

Abstract
We address here whether there is cellular memory of a transcriptional enhancer once it has served its purpose to establish an active chromatin state. We have previously shown that the mouse Igκ gene's downstream enhancers, E3′ and Ed, are essential but play redundant roles for establishing transcriptional activity in the locus during B cell development. To determine whether these enhancers are also necessary for the maintenance of transcriptional activity, we conditionally deleted E3′ in mature B cells that possessed Ed−/− alleles. Upon E3′ deletion, the locus became rapidly silenced and lost positive histone epigenetic marks, and the mature B cells partially dedifferentiated, induced RAG-1 and -2 along with certain other pro-B cell makers, and then redifferentiated after triggering Igλ gene rearrangements. We conclude that the Igκ gene's downstream enhancers are essential for both the establishment and maintenance of transcriptional activity and that there is no cellular memory of previous transcriptional activity in this locus. Furthermore, upon enhancer loss, the mature B cells unexpectedly underwent reversible retrograde differentiation. This result establishes that receptor editing can occur in mature B cells and raises the possibility that this may provide a tolerance mechanism for eliminating autoreactive B cells in the periphery.
INTRODUCTION
During B cell development, the mouse IgH and IgL loci become activated in a stepwise fashion for gene rearrangement (1). The IgH gene rearranges first, by sequential D-J and then by V-(D)J joining, leading to the pro- and pre-B cell stages of development, respectively. The Igκ locus undergoes rearrangement next in pre-B cells, where a Vκ gene is joined to a Jκ region. If Igκ V-J joining is productively unsuccessful because of out-of-reading frame recombination junctions, then the Igλ locus becomes activated for rearrangement and expression, which in wild-type (WT) mice accounts for production of only approximately 5% of the total IgL chains (2).
In order to characterize chromatin structure-function relationships in a model system, research in our laboratory has focused on the mouse Igκ locus because it offers the opportunity to identify changes in chromatin structure that may precede gene rearrangement and transcriptional activation during B lymphocyte differentiation, as well as to visualize chromatin remodeling events that are linked to gene activation (reference 3 and references therein). Rearrangement of the Igκ locus deposits a Vκ gene carrying its own promoter into a chromatin domain containing three powerful enhancers: an intronic enhancer (Ei) within the transcription unit (4), and two enhancers downstream of the transcription termination region, termed E3′ and Ed (3, 5). The results of chromosome conformation capture experiments have revealed that in activated B cells, the three enhancers exhibit all possible pairwise interactions with themselves and rearranged Vκ gene promoters, with the looping out of the intervening DNA sequences (6). However, in unstimulated B cells, rearranged Vκ gene promoters only form complexes with either Ei or E3′, but not with Ed. This results in a basal transcriptional state in the locus (7).
The functions of the Igκ gene's enhancers in B lymphocytes have been previously studied by creating single or pairwise enhancer-targeted deletions. These experiments revealed that Ei and E3′ each play quantitative roles in Igκ gene rearrangement (8, 9), while deletion of both Ei and E3′ eliminates rearrangement (10). In addition, E3′ and Ed each play quantitative roles in rearranged gene transcription (8, 11), while deletion of both E3′ and Ed abolishes Igκ gene transcription (12). These results reveal that these enhancers play partially overlapping compensatory roles in this locus.
While it seems clear that enhancers are required to initiate an active chromatin state, whether they are required continuously to maintain the active state once established is an interesting question (13). This question has been addressed in the human β-globin locus and mouse IgH gene by deleting these genes' locus control region, intronic Eμ or far downstream enhancers. The results of these studies revealed that transcription ceased in each case upon deletion of these enhancers (14–16). However, transformed cell lines were used in each of these investigations, and many rounds of DNA replication ensued after enhancer deletion before the transcriptional consequences of such deletions were assayed. Hence, the effects of enhancer deletion in the absence of ongoing DNA replication in a setting that resembles the in vivo condition more closely remains unresolved by these studies. In contrast, when the E4p CD4 T cell enhancer was conditionally deleted in mature CD4+ T cells, CD4 expression was stably maintained through several rounds of division, indicating that E4p was no longer needed to maintain transcriptional activity (17). Here we address whether the Igκ gene's downstream enhancers are necessary for both the establishment and maintenance of transcription in the locus. We took advantage of the observations that E3′ and Ed are essential for establishing transcriptional activity (12) but that B cell development and rearranged Igκ gene transcription are nearly normal in Ed−/− mice (11) by conditionally deleting E3′ in vivo in mature B cells that possessed Ed−/− alleles. We found that the locus rapidly became silenced and lost positive epigenetic histone marks upon E3′ deletion even in the absence of DNA replication, indicating that the downstream enhancers are required for both the establishment and maintenance of transcriptional activity in this system. These results represent the first example demonstrating that an enhancer's continuous presence is essential in vivo to maintain gene activity in nonreplicating chromatin.
