

Abstract
Nonalcoholic steatohepatitis (NASH) is a liver disorder that still demands improved treatment. Understanding the pathogenesis of NASH will help to develop novel approaches to prevent or treat this disease. In this study, we revealed a novel function of the aryl hydrocarbon receptor (AhR) in NASH. Transgenic or pharmacological activation of AhR heightened animal sensitivity to NASH induced by the methionine- and choline-deficient (MCD) diet, which was reasoned to be due to increased hepatic steatosis, production of reactive oxygen species (ROS), and lipid peroxidation. Mechanistically, the increased ROS production in AhR-activated mouse liver was likely a result of a lower superoxide dismutase 2 (SOD2) activity and compromised clearance of ROS. Activation of AhR induced tetrachlorodibenzo-p-dioxin (TCDD)-inducible poly(ADP-ribose) polymerase (TiPARP) gene expression, depleted NAD+, deactivated the mitochondrial sirtuin deacetylase 3 (Sirt3), increased SOD2 acetylation, and thereby decreased SOD2 activity. We also showed that Sirt3 ablation sensitized mice to NASH, whereas adenoviral overexpression of Sirt3 alleviated the NASH phenotype in AhR-transgenic mice. We conclude that activation of AhR sensitizes mice to NASH by facilitating both the “first hit” of steatosis and the “second hit” of oxidative stress. Our results suggest that the use of AhR antagonists might be a viable approach to prevent and treat NASH. Manipulation of the expression or activity of Sirt3 may also represent a novel approach to manage NASH.
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is part of the spectrum of the nonalcoholic fatty liver disease (NAFLD) characterized by steatosis, inflammation, and progressive fibrosis (1, 2). Sustained NASH may eventually progress to cirrhosis, liver failure, and hepatocellular carcinoma. Although the detailed mechanism for NASH remains to be defined, it is generally believed that the pathogenesis of NASH fits a two-hit model, in which the first hit is an initial metabolic disturbance that increases the influx of free fatty acids and de novo lipogenesis, whereas the second hit includes oxidative stress and lipid peroxidation (3, 4).
Oxidative stress refers to excessive formation of reactive oxygen species (ROS) and/or limited antioxidant defense. Oxidative stress plays a central role in the pathogenesis of NASH. Excessive fatty acid oxidation, due to the accumulation of fatty acids in the hepatocytes, induces oxidative stress in the mitochondria, peroxisomes, and microsomes (5). Overwhelming oxidative stress causes cell membrane lipid peroxidation and hepatic stellate cell activation, leading to the exhibition of the hallmarks of NASH, such as fibrosis, chronic inflammation, and apoptosis. Indeed, increased levels of serum oxidants and decreased antioxidant defense have been reported in NASH in both humans and rodents (6–10).
Mitochondria represent a principal source of cellular ROS as the result of inefficiencies in the flow of electrons along the electron transport chain (5). Superoxide (O2−), generated in the mitochondria during respiration, is primarily scavenged by superoxide dismutase 2 (SOD2) to hydrogen peroxide (H2O2), which is subsequently converted to water by catalase. The activity of SOD2 is tightly regulated by the mitochondrial sirtuin deacetylase 3 (Sirt3)-mediated deacetylation on its lysine residues (11, 12). Sirt3 deacetylates SOD2, increases SOD2 activity, and thus enhances the scavenging of superoxide (11–13).
The aryl hydrocarbon receptor (AhR) is a ligand-activated xenobiotic receptor that is expressed in most tissues (14, 15). Activated AhR forms a heterodimer with the AhR nuclear translocator (ARNT) and binds to xenobiotic response elements in the promoter regions of the target genes, such as the cytochrome P450 1A1 (CYP1A1), CYP1A2, and CYP1B1 (16). AhR can be activated by numerous environmental contaminants, including tetrachlorodibenzo-p-dioxin (TCDD), benzopyrene, and coplanar polychlorinated biphenyls (16). More recent studies showed that AhR can also be activated by endogenous ligands (17–19). We have previously reported that transgenic or pharmacological activation of AhR led to spontaneous fatty liver disease by transcriptionally activating the fatty acid translocase FAT/CD36 and increasing the fatty acid uptake (20). The steatotic effect of AhR was also independently reported by others (21). We and others have also reported that AhR induced cellular oxidative stress and increased lipid peroxidation (20–22), another risk factor for NASH. In human populations, dioxin exposure has been reported to be associated with increased incidence of fatty liver (23). However, it remains to be determined whether and how activation of AhR sensitizes rodents or humans to NASH.
