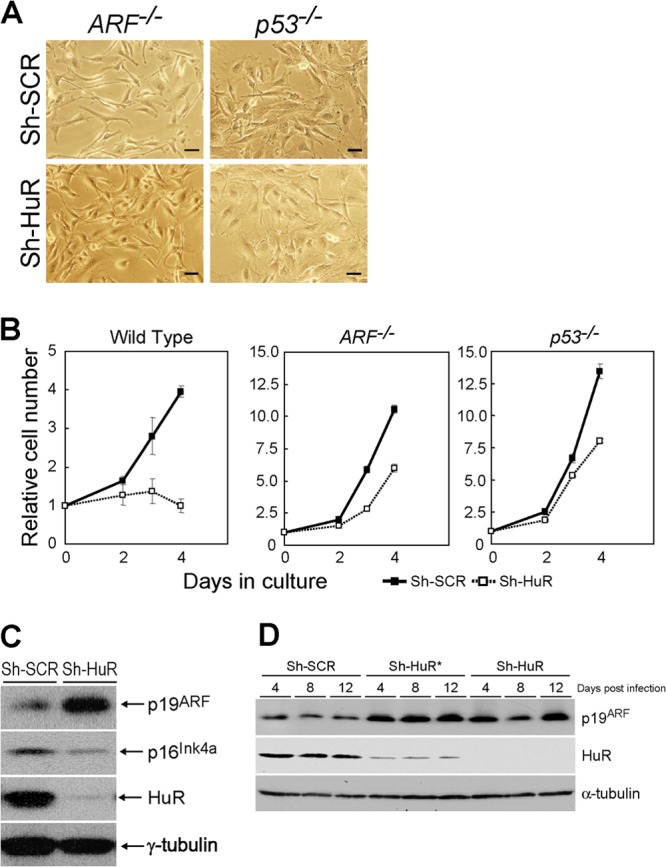
Senescence in HuR-depleted MEFs is dependent on the ARF-p53 pathway. (A) MEFs prepared from ARF- or p53-null animals were infected with control or sh-HuR retroviruses. Infected cells were stained for SA-β-Gal. Bars, 25 μm. (B) Control or sh-HuR MEFs with the indicated genotypes were analyzed for their growth rates. Error bars represent means ± SE of results from triplicate wells. (C) Cell lysates were prepared from wild-type MEFs infected with control or sh-HuR retroviruses. The expression of the indicated proteins was analyzed by immunoblotting. γ-Tubulin was used as a loading control. (D) Wild-type MEFs were infected with two independent sh-HuR retroviruses that target different regions of HuR mRNA. Cell lysates were prepared at 4, 8, and 12 days postinfection and analyzed for the expression of the indicated proteins by immunoblotting.
p19ARF is induced in HuR knockdown cells.
During cellular senescence, p19ARF and p16Ink4a levels are increased and lead the activating signals to p53 and pRb, respectively (41). Since senescence induced by HuR depletion requires ARF (Fig. 1C and 2A), we checked the levels of these proteins in control and sh-HuR MEFs. Loss of HuR expression led to a significant increase in p19ARF levels (Fig. 2C), while p16Ink4a levels were not increased in these cells. Similar results were obtained using another shRNA, one that targets different regions of the HuR gene (33), confirming that increased levels of p19ARF were not due to the off-target effects of sh-HuR (Fig. 2D). On the other hand, overexpression of HuR did not affect either the cells' replicative life span or p19ARF levels (Fig. 3A and B).
Fig 3.
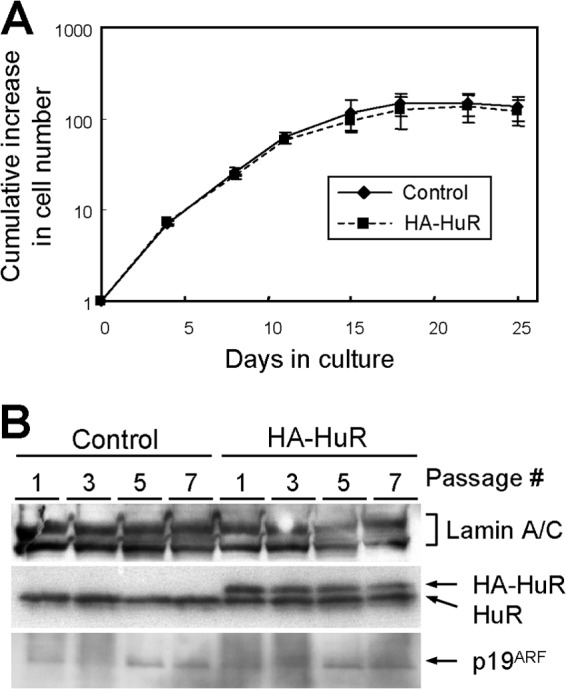
HuR overexpression does not affect p19ARF levels or replicative life spans. (A) Wild-type MEFs were infected with retroviruses encoding GFP (control) or HA-HuR. Infected cells were cultured by the NIH 3T3 protocol. Error bars represent SEM of results from triplicate wells. (B) Cell lysates were prepared at the indicated passage numbers. The expression of the indicated proteins was analyzed by immunoblotting. Lamin A/C was used as a loading control.
HuR does not affect ARF mRNA or protein stability.
The above results indicate that, unlike in HDFs, HuR regulates the expression of p19ARF but not of p16Ink4a in MEFs. To gain insights into how HuR regulates p19ARF expression, we first compared ARF mRNA levels in control and sh-HuR MEFs. Real-time PCR analysis revealed no increase in ARF or Ink4a mRNA levels in the presence or absence of HuR (Fig. 4A), implying that HuR was not involved in the transcriptional regulation of these genes. We next checked whether HuR could affect the stability of these mRNAs. Cells were treated with actinomycin D to block de novo mRNA synthesis, and the remaining mRNA was chased by real-time PCR. Although HuR has been shown to negatively regulate Ink4a mRNA stability in human fibroblasts (25), there was no significant difference in the levels of stability of ARF mRNA in MEFs (Fig. 4B). Likewise, we observed no difference in the ratios of cytoplasmic to nuclear ARF and Ink4a mRNA between these cells; therefore, it is unlikely that HuR regulates the nuclear export of these mRNAs (Fig. 4C). We also compared levels of protein stability in these cells with a cycloheximide chase but did not observe changes in p19ARF stability (Fig. 4D and E).
Fig 4.