Repeated rearrangements that alter the specificity of the B cell receptor (BCR) to avoid autoreactivity are referred to as receptor editing (18). It has been demonstrated that receptor editing is an important mechanism for the maintenance of immune tolerance at early stages of B cell ontogeny in the bone marrow. If a developing B cell expresses a BCR that recognizes an autoantigen, it signals reexpression of the RAG-1 and -2 genes that triggers further gene rearrangements. Receptor editing to produce nonautoreactive BCRs can be accomplished by repeated Vκ rearrangements and by inactivation of rearranged autoreactive Igκ genes by RS rearrangements, which leads to isotype switching (i.e., from κ to λ light chains). Although continued receptor editing has been reported also to occur in mature B cells, which in some cases has been referred to as receptor revision (19, 20), some of these observations have been explained by the existence of immature B cells in the periphery (21). Hence, whether receptor editing can occur in mature B cells has remained controversial. Unexpectedly, however, the mature B cells possessing the lost enhancers studied here partially dedifferentiated, induced RAG-1 and -2 along with certain other pro-B cell makers, and then redifferentiated after triggering Igλ gene rearrangements. These results reveal that continued receptor editing can occur in mature B cells and raise the possibility that this may serve as one mechanism of peripheral tolerance against autoantigens.
MATERIALS AND METHODS
Animal models.
Tamoxifen-inducible Cre mice, named ROSA26CreER(T) (22), were purchased from The Jackson Laboratory (stock no. 004847). Mice bearing a CD23-Cre knocked-in gene were kindly provided by Meinrad Busslinger of the Research Institute of Molecular Pathology, Vienna, Austria (23). Ed−/− mice were previously established in our laboratory (11). Mice bearing loxP sites flanking E3′ and an Ed deletion (E3′F/+ Ed−/+) were used to establish double enhancer knockout mice in a previous study (12). In the present study, these mice were backcrossed with CD23-Cre mice to obtain heterozygous E3′F/+ Ed−/+ CD23-Cre mice. Homozygous E3′F/F Ed−/− CD23-Cre mice were obtained by breeding E3′F/+ Ed−/+ with E3′F/+ Ed−/+ CD23-Cre mice. A similar strategy was used to obtain E3′F/F Ed−/− ROSA26CreER(T) mice. All mouse strains were used in accordance with protocols approved by the University of Texas (UT) Southwestern Medical Center Institutional Animal Care and Use Committee (IACUC).
Flow cytometry.
Single-cell suspensions were prepared from bone marrow and spleens of 8- to 14-week-old mice. Samples were stained with antibodies and analyzed using a FACSCalibur with CellQuest software (BD Bioscience, San Diego, CA). For IgL isotype analysis, splenic cells were first stained with anti-FcγII/III (catalog no. 553142BD; BD Bioscience) (1 μg/ml) at 4°C for 30 min. After the cells were washed, they were stained with anti-mouse Igκ-PE (anti-mouse Igκ conjugated to phycoerythrin) (catalog no. 559940; BD Bioscience) (0.4 μg/ml), anti-mouse Igλ1-3-FITC (anti-mouse Igλ1-3 conjugated to fluorescein isothiocyanate) (catalog no. 553434; BD Bioscience) (5 μg/ml), and anti-mouse B220-PE-Cy5.5 (anti-mouse B220 conjugated to phycoerythrin and Cy5.5) (catalog no. 553092; BD Bioscience) (0.5 μg/ml) at 4°C for 30 min. Allophycocyanin (APC)-conjugated antibody specific for μ chain (catalog no. 550676; BD Bioscience) (1 μg/ml), δ chain (catalog no. 17-5993; eBioscience) (0.5 μg/ml), or B cell-activating factor receptor (BAFF-R) (catalog no. 134105; Biolegend) (1 μg/ml) were used in four-color fluorescence-activated cell sorting (FACS) to detect their expression on B220, Igκ, and Igλ labeled cells. Other reagents used were as follows: anti-CD43-PE (BD Bioscience), anti-CD25-APC (BD Bioscience), anti-λ5-biotin (BD Bioscience), anti-preBCR-biotin (BD Bioscience), streptavidin-APC (BD Bioscience), and Alexa Fluor 647-conjugated anti-VλX monoclonal antibody (10C5) (originally from P. A. Cazenave [Pasteur Institute, Paris, France] [24]) kindly provided by Martin Weigert (25).