In this study, we showed activation of AhR-sensitized mice to methionine- and choline-deficient (MCD) diet-induced NASH, which may have been accounted for by the decreased SOD2 activity and increased ROS production in the liver. Mechanistically, we found Sirt3 activity was inhibited by AhR, whereas hepatic overexpression of Sirt3 alleviated MCD diet-induced NASH in AhR-activated mice.
MATERIALS AND METHODS
Animals, diet and drug treatment, and histology.
The creation of the tetracycline-inducible CA-AhR TG mice has been previously described (20). The TG mice and their wild-type (WT) controls used in this study have been backcrossed to C57BL/6J for at least 6 generations. When necessary, doxycycline (DOX; 2 mg/ml) was given in drinking water for 2 weeks to silent transgene expression. The Sirt3−/− mice (stock number 012755) (24) were purchased from the Jackson Laboratory (Bar Harbor, ME). For diet treatment, mice received the MCD diet (catalog number TD.90262) or the control amino acid diet (catalog number TD.90149) from Harlan (Denver, CO). Mice were treated with the MCD diet for 4 to 6 weeks in most of the experiments, except that when TCDD-treated WT mice or Ad-Sirt3 mice were used, mice were treated with the MCD diet for 2 weeks only. This is because an extended MCD diet treatment in TCDD-treated mice was partially lethal, whereas the 2-week treatment was chosen for the Ad-Sirt3 mice for optimal adenoviral expression of Sirt3 (data not shown). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was purchased from Biomol (Plymouth Meeting, PA). When necessary, mice received a single gavage of vehicle (corn oil) or TCDD (10 μg/kg) by using a plastic-coated mouse-feeding needle. For histology, tissues were fixed in 4% formaldehyde, embedded in paraffin, sectioned at 5 μm, and stained with H&E. The use of mice in this study has complied with relevant federal guidelines and institutional policies.
Measurements of SOD activity and levels of H2O2, O2−, and MDA.
SOD activity was measured as the inhibition of nitroblue tetrazolium (NBT) reduction in a xanthine-xanthine oxidase system (12, 25). Briefly, liver tissues were homogenized in 50 mM potassium phosphate buffer (pH = 7.8, with 1.34 mM diethylenetriaminepentaacetic acid) and then centrifuged at 12,000 × g for 10 min. The tissue lysate supernatants were collected for the SOD activity assay. This total SOD assay is based on the competition between SOD and an indicator molecule (NBT) for superoxide production from xanthine and xanthine oxidase. Incubation with 5 mM sodium cyanide for at least 45 min was used to inhibit SOD1 activity in order to selectively measure the SOD2 activity. The difference between total SOD and SOD2 activity is considered to be the SOD1 activity. The H2O2 levels were measured by using the Amplex red hydrogen peroxide assay kit from Invitrogen as described by others (26). The O2− level in primary hepatocytes was measured by electron paramagnetic (EPR) spectrometry using the cell-permeable hydroxlymine spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) (Alexis). In brief, 50-μl cell suspensions were placed into a temperature- and gas-controlled Bruker eScan tabletop EPR spectrometer cavity and analyzed for 10 min at 37°C and 21% O2. Values represent the relative signal intensity of the downfield peak of 5 signal-averaged scans from 9 to 10 min. The EPR instrument settings were as follows: field sweep, 50 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 2 G; conversion time, 327 ms; time constant, 655 ms; and receiver gain, 1 × 105. To minimize the effects of adventitious metals, all buffers were treated with Chelex resin and contained 25 μM deferoxamine. The addition of purified CuZnSOD was used to validate O2− as the radical responsible for the observed signal as previously described (27). The method to measure the liver concentration of malondialdehyde (MDA) was previously described (20).
Immunoprecipitation and Western blot analysis.
Livers were homogenized and mitochondria were isolated as previously described (24). Mitochondria were lysed in ice-cold RIPA buffer containing 10 mM nicotinamide and 1 μM trichostatin A. The lysates were immunoprecipitated with an anti-SOD2 antibody (catalog number sc-133254) from Santa Cruz (Santa Cruz, CA) overnight and immunoblotted with an antiacetylation antibody (catalog number 9681) from Cell Signaling (Danvers, MA). The Sirt3 antibody (catalog number AP6242a) was purchased from ABGENT (San Diego, CA).
Hepatic overexpression of Sirt3 by adenoviral transduction.
Adenovirus overexpressing Flag-Sirt3 (Ad-Sirt3) was kindly provided by Bert Vogelstein from the Johns Hopkins University. The control adenovirus (Ad-Ctrl) contained the green fluorescence protein. Adenoviruses were injected through tail veins at the dose of 4 × 109 PFU per mouse. Infected mice were fed the MCD diet for 2 weeks before NASH phenotype analysis.