HuR does not affect ARF mRNA transcription, stability, nuclear export, or p19ARF turnover. (A) The expression of ARF and Ink4a mRNA in control and sh-HuR MEFs was analyzed by real-time PCR. mRNA in each sample was normalized to 18S rRNA. (B) Control or sh-HuR retrovirus-infected MEFs were treated with actinomycin D for the indicated periods. Total RNA was extracted at each time point, and ARF and Ink4a mRNA levels relative to the 18S rRNA level were analyzed by real-time PCR. (C) Nuclear and cytoplasmic fractions were prepared from control and sh-HuR MEFs. (Upper blot) Samples were analyzed by immunoblotting using Lamin (for the nuclear marker) and α-tubulin (for the cytoplasmic marker) antibodies. (Lower blot) RNA was isolated from these fractions. ARF and Ink4a mRNA levels were quantified by real-time PCR and normalized to the 18S rRNA level in each fraction, and the ratios of cytoplasmic mRNA to nuclear mRNA were determined. (D) Wild-type MEFs infected with control or sh-HuR retroviruses were treated with cycloheximide (CHX) for the indicated periods. Cell lysates were prepared, and p19ARF levels were analyzed by immunoblotting. (E) The intensity of the p19ARF band in each sample was determined using ImageJ and plotted. (F) HuR does not affect Ago2 association with ARF mRNA in mouse cells. Lysates of MEFs expressing sh-SCR or sh-HuR were immunoprecipitated using Ago2 or a control antibody. RNA recovered from the immune complex was analyzed using real-time PCR. Error bars represent SEM of results from triplicate samples.
In human cells, HuR has been shown to destabilize Ink4a mRNA by recruiting RISC to it (25). We therefore wished to determine if this was also the case with ARF regulation in MEFs. Lysates from sh-SCR and sh-HuR MEFs were immunoprecipitated using Ago2 antibodies, and RNAs recovered from immune complexes were subjected to real-time PCR analysis for ARF. ARF mRNA was enriched in the Ago2 immune complex from sh-SCR cells, suggesting that RISC is also involved in ARF mRNA regulation (Fig. 4F). Nonetheless, we did not observe any decrease in the RISC-ARF mRNA interaction in HuR-depleted cells. Thus, unlike in human cells, RISC is not involved in HuR-mediated ARF mRNA regulation.
HuR translationally regulates p19ARF expression.
Next, we checked the possibility that HuR affects the translation of ARF mRNA since it has been well established that HuR regulates the translation of its target mRNAs (13, 40). MEFs were infected with GFP or GFP fused to ribosomal protein L10a (GFP-L10a) together with sh-SCR or sh-HuR retroviruses. Cytoplasmic lysates were immunoprecipitated using GFP antibody to purify ribosome-mRNA complexes (32, 42). Immunoblotting confirmed that GFP-L10a proteins were specifically enriched in immunoprecipitated complexes, and equivalent amounts of GFP-L10a were obtained from control and sh-HuR cells (Fig. 5A). RNAs were then recovered from immune complexes and subjected to real-time PCR analysis. ARF mRNA was significantly enriched in the ribosome complex in sh-HuR cells; the amount of ribosome-associated ARF mRNA was more than five times higher than that of the control, while no change in ribosome association with Ink4a mRNA was observed under these conditions. To further validate the ARF mRNA-ribosome association, cytoplasmic lysates were fractionated into polysome/nonpolysome fractions by sucrose gradient sedimentation (Fig. 5B). RNAs were recovered from each fraction, and ARF and Ink4a mRNAs were analyzed by real-time PCR. As in the GFP-L10a immunoprecipitation experiment (Fig. 5A), we observed more ARF mRNA in the polysome fractions of HuR-depleted cells than in the nonpolysome fractions (Fig. 5C). Together, these results indicate that HuR specifically represses p19ARF expression by inhibiting mRNA-ribosome association.
Fig 5.

HuR regulates the translation of ARF mRNA. (A) Wild-type MEFs were infected with sh-SCR or sh-HuR retroviruses together with GFP or GFP-L10 retroviruses. Cytoplasmic lysates were immunoprecipitated using GFP antibody. Immunoblotting using GFP antibody indicated that equal amounts of GFP-L10 protein were immunoprecipitated. RNAs were extracted from immune complexes and subjected to real-time PCR analysis. Amounts of ARF or Ink4a mRNA in each sample were normalized to 18S rRNA in the complex. Data are representative of three independent experiments. Error bars represent SEM of results from triplicate samples. (B) Cytoplasmic lysates prepared from MEFs infected with sh-SCR or sh-HuR retroviruses were fractionated by sucrose density gradient centrifugation. Samples were manually separated into 120 fractions, and the relative values of optical densities at 254 nm were plotted (graphs). Ten fractions were pooled, and 28S and 18S rRNAs and ribosomal proteins (L10, L11, and S6) were visualized by ethidium bromide staining and immunoblotting, respectively. (C) The amount of ARF or Ink4a mRNA in each fraction was analyzed using real-time PCR.
We next sought to investigate if HuR affects ARF mRNA localization. To this end, ARF mRNA, including both its 5′- and its 3′UTR, was conjugated to tandem MS2-binding sequences (MS2 tag in Fig. 6A) (43) and coexpressed with MS2-EGFP fusion protein with a nuclear localization signal (MS2-EGFP-NLS) in ARF and p53 double-knockout (DKO) MEFs expressing sh-SCR or sh-HuR (30). In the absence of ARF mRNA, the GFP signal was observed only in the nucleus, irrespective of HuR status (Fig. 6B). In cells expressing MS2-tagged ARF mRNA, the GFP signal was also observed in the nucleus, indicating that the majority of ARF mRNA remains in the nucleus. Interestingly, we observed that ARF mRNA (full length) specifically accumulated in a subnuclear compartment when HuR was depleted. This subnuclear compartment represented nucleoli, since the GFP signal colocalized with p19ARF (Fig. 6C). Thus, HuR also regulates the nuclear trafficking of ARF mRNA, which may contribute to translational regulation (44).
Fig 6.

Loss of HuR relocalizes ARF mRNA to nucleoli. (A) DNA constructs for the expression of MS2-tagged ARF mRNA (full length or Δ5′UTR). CMVp, cytomegalovirus promoter. (B) Sh-SCR- or sh-HuR-expressing ARF p53 DKO cells were transiently transfected with MS2-GFP-NLS plasmids. Where indicated, cells were cotransfected with MS2-tagged ARF (full length or Δ5′UTR)-expressing plasmids. Three days later, cells were fixed in paraformaldehyde and stained with DAPI. Bars, 10 μm (C) Sh-HuR-expressing ARF p53 DKO cells were transiently transfected with MS2-GFP-NLS together with MS2-tagged ARF (full length)-expressing plasmids. Cells were stained using p19ARF antibody and DAPI.
HuR associates with ARF mRNA in living cells.