Cell purification and culture.
Splenic single-cell suspensions were incubated with an optimal concentration of biotinylated anti-B220 (BD Bioscience). Then magnetic activated cell sorting (MACS) MS separation columns (Miltenyi Biotec, Auburn, CA) were used to obtain B cells. B220+ Igκ+ Igλ−, B220+ Igκ− Igλ+, and B220+ Igκ− Igλ− B cells from E3′F/F Ed−/− CD23-Cre mice were sorted on a MoFlo machine (Dako Cytomation, Carpinteria, CA). Sorted B220+ Igκ− Igλ− B cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, l-glutamine, 50 mM β-mercaptoethanol, and 66 ng/ml BAFF at 106 cells/ml for 24 h at 37°C.
Real-time PCR for transcript levels and Vλ-Jλ1 and RS rearrangement assays.
The primer sequences used in these assays are listed in Table 1. Total RNA was reverse transcribed into cDNA using the SuperScript VILO cDNA synthesis kit (Invitrogen). Real-time PCR was performed with primers specific for Igκ, IgH, RAG-1, RAG-2, VpreB, λ5, Pax-5, TdT, BLIMP, and IL-7R. Transcript levels were calculated using the ΔCT method and normalized to the cDNA levels of the mouse hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene. For Vλ-Jλ1 gene rearrangement assays in DNA isolated from purified cells, real-time PCR was performed with the Vλ-f and Vλ-r primers (f stands for forward and r stands for reverse) (26). For VRS rearrangement, VκD and RS101 primers were used. For IRS rearrangement, real-time PCR was performed with primers IRS-f and IRS-r. The RS rearrangement level in E3′F/F Ed−/− CD23-Cre Igλ+ was compared with those of E3′F/F Ed−/− Igλ+ cells in which the RS rearrangement level was set at 100%. DNA levels were normalized to the levels of a β-actin genomic region.
Table 1.
Primers for real-time PCR assays
| Gene or primer | Primer sequence |
|---|---|
| Genes | |
| HPRT | 5′-GGGGGCTATAAGTTCTTTGC-3′ |
| 5′-TCCAACACTTCGAGAGGTCC-3′ | |
| Igκ | 5′-GGCTGCAGSTTCAGTGGCAGTGGRTCWGGRAC-3′ |
| 5′-AGGACGCCATTTTGTCGTTCACT-3′ | |
| IgH | 5′-CTGCCCAGCACCATTTCCTT-3′ |
| 5′-TCTGTGGTGAAGCCAGATTC-3′ | |
| RAG-1 | 5′-ATGGCTGCCTCCTTGCCGTCT-3′ |
| 5′-GTATCTCCGGCTGTGCCCGTC-3′ | |
| RAG-2 | 5′-ATGTCCCTGCAGATGGTAACA-3′ |
| 5′-TAAATCTTATCGGAAAGCTCA-3′ | |
| λ5 | 5′-GGGTCTAGTGGATGGTGTCCACCACATAC-3′ |
| 5′-GAAGATTCTTTAAGGAAGGCAGGAACAGAGTG-3′ | |
| VpreB | 5′-TCCTGCTCATGCTGCTGGCCTATCTCACAG-3′ |
| 5′-CAGAGATGCTCAGATACCCCAGGTTCCTGGTC-3′ | |
| TdT | 5′-GTGGCTTCCACGCCTTATGACATCCGATTTCG-3′ |
| 5′-TCCCATGCATTCGATGAGCCAGGAGATGTC-3′ | |
| BLIMP | 5′-GACTGGTTACAAGACTCTTCCTTACCCTCTG-3′ |
| 5′-GTGGGTCTTGAGATTGCTTGTGCTGCTAAATC-3′ | |
| IL-7R | 5′-GCTCTGGGTAGAGCTTTCGCTATAGTTTTCTG-3′ |
| 5′-TGCAGCTTGTTAAGAGTTAGGCATTTCACTCG-3′ | |
| Primers | |
| Vλ-f | 5′-GCCATTTCCCAGGCTGTTGTGACTCAGG-3′ |
| Vλ-r | 5′-ACTCACCTAGGACAGTCAGTTTGGTTCC-3′ |
| Jκ1-2 | 5′-GACAACGGAAGAAAGAGACTTTGGA-3′ |
| VκD | 5′-GGCTGCAGSTTCAGTGGCAGTGGRTCWGGRAC-3′ |
| RS101 | 5′-ACATGGAAGTTTTCCTGGGAGAATAT-3′ |
| IRS-f | 5′-ATGTGAACCCCCGCGGTAGCAT-3′ |
| IRS-r | 5′-GGAACATGGAAGTTTTCCTGGGAG-3′ |
ChIP assays.