Measurement of NAD+.
NAD+ and NADH were measured by fluorescence in the liver homogenates (250 mg [wet weight] in 1 ml of phosphate-buffered saline [PBS]) (28). Fifty microliters of samples was added to 50 μl of 7% perchloric acid, vortexed for 30 s, sonicated, and then incubated for 30 min at 60°C to destroy the endogenous enzyme activity. After centrifugation (13,000 rpm for 3 min), the supernatants were neutralized and 2-μl and 4-μl aliquots were added to black polystyrene 96-well plates from Corning (Vernon Hills, IL). Reaction mixtures (130-μl total volume) contained cycling assay buffer (5 mM Tris, 5 mM MgCl2, 50 mM KCl), 54 μM resazurin, 0.4 unit/ml lactate dehydrogenase, and 2.25 mM lactate, with the pH adjusted to 7.5. The reactions of transforming resazurin to resorufin were initiated with 20 μl of diaphorase per well for a final concentration of 0.7 unit/well. Standard curves for NAD+ were included in each assay. Resorufin fluorescence was measured immediately after adding diaphorase using a plate reader (excitation, 530 nm; emission, 580 nm) at 30-s intervals for 12.5 min. Values were corrected for dilutions and normalized to the protein contents.
Measurement of Sirt3 activity.
The Sirt3 activity was assessed by fluorometric measurement of deacetylation of a Sirt3 peptide substrate by using the CycLex SIRT3 deacetylase kit (catalog number CY-1153) from CycLex (Nagano, Japan).
Gene expression analysis by real-time PCR.
Reverse transcription (RT) was performed with random hexamer primers and Superscript RT III enzyme from Invitrogen. SYBR green-based real-time PCR was performed with the ABI 7300 system (29). Data were normalized against the control cyclophilin.
Statistical analysis.
Data are expressed as the means ± standard deviations (SD). One-way analysis of variance (ANOVA) Tukey's test was performed for statistical analysis using GraphPad Prism version 4.0. P values of less than 0.05 were considered statistically significant.
RESULTS
Genetic activation of AhR sensitized mice to MCD diet-induced NASH.
We have previously reported the creation of tetracycline-inducible constitutive active AhR (CA-AhR)-transgenic mice expressing CA-AhR in the liver under the control of the fatty acid binding protein (FABP) gene promoter (20). CA-AhR was constructed by deleting the minimal ligand-binding domain of AhR (30). As outlined in Fig. 1A, in mice carrying both the tetracycline response element (TetRE)-CA-AhR and FABP-tetracycline transcriptional activator (tTA) transgenes, tTA expressed in the liver will bind to TetRE and induce the expression of CA-AhR, whereas treatment of mice with doxycycline (DOX) will dissociate tTA from TetRE and thus silence the expression of CA-AhR. The expression of the CA-AhR transgene and its silencing by DOX was confirmed by real-time PCR (Fig. 1A). The expression of CA-AhR was largely unaffected in transgenic (TG) mice treated with the MCD diet (Fig. 1A). The expression of the endogenous AhR was indistinguishable between wild-type (WT) and TG mice (data not shown).
Fig 1.

Transgenic activation of AhR sensitized mice to MCD diet-induced NASH. (A) Top, schematic representation of the liver-specific “Tet-off” CA-AhR transgenic system. DOX, doxycycline; FABP, fatty acid binding protein; PCMV, minimal CMV promoter; TetRE, tetracycline responsive element; tTA, tetracycline transcriptional activator. Bottom, the expression of CA-AhR was confirmed by transgene-specific real-time PCR. The cycle numbers are labeled. (B to G) WT and TG mice were fed with the MCD diet for 6 weeks in the presence or absence of DOX before being analyzed for hepatic triglyceride level (B); liver section Picro-sirius red staining, with the arrowhead indicating the collagen deposition (C); liver section H&E staining, with the arrows indicating the neutrophil infiltration (D); hepatic expression of fibrogenic genes (E) and inflammatory genes (F); and serum ALT levels (G). n = 6 for each group. *, P < 0.05.