Since our results suggest that HuR regulates the translation of ARF mRNA, we next sought to determine if HuR associates with ARF mRNA. Wild-type MEFs were infected with control (GFP) or HA-HuR expression retroviruses, and HA-HuR complexes were immunoprecipitated using HA antibody. As previously reported, β-actin, c-myc, and p53 mRNAs were specifically enriched in HuR complexes (Fig. 7A) (16, 20, 26). Under these conditions, we could not detect ARF mRNA in the HuR immune complex. We performed similar experiments with endogenous HuR proteins and failed to detect binding of HuR to ARF mRNA (Fig. 7B). We next employed a UV cross-linking and immunoprecipitation (CLIP) assay, which is a more sensitive method to detect protein-RNA interaction. MEFs were irradiated with UV to covalently cross-link protein-RNA complexes prior to lysate preparation and immunoprecipitated using HuR or control antibodies. Although we could not detect the HuR and ARF mRNA interaction with the standard RNA immunoprecipitation protocol (Fig. 7A and B), ARF mRNA was slightly enriched in HuR immune complexes following UV cross-linking (Fig. 7C). Hence, it is likely that ARF mRNAs form an extremely fragile or transient complex in living cells, unlike other HuR ligands.
Fig 7.
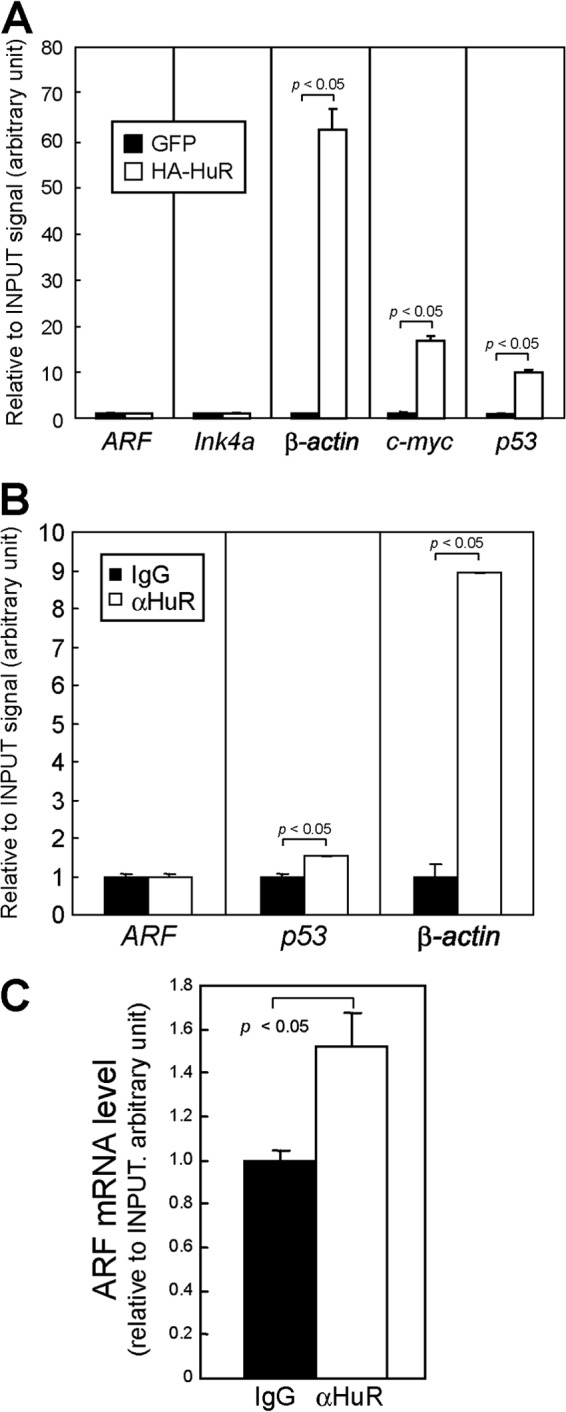
HuR weakly associates with ARF mRNA in living cells. (A) Lysates of (wild-type) MEFs expressing HA-tagged HuR proteins were immunoprecipitated using HA antibody. RNA extracted from the immune complex was analyzed by real-time PCR. Error bars represent SEM (n = 3). (B) Lysates prepared from wild-type MEFs were immunoprecipitated using control (IgG) or HuR antibodies. RNAs were extracted from immune complexes and subjected to real-time PCR analysis. p53, c-myc, and β-actin were used as positive controls. (C) Lysates were prepared from UV-cross-linked MEFs and immunoprecipitated using control or HuR antibodies. RNAs were recovered from the immune complex following proteinase K treatment and analyzed by real-time PCR for ARF mRNA levels. Error bars represent SEM of results from triplicate samples.
HuR regulates the translation of ARF mRNA through its 5′UTR.
To find the region responsible for HuR in ARF mRNA, we expressed exogenous ARF mRNA that included the open reading frame (ORF) and both the 5′- and 3′UTRs (full-length ARF), the ORF and 5′UTR (Δ3′UTR ARF), or the ORF and 3′UTR (Δ5′UTR ARF) in NIH 3T3 cells (ARF and Ink4a null) expressing sh-SCR or sh-HuR. These cells expressed comparable amounts of exogenous ARF mRNA (Fig. 8A). Under these conditions, p19ARF levels were increased in the absence of HuR expression, and this effect was more prominent in full-length ARF mRNA cells and in Δ3′UTR cells than in Δ5′UTR cells (Fig. 8B). p19ARF expression from Δ5′UTR ARF mRNA was also slightly increased in HuR-depleted cells. However, this likely reflects the larger amount of ARF mRNA in these cells (Fig. 8A), since the effect of HuR knockdown was diminished when p19ARF levels were normalized to ARF mRNA levels in each sample (Fig. 8C). Consistently with the above results, we observed more ribosome association with full-length and Δ3′UTR ARF mRNAs than with Δ5′UTR mRNA in the absence of HuR (Fig. 8D). Furthermore, CLIP analysis revealed that the 5′UTR is required for HuR association (Fig. 8E). Together, these results strongly suggest that HuR regulates the translation of ARF mRNA through its 5′UTR. Consistently with this notion, ARF mRNA localized to nucleoli irrespective of HuR status when the 5′UTR was deleted (Fig. 6B, lowest panels). However, this region by itself did not respond to HuR when it was conjugated to the luciferase reporter (data not shown), suggesting that the ORF region also contributes to regulation or that there are more-stringent requirements for the RNA secondary structure. We also performed similar experiments using full-length Ink4a mRNA or Ink4a mRNA lacking both the 5′ and 3′UTRs (ORF). Consistently with the above results indicating that HuR does not increase p16Ink4a levels in MEFs and that ARF mRNA does not share the 5′UTR with Ink4a, knockdown of HuR did not affect p16Ink4a expression from these mRNAs, further confirming that the effect of HuR is specific to ARF in this locus (Fig. 9).
Fig 8.