Chromatin immunoprecipitations (ChIPs) were performed according to Millipore's protocol with antibodies against acetylated histone H3 (catalog no. 06-599; Millipore, Billerica, MA), H3K4me3 (histone H3 with trimethylation of lysine 4) (catalog no. 9727; Cell Signaling, Boston, MA), or H3K27me3 (histone H3 with trimethylation of lysine 27) (catalog no. 07-449; Millipore). Results were quantitated by real-time PCR, and the signals from IP samples were referenced to their respective inputs to normalize for differences in primer efficiencies. A degenerate Vκ primer (VκD) and another primer, Jκ1-2, which is located between Jκ1 and Jκ2, were used to detect histone modification of rearranged Vκ-Jκ1 genes. GAPDH was treated as a negative control, and its enrichment fold was considered 1. The enrichment of every test fragment was referenced to GAPDH. The real-time PCR assays were repeated two or three times, and the results were averaged.
RESULTS
Overall experimental strategy.
A rearranged, transcriptionally active Igκ locus in B cells possesses three powerful transcriptional enhancers, Ei, E3′, and Ed, well downstream of Vκ gene promoters (Fig. 1A). We have shown previously that E3′ and Ed are essential for the establishment of Igκ gene expression. In double enhancer knockout mice, both germ line and rearranged Igκ gene transcription is completely shut off, and B cell development is partially blocked at the pre-B cell stage (12). However, B cell development and rearranged Igκ gene transcription are nearly normal in Ed−/− mice, whose major defect is in T cell-dependent stimulated transcription, which nevertheless is still about 70% of the level of WT mice (11). Therefore, to determine whether E3′ and Ed are also necessary for the maintenance of transcriptional activity, we developed experimental systems to investigate the effects of deleting E3′ in cells that possessed Ed−/− alleles. For this purpose, we took advantage of a targeted, germ line-transmittable Igκ locus that we had previously created through bacterial artificial chromosome (BAC) recombineering and site-directed integration technology (12). Unlike the endogenous locus (Fig. 1B), the targeted allele possessed E3′ and a Neo gene each flanked by LoxP sites, with Ed replaced by a Puro gene flanked by Flp recombination target (Frt) sites (Fig. 1C). After the appropriate breeding with Flp-expressing mice (27), we obtained a mouse strain with a floxed E3′ element in which Ed had been deleted, termed E3′F/F Ed−/− (Fig. 1D). Although these mice still possessed a Neo gene downstream of E3′, we found that this Neo gene had no significant deleterious effects on B cell development, Igκ gene rearrangement, or rearranged Igκ gene transcription levels (see below). As shown in Fig. 1E, we developed three distinct animal models for the studies reported here by breeding these E3′F/F Ed−/− mice with either germ line-expressing Cre mice (28) or with tamoxifen-inducible Cre mice (22) or with mice specifically expressing Cre in mature B cells (23). Successful deletion of E3′ in each of these cases generates E3′−/− Ed−/− mice, which have single LoxP and Frt sites in place of E3′ and Ed, respectively (Fig. 1F). The control Southern blotting experiments shown in Fig. 1G establish that both the LoxP- and Frt-flanked enhancers are efficiently deleted from the appropriate alleles upon exposure to the appropriate recombinases in the germ line. However, the goals of our experiments to be described below are to delete E3′ from E3′F/F Ed−/− alleles after the locus has been established to become transcriptionally active and to test the outcomes of such events.