To determine the in vivo role of AhR in NASH, we fed WT and TG mice with the amino acid control diet or MCD diet for 6 weeks. The MCD diet is a widely used dietary model of NASH that produces key features of NASH, including steatosis, hepatic inflammation, and fibrosis (31). The MCD diet alone had little effect on the expression of the endogenous AhR and its typical target genes (data not shown). We found that the hepatic triglyceride level was higher in TG mice than in WT mice (Fig. 1B), consistent with our reported steatotic role of AhR (20). Increased collagen deposition (Fig. 1C) and neutrophil infiltration (Fig. 1D) in the liver of TG mice were shown by Picro-sirius red staining and H&E staining, respectively. The transgenic sensitization of NASH was further confirmed by gene expression analysis. This included the induction of the fibrogenic genes Col1a1, Col1a2, TGF-β, and α-SMA (Fig. 1E), as well as the induction of proinflammatory genes TNF-α, IL-1β, and IL-6 (Fig. 1F). These phenotypes were transgene dependent, because they were normalized in TG mice treated with DOX (Fig. 1B to F). Interestingly, despite their heightened NASH phenotypes in fibrosis and inflammation, the serum level of alanine aminotransferase (ALT) was not higher in TG mice than in WT mice (Fig. 1G).
Pharmacological activation of AhR sensitized mice to MCD diet-induced NASH.
We treated WT C57BL/6J mice with a single dose of the AhR ligand TCDD (10 μg/kg) and then fed the mice with a control diet or the MCD diet for 2 weeks. Under the control diet, this dose of TCDD did not cause apparent liver toxicity, as suggested by a minimum change in the serum ALT level, but was effective in inducing AhR target genes (data not shown). In contrast, when mice were fed with the MCD diet, TCDD treatment significantly increased the serum ALT level (Fig. 2A). The TCDD-treated mice also showed increased hepatic steatosis (Fig. 2B), neutrophil infiltration (Fig. 2C), fibrogenic gene expression (Fig. 2D), and inflammatory gene expression (Fig. 2E).
Fig 2.

Pharmacological activation of AhR sensitized mice to MCD diet-induced NASH. (A to E) WT mice were injected with a single dose of TCDD (10 μg/kg) or vehicle and then fed with the MCD diet for 2 weeks before being analyzed for serum ALT level (A); hepatic triglyceride content (B); liver section H&E staining, with the arrows indicating the neutrophil infiltration (C); and hepatic expression of fibrogenic genes (D) and inflammatory genes (E). n = 5 for each group. *, P < 0.05; **, P < 0.01. TCDD versus vehicle.
Activation of AhR increased ROS production, compromised SOD2 activity, and increased lipid peroxidation.
Oxidative stress plays a central role in the pathogenesis of NASH (3, 4). To understand the mechanism by which AhR sensitized mice to NASH, we measured the ROS levels in AhR-activated mice. The H2O2 level was increased in the MCD diet-fed TG mice (Fig. 3A) and TCDD-treated WT mice (Fig. 3B). Treatment of WT primary mouse hepatocytes with TCDD also increased the O2− production (Fig. 3C). The increased ROS level in AhR-activated mice may have been accounted for by decreased SOD activity. Interestingly, it is the activity of the mitochondrial SOD2, but not SOD1, that was significantly decreased by AhR activation (Fig. 3D and E). Lipid peroxidation is often used as a surrogate marker for oxidative stress. Consistent with their increased ROS production, the MCD diet-fed and AhR-activated mice showed increased liver concentrations of malondialdehyde (MDA), an end product of lipid peroxidation (Fig. 3F and G). The transgenic phenotypes were normalized upon DOX treatment.
Fig 3.

Activation of AhR increased ROS production, compromised SOD2 activity, and increased lipid peroxidation. Mice were the same as described in the legends to Fig. 1 and 2. (A and B) Hepatic H2O2 levels in WT and TG mice treated without or with DOX (A) or WT mice treated with vehicle or TCDD (B). (C) Superoxide (O2−) levels in primary hepatocytes treated with vehicle or TCDD. (D and E) Total SOD, SOD2, and SOD1 activities in WT and TG mice treated without or with DOX (D) or WT mice treated with vehicle or TCDD (E). (F and G) Hepatic concentrations of malondialdehyde (MDA) in WT and TG mice treated without or with DOX (F) or WT mice treated with vehicle or TCDD (G). *, P < 0.05.
Activation of AhR increased SOD2 acetylation and decreased Sirt3 activity.