HuR regulates p19ARF expression through the 5′UTR of ARF mRNA. (A) NIH 3T3 cells expressing sh-SCR or sh-HuR were transfected with plasmids bearing full-length ARF, including the 5′- and 3′UTRs (full length), ARF lacking the 3′UTR (Δ3′UTR), or ARF lacking the 5′UTR (Δ5′UTR) together with GFP expression plasmids. Three days later, total RNA was extracted and exogenous ARF expression was analyzed by real-time PCR. Values were normalized to GFP mRNA levels in each sample. (B) The cells from panel A were analyzed by immunoblotting for expression of p19ARF and GFP. (C) p19ARF levels in panel B were quantified using ImageJ, and the p19ARF level and ARF mRNA level in each sample were calculated. (D) NIH 3T3 cells expressing sh-SCR or sh-HuR were transfected with ARF expression plasmids (full length, Δ3′UTR, or Δ5′UTR) together with GFP-L10 plasmids. Three days later, cytoplasmic lysates were prepared and immunoprecipitated using GFP antibody to purify RNA-protein complexes, including GFP-L10. RNAs were recovered from immune complexes and subjected to real-time PCR analysis for ARF mRNA. Values were normalized to input signals in each sample. (E) 293T cells were transfected with ARF expression plasmids that express full-length or mutant ARF mRNA that lacks the 5′UTR (Δ5′UTR) together with HA-HuR expression plasmids. Forty-eight hours later, cells were subjected to UV cross-linking and immunoprecipitated using control or HA antibodies. Recovered RNA was analyzed by real-time PCR. Error bars represent SEM of results from triplicate samples.
Fig 9.
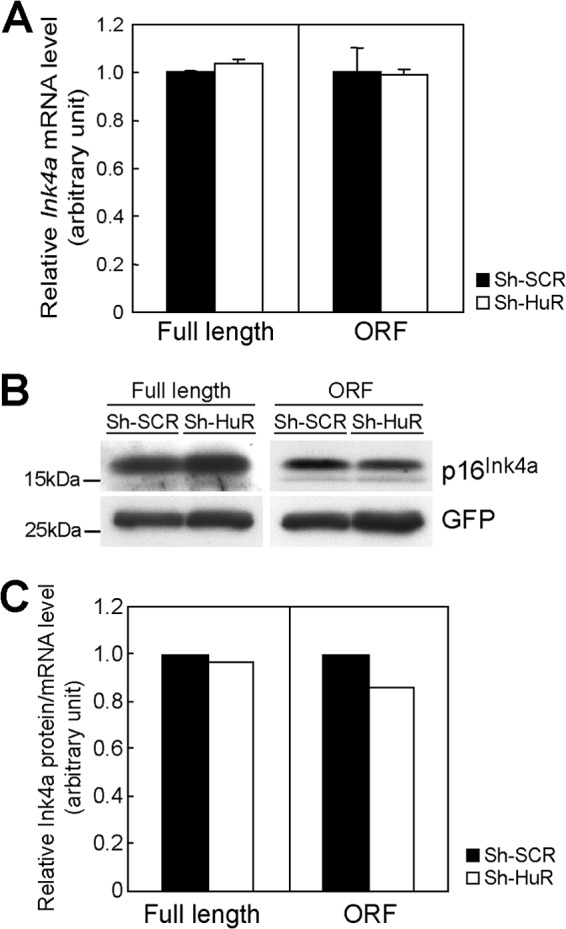
HuR does not affect Ink4a translation. (A) NIH 3T3 cells expressing sh-SCR or sh-HuR were transfected with plasmids bearing full-length Ink4a, including its 5′- and 3′UTRs (full length), or Ink4a lacking its 5′- and 3′UTRs (ORF) together with GFP expression plasmids. Three days later, RNAs were extracted and the expression of exogenous Ink4a mRNA was analyzed by real-time PCR. Values were normalized to GFP expression levels in each sample. (B) The expression of p16Ink4a and GFP was analyzed by immunoblotting. (C) p16Ink4a levels were quantified and normalized to Ink4a expression levels. Error bars represent SEM of results from triplicate samples.
Nucleolin interacts with ARF mRNA in nucleoli and is required for p19ARF expression in HuR knockdown cells.
We next sought a possible mediator of p19ARF expression in HuR knockdown cells. The nucleolar RNA-binding protein nucleolin has been shown to bind to several mRNAs involved in the cellular stress response, and the binding of nucleolin enhances the translation of their target mRNAs (45). Moreover, microarray analysis of mRNA in the nucleolin complex has revealed that CDKN2A (Ink4a and ARF) mRNA physically associates with nucleolin in HeLa cells (45). Because ARF mRNA localized to nucleoli upon HuR depletion, we tested if nucleolin interacts with the nucleolar ARF mRNA in HuR knockdown cells. While no ARF mRNA was detected in the nucleolin complex of control cells, it was significantly enriched in the absence of HuR expression (Fig. 10A). The interaction of nucleolin with ARF mRNA does not require 5′UTR; therefore, relocalization of ARF mRNA to the nucleolus seems sufficient for the interaction (Fig. 10B). Thus, HuR impedes the nucleolar localization of ARF mRNA by binding to its 5′UTR, thereby inhibiting the interaction of ARF mRNA with nucleolin. Next, we examined whether nucleolin is required for p19ARF expression in HuR knockdown cells. For this purpose, siRNA targeting nucleolin mRNA was transfected into sh-SCR- or sh-HuR-expressing MEFs. Although the effect of siRNA on nucleolin level was limited, p19ARF induction was suppressed to basal levels in HuR knockdown cells (Fig. 10C), suggesting that nucleolin is required for p19ARF induction in HuR-depleted cells.
Fig 10.