Fig 1.

Strategy for creating and testing conditional E3′−/− Ed−/− knockout mice. (A) Schematic diagram depicting a rearranged Igκ locus. The coordinates of Ei, E3′, Ed, and HS10 in the NCDI37/mm9 mouse chromosome 6 sequence are 70,675,570 to 70,676,084, 70,685,250 to 70,686,058, 70,693,704 to 70,694,944, and 70,713,758 to 70,715,084, respectively. MEi, T, and Rpia correspond to the matrix association region intronic enhancer element, the transcription termination region, and the ribose 5′-phosphate isomerase gene, respectively. The bracket below the schematic diagram demarcates the relevant region targeted in these studies. (B) Endogenous Igκ locus segment possessing E3′ and Ed. The positions of probes A and B are indicated by small black boxes above the map. BamHI restriction sites (B) are indicated below the map. (C) Targeted allele of the Igκ locus. The targeting construct was made using BAC recombineering as previously described (12). E3′ was replaced with a Neo+ E3′ cassette flanked by loxP sites, and Ed was replaced with a Puro gene flanked by Frt sites. (D) The Puro gene was deleted by breeding the mice with a Flp-expressing strain (28) to obtain E3′F Ed− mice. It should be emphasized that despite the fact that the Neo gene is still present adjacent to E3′ in this conditional allele, B cell development and transcription of rearranged Igκ genes are very similar to those in Ed−/− mice (see Fig. 2A, top panels). (E) Experimental strategy used to delete E3′ in the mouse germ line (left segment of the flow chart), in any cell type upon tamoxifen treatment (middle segment of the flow chart), or specifically in mature B cells (right segment of the flow chart). (F) Structure of the conditional allele after deletion of E3′ by expressing Cre recombinase. (G) Southern blotting of BamHI-digested mouse tail DNA. The mouse genotypes studied are indicated above the lanes. The locations of probes A and B are shown above the map in panel F. The left blot demonstrates as a control that the E3′F/F allele is successfully deleted upon breeding such mice with mice expressing Cre recombinase in the germ line (27). The right blot demonstrates that the Puro gene was successfully deleted upon breeding mice with mice expressing Frt recombinase in the germ line (28).
Inducible enhancer deletion results in loss of Igκ gene expression and positive epigenetic histone marks in mature B cells.
As our initial approach to test for the importance of the Igκ gene's downstream enhancers on continued gene expression following their deletion, we bred E3′F/F Ed−/− mice with those harboring a tamoxifen-inducible Cre [ROSA26CreER(T) (22)] to obtain a homozygous E3′F/F Ed−/− ROSA26CreER(T) line. It is known that the lifetime of splenic mature B cells can be from several weeks to months (29), so we sought to delete E3′ in such nonreplicating mature B cells for the subsequent analysis of Igκ gene expression in these cells. We reasoned that if the enhancer's function is memorized, mature B cells that lost E3′ should still be able to express Igκ on their cell surface. These mice were treated with tamoxifen for two consecutive days, and Igκ gene expression on splenic B cells was analyzed by FACS. As shown in Fig. 2A (bottom panels), after exposure to tamoxifen, the percentages of Igκ+ B cells dropped dramatically with time, and only 8% of the splenic cells were Igκ+ B cells by day 7. Moreover, such treatment did not affect expression in control genetic lines, nor did it change B cell populations in the splenic B cells of WT mice (Fig. 2A, top panels). Southern blotting revealed that more than 90% of mature B cells and bone marrow cells had lost E3′ 5 days after initial Cre induction (Fig. 2B). We also analyzed Igκ mRNA levels by real-time PCR assays and found that Igκ gene expression was significantly reduced upon E3′ deletion after tamoxifen injection relative to expression in controls (Fig. 2C). In addition, the size of the spleen and total B cell splenic cell number were significantly reduced upon E3′ deletion even after 3 days of initial Cre induction relative to controls (Fig. 2D). It is known that BCR signaling is required for survival of mature B cells (30). Hence, loss of Igκ gene expression leads to cell death in the spleen. We conclude that the Igκ gene's downstream enhancers are not only essential for the establishment of locus transcriptional activity, but they are also required for its maintenance even in the absence of DNA replication.