The SOD2 activity is tightly regulated by acetylation on its lysine residues (11, 12). We went on to measure the SOD2 acetylation by SOD2 immunoprecipitation followed by Western blotting using an anti-acetyl-lysine antibody. Indeed, transgenic (Fig. 4A) or pharmacological (Fig. 4B) activation of AhR increased SOD2 acetylation. Sirt3 is a mitochondrial NAD+-dependent deacetylase responsible for SOD2 deacetylation (11, 12). The Sirt3 expression was not affected in AhR-activated mice (Fig. 4C). Instead, the level of NAD+, which is required for the deacetylase activity of Sirt3, was decreased in the liver of TG mice and TCDD-treated WT mice (Fig. 4D). The decreased NAD+ level in AhR-activated mice may have been accounted for by the upregulation of the TCDD-inducible poly(ADP-ribose) polymerase (TiPARP) gene (Fig. 4E), an AhR target gene known to deplete NAD+ (28). Finally, we showed the activity of Sirt3 was decreased in both TG mice and TCDD-treated WT mice (Fig. 4F). These results together suggested that activation of AhR inhibited SOD2 activity by inhibiting Sirt3-mediated SOD2 deacetylation.
Fig 4.
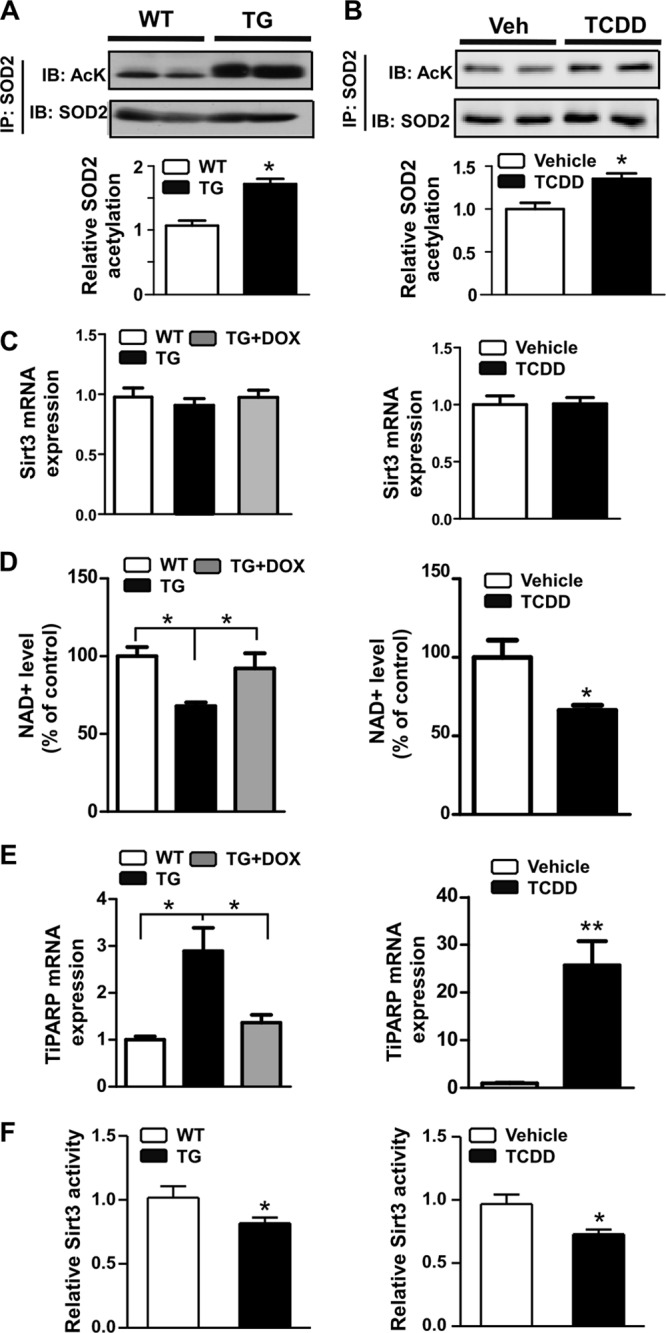
AhR activation increased SOD2 acetylation and decreased Sirt3 activity. (A and B) SOD2 acetylation in TG mice (A) and TCDD-treated WT mice (B), as determined by immunoprecipitation and immunoblotting. The bottom rows in panels A and B are the quantifications of the Western blot results. (C) Hepatic Sirt3 mRNA expression in TG mice (left) and TCDD-treated WT mice (right); (D) NAD+ level in TG mice and TCDD-treated WT mice; (E) hepatic TiPARP mRNA expression in TG mice and TCDD-treated WT mice; (F) hepatic Sirt3 activity in TG mice (left) and TCDD-treated WT mice (right). *, P < 0.05; **, P < 0.01.
Sirt3 ablation sensitized mice to MCD diet-induced NASH.