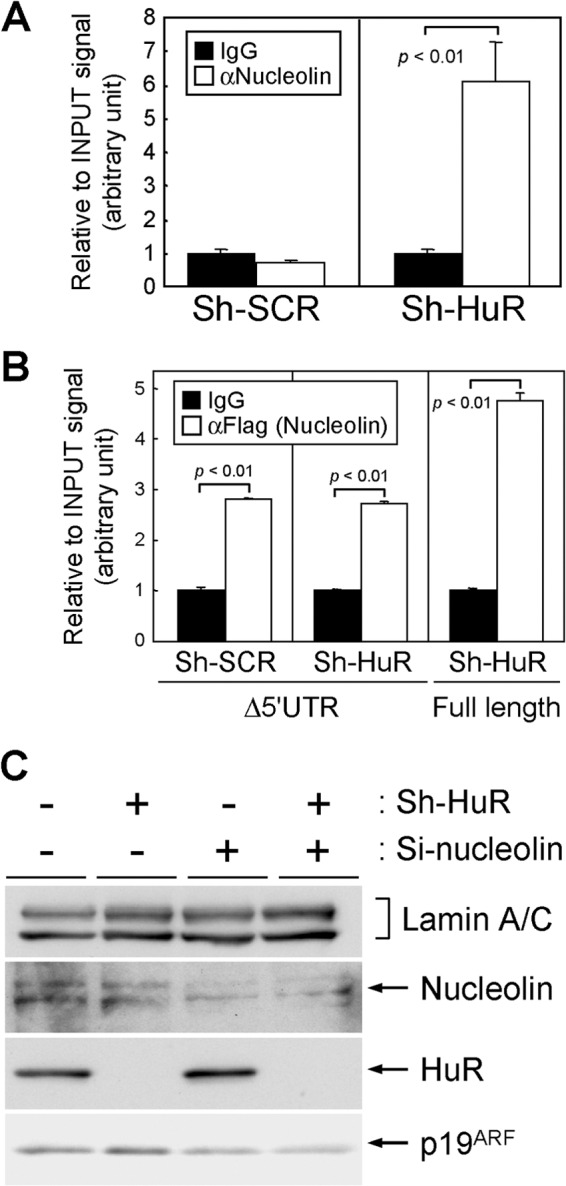
Nucleolin associates with ARF mRNA and mediates the p19ARF induction in HuR knockdown cells. (A) Lysates prepared from MEFs expressing sh-SCR or sh-HuR were immunoprecipitated using control (IgG) or nucleolin antibodies. RNAs were recovered from the immune complexes and analyzed by real-time PCR. (B) NIH 3T3 cells expressing sh-SCR or sh-HuR were transiently transfected with ARF (Δ5′UTR or full length)-expressing plasmids together with Flag-tagged nucleolin-expressing plasmids. Two days later, lysates were prepared and immunoprecipitated using control or Flag tag (M2) antibodies. RNAs in the immune complexes were analyzed by real-time PCR. Error bars represents SEM of results from triplicate samples. (C) MEFs (P2) expressing sh-SCR or sh-HuR were transfected with siRNA targeting nucleolin. Two days later, lysates were prepared and the expression of the indicated proteins was analyzed by immunoblotting. Lamin A/C was used as a loading control.
Loss of HuR inhibits adipocytic differentiation in a p19ARF-dependent manner.
A recent report by Minamino and colleagues has shown that senescence in adipose tissue results in decreased insulin sensitivity, thereby leading to type 2 diabetes mellitus (46). Hence, we investigated whether HuR-mediated p19ARF regulation had any effect on adipocyte function. To this end, we first tested whether loss of HuR could affect adipocyte differentiation in vitro. Wild-type MEFs expressing sh-SCR or sh-HuR were differentiated into adipocytes in the presence of insulin, dexamethasone, and 3-isobutyl-1-methylxanthine (IBMX). Oil Red O staining revealed that HuR depletion suppressed adipocytic differentiation in wild-type MEFs (Fig. 11A). It has been reported that HuR directly binds to C/EBPβ mRNA to regulate its expression (47). We therefore checked if HuR depletion affected the expression of genes required for adipocyte differentiation. As shown in Fig. 11B, C/EBPβ expression was slightly diminished in HuR-depleted cells. Nonetheless, levels of expression of its downstream C/EBPα and PPARγ genes were still comparable to those in control cells, suggesting that defects in adipogenesis in the absence of HuR were not attributed to altered expression of adipocyte-related genes. We then checked whether the adipocyte phenotype was dependent on ARF. In sharp contrast, HuR knockdown had virtually no effect on adipocyte differentiation in ARF knockout MEFs (Fig. 11C).
Fig 11.

Adipogenesis is impaired in HuR-depleted wild-type MEF. (A) Wild-type MEFs were infected with sh-SCR or sh-HuR retroviruses. Selected cells were stimulated to differentiate them into adipocytes in the presence of insulin, 3-isobutyl-1-methylxanthine (IBMX), and dexamethasone for 10 days and stained with Oil Red O. Bars, 50 μm. (B) Wild-type MEFs with sh-SCR or sh-HuR were cultured in adipocyte differentiation medium for the indicated periods. The expression of C/EBPα, -β, -δ, and PPARγ were analyzed by immunoblotting. (C) ARF-null MEFs with sh-SCR or sh-HuR were stimulated to differentiate them for 10 days and stained with Oil Red O. Bars, 50 μm. (D and E) Wild-type (D) and ARF-null (E) MEFs expressing sh-SCR or sh-HuR were stimulated to differentiate them for 0, 1, and 2 days. Cells were pulse-labeled with EdU for 45 min and stained for EdU. EdU-positive and -negative cells in microscopic fields were counted. Data are representative of two independent experiments. Error bars represent SEM of results from five microscopic fields.
Given that p19ARF activates the p53-dependent cell cycle checkpoint, we were prompted to check the possibility that p19ARF affects clonal expansion during the initial stage of adipogenesis. Cells were stimulated to differentiate and were pulse-labeled with 5-ethynil-2′-deoxyuridine (EdU) to assess cell cycle reentry. EdU staining showed a significant reduction in cell cycle reentry in HuR-depleted wild-type MEFs (Fig. 11D). In contrast, S-phase entry was not affected by sh-HuR in the absence of ARF (Fig. 11E). These results suggest that defective adipogenesis in HuR-depleted cells can be attributed to p19ARF-dependent cell cycle arrest or senescence.
Adipose-specific HuR knockout accelerates age-dependent insulin resistance.
Our above results indicated that the loss of HuR enhanced the translation of ARF mRNA, thus inducing p19ARF-dependent cellular senescence, and that HuR may affect adipocyte function through p19ARF. To explore the impact of HuR-mediated translational regulation of the ARF gene in vivo, we generated adipose tissue-specific HuR knockout mice (HuRfl/fl; AP2-CRE) (Fig. 12A and B) (10). ARF mRNA levels were low in the adipose tissue of young animals (1 to 3 months old) of both genotypes but significantly increased in older animals (6 to 9 months old) (Fig. 12C), which is consistent with previous reports indicating that ARF expression increases in many tissues as animals age (48). p19ARF was still hardly detectable in the adipose tissue of older HuRfl/fl mice (Fig. 12D). However, we detected p19ARF in a certain population of older HuRfl/fl; AP2-CRE mouse adipose tissue (Fig. 12E and F). Changes in SA-β-Gal activity were difficult to detect; however, PAI-1 levels were significantly increased in HuR knockout adipose tissue (Fig. 12G). We subsequently tested if HuR loss in adipose tissue affected insulin-mediated glucose homeostasis, which is one of the major functions of this tissue. There was little difference in insulin sensitivity among both genotypes when animals were at a young age; however, in older animals, adipose-specific HuR deletion significantly accelerated insulin resistance (Fig. 13A). Similar results were obtained by the glucose tolerance test (Fig. 13B). So far, we have not been able to confirm that this effect is ARF dependent, because ARF-null animals develop tumors by this age (49). However, the timing of the onset of insulin resistance correlates well with that of p19ARF appearance in adipose tissue. Hence, these results suggest that HuR is required to repress p19ARF expression in adipose tissue, thereby inhibiting adipose senescence, which can lead to insulin resistance.