Fig 2.

E3′ and Ed are required for Igκ gene expression in splenic B cells of conditional E3′F/F Ed−/− mice harboring a tamoxifen-inducible Cre. Mice were injected with tamoxifen (2 mg/mouse/day at day 0 and day 1, consecutively). Samples were taken at day 0 before injection and at the indicated days thereafter. (A) Analysis of Igκ+ or Igλ+ splenic B cells of E3′F/F Ed−/− ROSA26CreER(T) mice before (day 0) or 3, 5, or 7 days after tamoxifen injection (bottom panels). Igκ+ or Igλ+ splenic B cells of control E3′F/F Ed−/− or Ed−/− ROSA26CreER(T) mice were also analyzed after 7 days of tamoxifen injection, as well as Igκ+ expression in WT ROSA26CreER(T) mice in spleen (SP) and bone marrow (BM) before (day 0) or 5 days after tamoxifen injection (top panels). Data are representative of independent FACS analyses from at least 4 mice. (B) Southern blotting was performed to assay for the loss of E3′ in both spleen (SP) and bone marrow (BM) before or 5 days after tamoxifen (Tamo) injection. (C) Splenic B cells (B220+) were isolated from E3′F/F Ed−/− ROSA26CreER(T) mice at the indicated times after tamoxifen injection, and real-time PCR assays were performed to measure the levels of Igκ gene transcripts. Splenic B cells from WT mice, control E3′F/F Ed−/−, or Ed−/− ROSA26CreER(T) mice were analyzed similarly. (D) Total splenic B cell numbers in the indicated genetic lines of mice before or after tamoxifen injection. Double asterisks in panels C and D indicate P < 0.01 with respect to a comparison with E3′F/F Ed−/− mice (Student's t test). (E) Splenic B cells (B220+) were isolated from E3′F/F Ed−/− ROSA26CreER(T) or control Ed−/− ROSA26CreER(T) mice at the indicated times after tamoxifen injection, and real-time PCR ChIP assays were performed to measure the levels of H3Ac, H3K4me3, and H3K27me3 in rearranged Vκ-Jκ1 regions. Fold enrichment refers to the sequence abundance in the immunoprecipitated sample divided by the corresponding sequence abundance in input DNA relative to a control GAPDH gene. Data are presented as means plus standard deviations (SD) (error bars) (for H3Ac, n = 3, day 0 versus day3, P < 0.01 [∗∗]; for H3K4me3, day 0 versus day 5, P < 0.05 [∗]). There are no statistically significant differences for H3K27me3 levels in the different samples. P values were determined by one-way analysis of variance (ANOVA) using SPSS 11.5 software.
Because epigenetic histone posttranslational modifications are associated with enhancer function (17), we isolated splenic B cells at different times after initial Cre induction and measured by ChIP assays the levels of the positive histone marks H3Ac (histone H3 acetylation) and H3K4me3, and the negative histone mark H3K27me3, which were associated with rearranged Vκ gene promoters and the 5′ segments of their corresponding transcription units. For this purpose, we utilized a degenerate PCR assay to amplify Vκ-Jκ1 regions in ChIPed samples. Five days after initial Cre induction, H3Ac and H3K4me3 levels in Vκ-Jκ1 regions were 20% and 40%, respectively, compared to the levels on day 0, while H3K27me3 levels remained unchanged (Fig. 2E). No changes in these modifications were observed in controls (Fig. 2E). We conclude that the continued presence of an essential downstream enhancer is required not only for the maintenance of Igκ gene expression but also for the maintenance of positive epigenetic marks.
Confirmation that an essential enhancer in the Igκ locus is indispensable for continued Igκ gene expression.