To further define the role of Sirt3 in NASH, we subjected the Sirt3−/− mice (24) to the MCD diet for 4 weeks. The lack of Sirt3 protein expression in Sirt3−/− mice was confirmed by Western blotting (Fig. 5A). Indeed, Sirt3 ablation sensitized mice to MCD diet-induced NASH. Specifically, compared to their WT counterparts, MCD diet-fed Sirt3−/− mice showed an increased serum ALT level (Fig. 5A), increased expression of fibrogenic genes (Fig. 5B), increased expression of inflammatory genes (Fig. 5C), increased hepatic triglyceride content (Fig. 5D), increased neutrophil infiltration (Fig. 5E), and a decreased SOD2 activity (Fig. 5F). The acetylation of SOD2 was increased in Sirt3−/− mice (Fig. 5G), consistent with the loss of Sirt3-mediated deacetylation activity.
Fig 5.

Sirt3 ablation sensitized mice to MCD diet-induced NASH. (A to E) WT and Sirt3−/− mice were fed the MCD diet for 4 weeks before being analyzed for serum ALT level (A); hepatic fibrogenic gene expression (B); inflammatory gene expression (C); hepatic triglyceride content (D); liver section H&E staining, with the arrows indicating the neutrophil infiltration (E); SOD activities (F); and SOD2 acetylation (G). The right section in panel G is the quantification of the Western blot results. The inset in panel A shows the lack of Sirt3 protein expression in Sirt3−/− mice, in which the composite image was derived from a single original image. *, P < 0.05. Sirt3−/− versus WT.
Overexpression of Sirt3 alleviated CA-AhR-transgenic mice from NASH.
We overexpressed Sirt3 in CA-AhR TG mice in order to determine whether overexpression of Sirt3 was sufficient to alleviate the NASH-sensitizing effect of AhR. The Sirt3 overexpression in the liver was achieved by infecting mice with an adenovirus encoding the Flag-tagged Sirt3. The expression of the transduced Sirt3 was confirmed by Western blotting (Fig. 6A). TG mice infected with the control virus (Ad-Ctrl) or Ad-Sirt3 were fed the MCD diet for 2 weeks before being analyzed for the NASH phenotype. In contrast to the sensitizing effect of Sirt3 ablation, Sirt3 overexpression alleviated TG mice from MCD diet-induced NASH. Specifically, compared to their Ad-Ctrl-infected counterparts, Ad-Sirt3-infected TG mice showed a decreased serum ALT level (Fig. 6B), decreased hepatic steatosis (Fig. 6C), decreased neutrophil infiltration (Fig. 6D), decreased expression of fibrogenic genes (Fig. 6E), decreased expression of inflammatory genes (Fig. 6F), increased SOD2 activity (Fig. 6G), and decreased production of H2O2 (Fig. 6H). The acetylation of SOD2 was decreased in the liver of Ad-Sirt3 mice (Fig. 6I), consistent with the increased Sirt3-mediated deacetylation activity.
Fig 6.

Overexpression of Sirt3 alleviated CA-AhR transgenic mice from NASH. Transgenic mice infected with the control virus (Ad-Ctrl) or Ad-Sirt3 were fed with the MCD diet for 2 weeks. (A) Western blotting to confirm the expression of Flag-Sirt3. The top and bottom panels were blotted with anti-Flag and anti-Sirt3 antibodies, respectively. The composite image of the top panel was derived from a single original image. (B to I) Serum ALT level (B); hepatic triglyceride content (C); liver section H&E staining, with the arrows indicating the neutrophil infiltration (D); hepatic fibrogenic gene expression (E) and inflammatory gene expression (F); SOD activities (G); hepatic H2O2 level (H); and SOD2 acetylation (I). The right section in panel I is the quantification of the Western blot results. *, P < 0.05. Ad-Sirt3 versus Ad-Ctrl.
DISCUSSION
In this study, we have revealed a novel function of AhR in NASH. The heightened sensitivity to NASH in AhR-activated mice was due to the combined effect of increased steatosis and increased ROS production (Fig. 7). The increased ROS level was likely a result of a decreased SOD2 activity and compromised ROS scavenging. Activation of AhR induced TiPARP gene expression, depleted NAD+, deactivated Sirt3, increased SOD2 acetylation, and thereby decreased SOD2 activity. Sirt3 ablation sensitized mice to NASH, whereas adenoviral overexpression of Sirt3 alleviated the NASH phenotype in AhR-activated mice.
Fig 7.
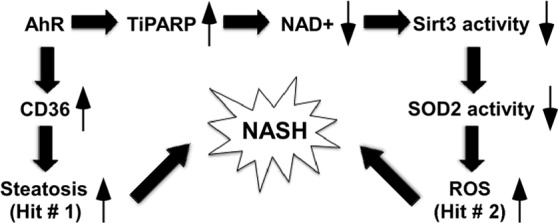
Summary of the effects of AhR on Sirt3 activity and NASH. We propose that activation of AhR sensitizes mice to NASH by facilitating both “hits”: the first hit of steatosis, and the second hit of oxidative stress.