Fig 12.

Adipose-specific HuR deletion accelerates the senescence of adipocytes. (A) Genotyping of adipose- and tail-derived genomic DNA. (B) HuR mRNA levels in adipose tissue from mice with the indicated genotypes were analyzed by real-time PCR. Values were normalized to 18S rRNA in each sample. (C) ARF mRNA was analyzed by real-time PCR. (D) Lysates were prepared from the adipose tissue of HuRfl/fl and HuRfl/fl; AP2-CRE mice. Expression of the indicated proteins was analyzed by immunoblotting. CDK4 was used as a loading control. Testis lysate from ARF−/− and ARF+/+ animals was used as the negative and positive controls for p19ARF, respectively. (E) Frozen sections of adipose tissue of HuRfl/fl and HuRfl/fl; AP2-CRE mice (9 months old) were immunostained using p19ARF and HuR antibodies. Sections were counterstained with DAPI. Bars, 20 μm. (F) Rates of p19ARF-positive cells in panel E were plotted. WAT, white adipose tissue. (G) PAI-1 mRNA levels were analyzed using real-time PCR.
Fig 13.
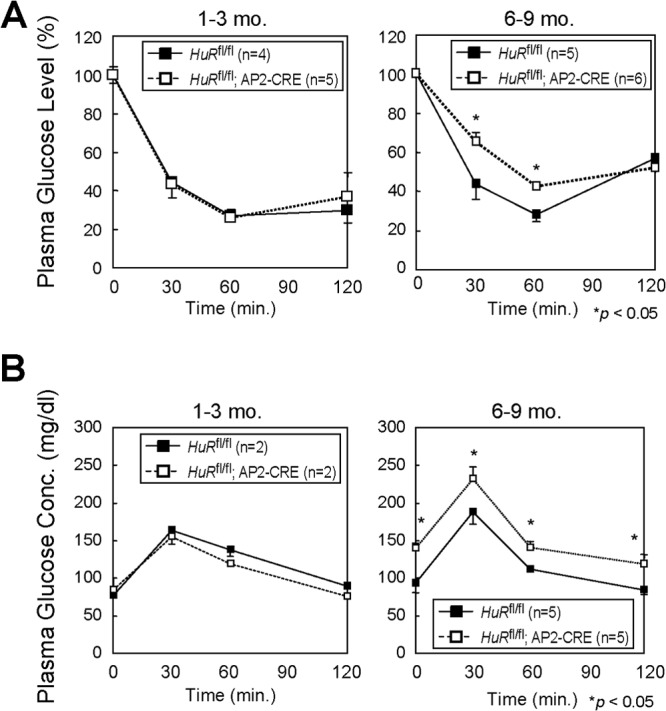
Adipose-specific HuR deletion accelerates age-dependent insulin resistance. Insulin tolerance tests (A) and glucose tolerance tests (B) were performed with HuRfl/fl and HuRfl/fl; AP2-CRE mice.
DISCUSSION
Our data show that HuR is downregulated in senescent mouse fibroblasts and that decreases in HuR contribute to senescence-associated growth arrest. It has been shown that HuR levels decline in human diploid fibroblasts during cellular senescence (22); therefore, the role of HuR in cellular senescence is likely to be evolutionally conserved. How HuR expression is controlled during senescence is unclear, but in human cells, it is attributed, at least in part, to increased expression of miR-519 during senescence (24). Whether HuR is regulated by miRNA in mouse senescence is unknown, but we did not observe a significant change in HuR mRNA levels in senescent MEFs (data not shown). Therefore, such posttranscriptional regulation may also contribute to the control of HuR levels in mouse cells.
Although HuR is implicated in senescence in both human and mouse cells, the mechanisms underlying them are different. The p16Ink4a-Rb pathway plays pivotal roles in cell cycle arrest during cellular senescence in human cells. In contrast, it has been well established that the p19ARF-p53 pathway is essential and that p16Ink4a is dispensable in the senescence of mouse cells. Consistently with these concepts, our results show that loss of HuR leads to increased expression of p19ARF, but not p16Ink4a, levels in MEFs. We further demonstrated that senescence caused by HuR loss can be abrogated by either ARF or p53 deletion. Therefore, under normal conditions in which cells express sufficient amounts of HuR protein, p19ARF expression is suppressed, thereby protecting cells from undergoing p53-dependent replicative senescence. Additionally, it has been proposed that HuR positively regulates the expression of Mdm2, which is a major E3 ligase for p53 protein, and is negatively regulated by p19ARF (50). Thus, HuR suppresses p53 activity by modulating the expression of multiple targets integrated into the p53 pathway. Although we do not formally exclude the possibility that p16Ink4a is also involved, it is conceivable that the p19ARF-p53 pathway is a major target of HuR to control the life span of mouse cells.
In human cells, HuR directly associates with ARE in the 3′UTR of Ink4a mRNA (25). This region is shared by ARF mRNA; therefore, it is possible that HuR also regulates p14ARF expression in human cells. Unlike in human cells, in MEFs, the loss of HuR has no influence on p16Ink4a levels, while p19ARF is increased. Consistently with these results, HuR associates with the 5′UTR of ARF mRNA, which is not shared by Ink4a. However, the interaction of HuR with ARF mRNA is weak and observed only after UV-mediated cross-linking. Therefore, it is likely that HuR forms a much more fragile complex with ARF mRNA than with other mRNAs. Alternatively, the effect of HuR may be indirect; HuR may target another factor(s) that regulates p19ARF expression. In this regard, it is worthy of note that HuR regulates the translation of β-catenin and Jun-B mRNAs by modifying the stability of linc-p21 RNA (51). This could be clarified by identifying ARF mRNA-interacting molecules. Additionally, HuR has been proposed to recruit RISC to human Ink4a mRNA independently of miRNA, thereby destabilizing it (25). Although the involvement of miRNA needs to be further clarified, it is possible that RISC-mediated regulation may also be involved in mice, since deletion of dicer-1 causes p19ARF-p53-dependent cellular senescence (52). Furthermore, we detected ARF mRNA in the Ago2 complex. Nonetheless, the interaction of Ago2 and ARF mRNA was not decreased upon HuR depletion, implying that RISC is not involved in HuR-mediated ARF regulation.