To confirm and extend the above results by another approach, we then bred E3′F/F Ed−/− mice with CD23-Cre mice, which express the Cre gene at the mature B cell stage (23). To evaluate the effects of E3′ deletion on B cell populations expressing surface Igκ chains, we compared the FACS patterns of B cells from the spleens and bone marrow of WT, CD23-Cre, Ed−/−, Ed−/− CD23-Cre, E3′F/F Ed−/−, or E3′F/F Ed−/− CD23-Cre mice with those from E3′−/− Ed−/− mice. As shown in Fig. 3A, mature splenic B cells from E3′F/F Ed−/− CD23-Cre mice exhibit greatly reduced Igκ gene expression compared to that of WT, CD23-Cre, Ed−/−, Ed−/− CD23-Cre, or E3′F/F Ed−/− control mice, while as expected, Igκ+ B cells were absent in samples from E3′−/− Ed−/− mice. However, in bone marrow, the numbers of Igκ+ B cells were very similar in samples from E3′F/F Ed−/− or E3′F/F Ed−/− CD23-Cre mice (Fig. 3A). We measured the efficiency of E3′ deletion in E3′F/F Ed−/− CD23-Cre mice by Southern blotting. The Cre activity was specific to splenic cells and not expressed in bone marrow, as we did not find significant deletion of E3′ in bone marrow B cells, whereas approximately 80% of the Igκ alleles of splenic B cells lacked E3′ (Fig. 3B). As expected, deletion of E3′ in Igκ− B cells was complete, but purified Igκ+ B cells from E3′F/F Ed−/− CD23-Cre mice still harbored one undeleted E3′F allele (Fig. 3B). The total number of B cells in the spleen was moderately reduced in E3′F/F Ed−/− CD23-Cre mice compared to controls (Fig. 3C). We also found that the number of Igλ+ splenic B cells in E3′F/F Ed−/− CD23-Cre mice was significantly greater than that of control mice and very close to that observed in E3′−/− Ed−/− mice (Fig. 3D), whereas the numbers of Igλ+ B cells in the bone marrow were very similar in these mouse lines (Fig. 3D). To confirm that the loss of Igκ gene expression occurred in mature B cells, but not in the bone marrow of E3′F/F Ed−/− CD23-Cre mice, FACS analysis was performed to examine markers of early B cell development. As shown in Fig. 3E, the amounts of immature B (B220low IgM+), pre-B (B220+ CD25+), and pro-B (B220+ C-Kit+) cells in the bone marrow in E3′F/F Ed−/− CD23-Cre mice and control mice were identical. In agreement with our previous findings, samples from E3′−/− Ed−/− mice exhibited increased numbers of pre-B (B220+ CD25+) cells (Fig. 3E) (12).
Fig 3.

E3′ and Ed are required for Igκ gene expression in splenic B cells of conditional E3′F/F Ed−/− mice harboring CD23-Cre. (A) FACS analysis was performed to measure the percentages of Igκ+ B cells in spleen (SP) or bone marrow (BM) of the indicated genetic lines of mice. Data are representative of independent FACS analyses from at least 4 mice of each genotype. In addition, we obtained essentially identical results with respect to Igκ gene expression in splenic B cells from E3′F/F Ed−/− mice that possessed or lacked the flanking Neo gene in the germ line or in samples from E3′F/F Ed−/− CD23-Cre mice that possessed or lacked the flanking Neo gene in the germ line. This rules out the possibility that the prior presence of the Neo gene in the targeted locus affects subsequent Igκ gene expression upon its deletion together with E3′. (B) Splenic cells (SP), bone morrow cells (BM), B220+ Igκ+ B cells (κ+B), or B220+ Igκ− B (κ−B) cells were isolated from spleen or bone marrow of the indicated genetic lines of mice. Southern blotting was performed to assay for the loss of E3′. (C) Total splenic B cell numbers in the indicated genetic lines of mice. Data are presented as means plus SD (n = 5, WT or E3′F/F Ed−/− versus E3′F/F Ed−/− CD23-Cre, P < 0.05 by Student's t test [∗]). (D) Percentages of Igλ+ B cells in spleen (SP) and bone marrow (BM) of the indicated genetic lines of mice. Data are representative of independent FACS analyses from at least 5 mice of each genotype. (E) FACS analysis was performed to measure the percentages of immature B, pre-B, and pro-B found in bone marrow of the indicated genetic lines of mice. Data are representative of independent FACS analyses from at least 3 mice of each genotype.
Diminished Igκ gene expression in mature B cells generates pro-B-like cells in the periphery.