We have previously reported that activation of AhR caused spontaneous hepatic steatosis primarily by the activation of CD36 and promotion of hepatic fatty acid uptake (20). However, hepatic triglyceride accumulation alone is believed to be not sufficient to trigger fibrosis and inflammation. Accumulating evidence suggests that oxidative stress is the necessary “second hit” to trigger liver injuries in NASH (4, 5). Indeed, NASH in both humans and rodents is associated with increased oxidative stress and decreased activities of antioxidant enzymes (6–10). SOD2 is one of the primary antioxidant enzymes in the mitochondria, which is the principal source of cellular ROS. SOD2 converts superoxide to hydrogen peroxide, which is subsequently converted to water by catalase and other peroxidases. Decreased expression or activity of SOD2 has been associated with NASH (6, 9). Our results showed that activation of AhR in the liver inhibited SOD2 activity and increased the ROS level. We conclude that activation of AhR sensitizes mice to NASH by facilitating both “hits”: the first hit of steatosis, and the second hit of oxidative stress (Fig. 7).
Sirt3 is the primary mitochondrial deacetylase (13). Sirt3 regulates the energy-producing mitochondrial proteins and enhances their enzymatic activities in response to nutrient distress (32, 33). Recent studies showed Sirt3 controls SOD2 activity through regulating its acetylation (11, 12). Sirt3−/− mice showed a decreased SOD2 activity and increased oxidative stress (11, 12). We found that activation of AhR decreased Sirt3 activity, which may have resulted from decreased levels of NAD+. This notion is consistent with the observation that activation of AhR decreased the NAD+ level by activating the TiPARP gene expression. TiPARP utilizes NAD+ for ADP-ribosylation reactions and thus contributes to NAD+ depletion.
The importance of Sirt3 in regulating oxidative stress and impacting the pathogenesis of NASH was further confirmed by our uses of the Sirt3 loss-of-function and gain-of-function models. We showed that the Sirt3−/− mice were more sensitive to MCD diet-induced NASH. The increased fibrosis and inflammation in Sirt3−/− mice phenocopied the sensitizing effect of AhR activation. Conversely, adenoviral overexpression of Sirt3 protected the CA-AhR TG mice from NASH by inhibiting hepatic steatosis and ROS production. The expression of CD36 was not significantly affected in MCD diet-fed Sirt3−/− mice or Ad-Sirt3 mice (data not shown), suggesting that the Sirt3 effect on NASH was independent of the CD36 regulation.
In summary, we showed AhR activation sensitized mice to NASH by inhibiting SOD2 activity, increasing ROS production, and increasing lipid peroxidation. Mechanistically, AhR inhibited SOD2 activity by decreasing the activity of Sirt3. Sirt3 ablation sensitized mice to NASH, whereas overexpression of Sirt3 was sufficient to alleviate the NASH-sensitizing effect of AhR. It remains to be determined whether the AhR−/− mice are protected from MCD diet-induced NASH. Our results suggest that AhR antagonists might be a viable approach to prevent and treat NASH. It is encouraging that major progress has been made in the identification and characterization of AhR antagonists (17, 18). Moreover, manipulation of the expression or activity of Sirt3, such as the use of exogenous NAD and Sirt3 inducers (34, 35), also represents a novel approach to manage NASH.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants DK083952 and ES014626 (to W.X.). J.H. was supported by the AHA Postdoctoral Fellowship 09POST2280546. E.E.K. was supported by AHA National Scientist Development Grant 10SDG3560005. W.X. is the Joseph Koslow Endowed Chair in Pharmaceutical Sciences at the University of Pittsburgh School of Pharmacy.