HuR exclusively affects translation in p19ARF expression. Loss of HuR enhances ribosome association with ARF mRNA. This translational activation is associated with the nucleolar accumulation of ARF mRNA. We found that ARF mRNA associates with nucleolin, which is required for p19ARF induction in HuR-depleted cells. Nucleolin associates with numerous mRNAs and shuttles between the nucleolus and the cytoplasm. The influences of nucleolin on target mRNA differ depending on the target transcript. A recent report by Abdelmohsen and colleagues demonstrated that nucleolin is required for ribosome binding and subsequent translation of its target mRNA (45). Consistently with our results, they also observed CDKN2A, as well as both ARF and Ink4a, among the nucleolin-associated mRNAs. Together with these observations, our data suggest that HuR-associated ARF mRNA is retained in the nucleoplasm and is not efficiently translated upon nuclear export (Fig. 14). However, in the absence of HuR, ARF mRNA localizes to the nucleolus, where it associates with nucleolin. As nucleolin enhances ribosome recruitment to its target mRNA (45), p19ARF synthesis is increased under these conditions. Interestingly, p53 mRNA also accumulates in the nucleoli upon DNA damage, when p53 mRNA translation is increased (44). Hence, nucleolar localization of mRNA may reflect general aspects of stress-dependent mRNA translation. It has recently been shown by Miceli and colleagues that oncogenic Ras activates the transcription of the ARF gene, as well as the translation of ARF mRNA through mTORC1 (53). In this context, it is noteworthy that mTORC1 activity can affect the binding of HuR to ornithine decarboxylase mRNA (54). Therefore, it would be interesting to see if mTORC1 and HuR cooperate in ARF mRNA regulation.
Fig 14.
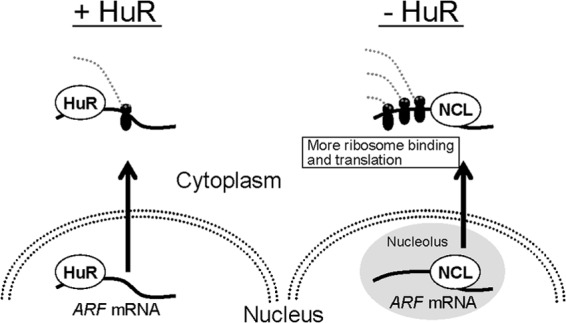
Model for ARF regulation by HuR and nucleolin. In the presence of HuR, ARF mRNA binds to HuR through the 5′UTR, and the mRNA localizes mainly to the nucleoplasm. The HuR-bound ARF mRNA is less efficiently translated. In the absence of HuR, ARF mRNA localizes to the nucleolus, where it associates with nucleolin (NCL). The nucleolin association facilitates ribosome binding, thereby enhancing the translation.
Cellular senescence is known to be involved in metabolic disorders as well as cancers. Among these, senescence in adipose tissue is associated with insulin resistance (46). HuR has also been shown to function in adipocytes by regulating C/EBPβ expression (55, 56). Our results reveal that, although C/EBPβ may be affected by HuR status, it has little effect on adipogenesis, which is consistent with a previous report that C/EBPβ-null MEFs are capable of undergoing adipogenesis (57). Instead, the function of HuR in adipogenesis depends largely on ARF, as HuR knockdown had virtually no effect on adipogenesis in ARF-null MEF or 3T3-L1 cells, in which the p53 pathway was inactivated by mdm2 amplification (Fig. 11 and data not shown) (58). Impaired adipogenesis is observed with concomitant reductions in clonal expansion during the initial stage of adipogenesis, which is alleviated in an ARF-null background. Hence, it is likely that adipogenic failure in HuR-depleted MEFs is attributed largely to p19ARF. There were no abnormalities in the shapes and sizes of adipose tissues in adipose-specific HuR knockout mice. However, these mice revealed progressive insulin resistance with age. The reason why this phenotype was not observed in young animals can be explained by differences in the levels of ARF mRNA among these animals. In young mice, increased translation by HuR loss does not lead to expression of sufficient amounts of p19ARF because of low ARF mRNA levels. However, in older animals, larger amounts of ARF mRNA and an increased rate of translation synergistically induced p19ARF in adipose tissue. SA-β-Gal activity was not as strong as in cultured cells, and we failed to quantitatively detect an increase in enzyme activity. Nonetheless, the senescence program is likely activated in those cells, since PAI-1 was significantly induced. It should be further clarified whether the phenotype is completely dependent on p19ARF or whether other adipocyte-related factors are involved. Also, it would be interesting to see if HuR is linked to metabolic disorders, such as type 2 diabetes mellitus. In this regard, it is noteworthy that there was strong linkage between the human ARF and Ink4a loci and the disease (59–61).
HuR is deregulated in many types of cancers (40), and there is no doubt that cellular senescence is a central tumor-suppressive mechanism in mammals. Hence, it is plausible that deregulated HuR activity and expression leads to an uncontrolled senescence program, thereby allowing cells to bypass senescence. This can be achieved by suppressing the activity of the p16Ink4a-Rb pathway and the p19ARF-p53 pathway in humans and mice, respectively. Moreover, HuR is downregulated in aged human tissues, which may contribute to an age-associated phenotype, such as decreased insulin sensitivity. Our data demonstrate a novel function of HuR in the maintenance of the cellular replicative life span and will lead to further understanding of the mechanism and biological roles of cellular senescence.
ACKNOWLEDGMENTS
We thank Leo Tsuda for providing the GFP-L10 construct, Takashi Funatsu for providing MS2-EGFP-NLS cDNA, and Noboru Motoyama for his valuable discussion and technical support.