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Vuppalanchi R, Chalasani N. 2009. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: selected practical issues in their evaluation and management. Hepatology 49:306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. 2008. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 14:72–81 [DOI] [PubMed] [Google Scholar]
- 3. Day CP, James OF. 1998. Steatohepatitis: a tale of two “hits”? Gastroenterology 114:842–845 [DOI] [PubMed] [Google Scholar]
- 4. Rolo AP, Teodoro JS, Palmeira CM. 2012. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 52:59–69 [DOI] [PubMed] [Google Scholar]
- 5. Koek GH, Liedorp PR, Bast A. 2011. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 412:1297–1305 [DOI] [PubMed] [Google Scholar]
- 6. Sreekumar R, Rosado B, Rasmussen D, Charlton M. 2003. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology 38:244–251 [DOI] [PubMed] [Google Scholar]
- 7. Sumida Y, Nakashima T, Yoh T, Furutani M, Hirohama A, Kakisaka Y, Nakajima Y, Ishikawa H, Mitsuyoshi H, Okanoue T, Kashima K, Nakamura H, Yodoi J. 2003. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J. Hepatol. 38:32–38 [DOI] [PubMed] [Google Scholar]
- 8. Koruk M, Taysi S, Savas MC, Yilmaz O, Akcay F, Karakok M. 2004. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 34:57–62 [PubMed] [Google Scholar]
- 9. Laurent A, Nicco C, Tran Van Nhieu J, Borderie D, Chereau C, Conti F, Jaffray P, Soubrane O, Calmus Y, Weill B, Batteux F. 2004. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology 39:1277–1285 [DOI] [PubMed] [Google Scholar]
- 10. Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, Sanisoglu SY, Erdil A, Ates Y, Aslan M, Musabak U, Erbil MK, Karaeren N, Dagalp K. 2005. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am. J. Gastroenterol. 100:850–855 [DOI] [PubMed] [Google Scholar]
- 11. Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. 2010. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12:662–667 [DOI] [PubMed] [Google Scholar]
- 12. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. 2010. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40:893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi T, Wang F, Stieren E, Tong Q. 2005. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280:13560–13567 [DOI] [PubMed] [Google Scholar]
- 14. Carver LA, Hogenesch JB, Bradfield CA. 1994. Tissue specific expression of the rat Ah-receptor and ARNT mRNAs. Nucleic Acids Res. 22:3038–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gu YZ, Hogenesch JB, Bradfield CA. 2000. The PAS superfamily: sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 40:519–561 [DOI] [PubMed] [Google Scholar]
- 16. Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. 2004. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 279:23847–23850 [DOI] [PubMed] [Google Scholar]
- 17. Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. 2008. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 18:207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nguyen LP, Bradfield CA. 2008. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21:102–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. 2011. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203 [DOI] [PubMed] [Google Scholar]
- 20. Lee JH, Wada T, Febbraio M, He J, Matsubara T, Lee MJ, Gonzalez FJ, Xie W. 2010. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139:653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu H, Cui W, Klaassen CD. 2011. Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced oxidative injury and steatohepatitis. Toxicol. Appl. Pharmacol. 256:122–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dalton TP, Puga A, Shertzer HG. 2002. Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact. 141:77–95 [DOI] [PubMed] [Google Scholar]
- 23. Lee CC, Yao YJ, Chen HL, Guo YL, Su HJ. 2006. Fatty liver and hepatic function for residents with markedly high serum PCDD/Fs levels in Taiwan. J. Toxicol. Environ. Health A 69:367–380 [DOI] [PubMed] [Google Scholar]
- 24. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27:8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schisler NJ, Singh SM. 1985. Tissue-specific developmental regulation of superoxide dismutase (SOD-1 and SOD-2) activities in genetic strains of mice. Biochem. Genet. 23:291–308 [DOI] [PubMed] [Google Scholar]
- 26. Mo L, Wang Y, Geary L, Corey C, Alef MJ, Beer-Stolz D, Zuckerbraun BS, Shiva S. 2012. Nitrite activates AMP kinase to stimulate mitochondrial biogenesis independent of soluble guanylate cyclase. Free Radic. Biol. Med. 53:1440–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik UZ, Hundley NJ, Romero G, Radi R, Freeman BA, Tarpey MM, Kelley EE. 2011. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radic. Biol. Med. 51:179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diani-Moore S, Ram P, Li X, Mondal P, Youn DY, Sauve AA, Rifkind AB. 2010. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 285:38801–38810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao J, He J, Zhai Y, Wada T, Xie W. 2009. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 284:25984–25992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McGuire J, Okamoto K, Whitelaw ML, Tanaka H, Poellinger L. 2001. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: delineation of overlapping repression and ligand binding functions within the PAS domain. J. Biol. Chem. 276:41841–41849 [DOI] [PubMed] [Google Scholar]
- 31. Varela-Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martinez-Chantar ML. 2009. Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int. J. Biochem. Cell Biol. 41:969–976 [DOI] [PubMed] [Google Scholar]
- 32. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. 2011. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U. S. A. 108:14608–14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. 2010. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 285:3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giralt A, Hondares E, Villena JA, Ribas F, Diaz-Delfin J, Giralt M, Iglesias R, Villarroya F. 2011. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 286:16958–16966 [DOI] [PMC free article] [PubMed] [Google Scholar]