This work was supported by grants from the Japanese Ministry of Education, Culture, Science, and Technology (grant 22790879) and the Japan Health Foundation.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Hayflick L, Moorhead PS. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25:585–621 [DOI] [PubMed] [Google Scholar]
- 2. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92:9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freund A, Orjalo AV, Desprez PY, Campisi J. 2010. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16:238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ben-Porath I, Weinberg RA. 2005. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 37:961–976 [DOI] [PubMed] [Google Scholar]
- 5. Sherr CJ. 2006. Divorcing ARF and p53: an unsettled case. Nat. Rev. Cancer 6:663–673 [DOI] [PubMed] [Google Scholar]
- 6. Campisi Jand D, d'Adda F. 2007. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8:729–740 [DOI] [PubMed] [Google Scholar]
- 7. Collado M, Blasco MA, Serrano M. 2007. Cellular senescence in cancer and aging. Cell 130:223–233 [DOI] [PubMed] [Google Scholar]
- 8. Good PJ. 1995. A conserved family of elav-like genes in vertebrates. Proc. Natl. Acad. Sci. U. S. A. 92:4557–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. 1996. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 271:8144–8151 [DOI] [PubMed] [Google Scholar]
- 10. Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, Harokopos V, Aidinis V, Hemberger M, Kontoyiannis DL. 2009. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol. Cell. Biol. 29:2762–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brennan CM, Steitz JA. 2001. HuR and mRNA stability. Cell. Mol. Life Sci. 58:266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doller A, Pfeilschifter J, Eberhardt W. 2008. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell. Signal. 20:2165–2173 [DOI] [PubMed] [Google Scholar]
- 13. Abdelmohsen K, Lal A, Kim HH, Gorospe M. 2007. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 6:1288–1292 [DOI] [PubMed] [Google Scholar]
- 14. Hinman MN, Lou H. 2008. Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 65:3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. 2001. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell. Biol. 21:721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dormoy-Raclet V, Menard I, Clair E, Kurban G, Mazroui R, Di Marco S, von Roretz C, Pause A, Gallouzi IE. 2007. The RNA-binding protein HuR promotes cell migration and cell invasion by stabilizing the beta-actin mRNA in a U-rich-element-dependent manner. Mol. Cell. Biol. 27:5365–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levy NS, Chung S, Furneaux H, Levy AP. 1998. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 273:6417–6423 [DOI] [PubMed] [Google Scholar]
- 18. Lopez de Silanes I, Gorospe M, Taniguchi H, Abdelmohsen K, Srikantan S, Alaminos M, Berdasco M, Urdinguio RG, Fraga MF, Jacinto FV, Esteller M. 2009. The RNA-binding protein HuR regulates DNA methylation through stabilization of DNMT3b mRNA. Nucleic Acids Res. 37:2658–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. 2004. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 23:3092–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 23:1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tominaga K, Srikantan S, Lee EK, Subaran SS, Martindale JL, Abdelmohsen K, Gorospe M. 2011. Competitive regulation of nucleolin expression by HuR and miR-494. Mol. Cell. Biol. 31:4219–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M. 2001. Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol. Cell. Biol. 21:5889–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. 2008. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. U. S. A. 105:20297–20302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. 2010. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany, NY) 2:333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang N, Yi J, Guo G, Liu X, Shang Y, Tong T, Cui Q, Zhan M, Gorospe M, Wang W. 2010. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol. Cell. Biol. 30:3875–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. U. S. A. 100:8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M. 2000. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 20:760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. 2001. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 413:83–86 [DOI] [PubMed] [Google Scholar]
- 29. Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. 2001. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413:86–91 [DOI] [PubMed] [Google Scholar]
- 30. Yamagishi M, Ishihama Y, Shirasaki Y, Kurama H, Funatsu T. 2009. Single-molecule imaging of beta-actin mRNAs in the cytoplasm of a living cell. Exp. Cell Res. 315:1142–1147 [DOI] [PubMed] [Google Scholar]
- 31. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12:2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. 2008. A translational profiling approach for the molecular characterization of CNS cell types. Cell 135:738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakamura H, Kawagishi H, Watanabe A, Sugimoto K, Maruyama M, Sugimoto M. 2011. Cooperative role of the RNA-binding proteins Hzf and HuR in p53 activation. Mol. Cell. Biol. 31:1997–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mu XC, Higgins PJ. 1995. Differential growth state-dependent regulation of plasminogen activator inhibitor type-1 expression in senescent IMR-90 human diploid fibroblasts. J. Cell. Physiol. 165:647–657 [DOI] [PubMed] [Google Scholar]
- 36. Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. 1993. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 8:2457–2467 [PubMed] [Google Scholar]
- 37. Palmero I, Pantoja C, Serrano M. 1998. p19ARF links the tumour suppressor p53 to Ras. Nature 395:125–126 [DOI] [PubMed] [Google Scholar]
- 38. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602 [DOI] [PubMed] [Google Scholar]
- 39. Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S. 1993. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 8:3313–3322 [PubMed] [Google Scholar]
- 40. Abdelmohsen K, Gorospe M. 2010. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 1:214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sherr CJ. 2001. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2:731–737 [DOI] [PubMed] [Google Scholar]
- 42. Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. 2008. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135:749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E. 2003. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr. Biol. 13:161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gajjar M, Candeias MM, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, Fahraeus R. 2012. The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21:25–35 [DOI] [PubMed] [Google Scholar]
- 45. Abdelmohsen K, Tominaga K, Lee EK, Srikantan S, Kang MJ, Kim MM, Selimyan R, Martindale JL, Yang X, Carrier F, Zhan M, Becker KG, Gorospe M. 2011. Enhanced translation by Nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res. 39:8513–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I. 2009. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15:1082–1087 [DOI] [PubMed] [Google Scholar]
- 47. Cherry J, Jones H, Karschner VA, Pekala PH. 2008. Post-transcriptional control of CCAAT/enhancer-binding protein beta (C/EBPbeta) expression: formation of a nuclear HuR-C/EBPbeta mRNA complex determines the amount of message reaching the cytosol. J. Biol. Chem. 283:30812–30820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al Regaiey K, Su L, Sharpless NE. 2004. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114:1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91:649–659 [DOI] [PubMed] [Google Scholar]
- 50. Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. 2009. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J. Clin. Invest. 119:3530–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. 2012. lincRNA-p21 suppresses target mRNA translation. Mol. Cell 47:648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, Lawrence JB, Imbalzano AN, Jones SN. 2008. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J. Cell Biol. 181:1055–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miceli AP, Saporita AJ, Weber JD. 2012. Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol. Cell. Biol. 32:348–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Origanti S, Nowotarski SL, Carr TD, Sass-Kuhn S, Xiao L, Wang JY, Shantz LM. 2012. Ornithine decarboxylase mRNA is stabilized in an mTORC1-dependent manner in Ras-transformed cells. Biochem. J. 442:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gantt K, Cherry J, Tenney R, Karschner V, Pekala PH. 2005. An early event in adipogenesis, the nuclear selection of the CCAAT enhancer-binding protein β (C/EBPβ) mRNA by HuR and its translocation to the cytosol. J. Biol. Chem. 280:24768–24774 [DOI] [PubMed] [Google Scholar]
- 56. Jones H, Carver M, Pekala PH. 2007. HuR binds to a single site on the C/EBPbeta mRNA of 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 355:217–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tanaka T, Yoshida N, Kishimoto T, Akira S. 1997. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 16:7432–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Berberich SJ, Litteral V, Mayo LD, Tabesh D, Morris D. 1999. mdm-2 gene amplification in 3T3-L1 preadipocytes. Differentiation 64:205–212 [DOI] [PubMed] [Google Scholar]
- 59. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S. 2007. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336 [DOI] [PubMed] [Google Scholar]
- 60. Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. 2007. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, McCarthy MI, Hattersley AT. 2007. Replication of genome-wide association signals in U.K. samples reveals risk loci for type 2 diabetes. Science 316:1336–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]