Interestingly, we have found that the loss of E3′ not only results in diminished expression of the Igκ gene in mature B cells, but it also generates a significant population of Igκ− Igλ− B cells. As shown in Fig. 4A, the number of Igκ − Igλ− B cells is much greater in E3′F/F Ed−/− CD23-Cre mice than in WT, CD23-Cre, Ed−/−, Ed−/− CD23-Cre, and control E3′F/F Ed−/− mice. It has been reported that a small number of splenic B cells can express IgλX, which is not detectable by the Igλ1,2,3 antibodies that we used in our above FACS assays (25). Therefore, we assayed for IgλX expression by FACS with a monoclonal antibody specific for this Igλ subtype and found no differences in IgλX expression between splenic B cells from WT, Ed−/− CD23-Cre, E3′F/F Ed−/−, or E3′F/F Ed−/− CD23-Cre mice (Fig. 4B), demonstrating that Igκ− Igλ− B cells from E3′F/F Ed−/− CD23-Cre mice indeed do not express any IgL on their cell surface. Next, we sorted these double negative and single positive B cells from E3′F/F Ed−/− CD23-Cre mice and analyzed selected gene transcript levels by real-time PCR. Unlike the single positive groups, these double negative B cells expressed significantly increased levels of RAG-1 and -2, and other transcripts that are characteristically expressed in pro-B cells, such as VpreB, λ5, TdT, and IL-7R (Fig. 4C). Comparison of these transcript levels with those of isolated pro-B cells from WT mice, however, revealed that RAG-1 and -2 levels were 8- to 10-fold higher in normal pro-B cells than in these double negative cells; whether this is because such cells are heterogeneous and only 10% of the population expresses the RAG genes at normal pro-B cell levels remains to be determined. In addition, although Igκ− Igλ− B cells express surrogate light chain gene transcripts, we did not detect by FACS increased surface expression of CD43, CD25, interleukin 7 receptor (IL-7R), pre-BCR, or λ5, compared to Igκ+ Igλ− or Igκ− Igλ+ cells from the same mice (Fig. 4D). However, the Igκ− Igλ− B cells expressed on their cell surface significant but lower levels of μ and δ chains compared to their counterpart groups, indicating that these cells arose from mature B cells, and not from immature B cells or pre-B cells (Fig. 4E). The continued expression of μ and δ chains in the absence of IgL chains is surprising but not unprecedented (reference 31 and references therein). We also found that the expression level of BAFF-R was comparable among all four groups, which is another marker expressed on mature B cells (Fig. 4E). We conclude that mature B cells from E3′F/F Ed−/− CD23-Cre mice partially dedifferentiate upon loss of Igκ gene expression.
Fig 4.

IgL double negative mature B cells from E3′F/F Ed−/− CD23-Cre mice express pro-B cell markers. (A) Three-color FACS analysis was performed to measure the B220, Igκ, and Igλ expression in splenic cells from the indicated genetic lines of mice. Shown are Igκ and Igλ expression in gated B220+ cells. Data are representative of independent FACS analyses from at least 4 mice of each genotype. (B) The expression of IgλX in splenic B cells from WT, Ed−/− CD23-Cre, or E3′F/F Ed−/− or E3′F/F Ed−/− CD23-Cre mice was analyzed by FACS. Data are representative of independent FACS analyses from 3 mice of each genotype. (C) B220+ Igκ+ Igλ−, B220+ Igκ− Igλ+, and B220+ Igκ− Igλ− B cells from E3′F/F Ed−/− CD23-Cre mice were sorted by FACS. Real-time PCR was performed to measure expression of the indicated transcript levels. Data are normalized to the levels in B220+ Igκ+ Igλ− cells taken as 1.0 and are presented as means plus SD (P < 0.05 ∗ and P < 0.01 ∗∗ compared to the value for B220+ Igκ+ Igλ− cells [n = 3]). (D) The expression of other early B cell markers on Igκ− Igλ−, Igκ+ Igλ−, and Igκ− Igλ+ B cells in the spleens of E3′F/F Ed−/−CD23-Cre mice were analyzed with FACS. Max, maximum. (E) Four-color FACS analysis was performed to measure the expression of μ chain, δ chain, and BAFF-R in the indicated single or double negative IgL groups of B220+ splenic cells of E3′F/F Ed−/− CD23-Cre mice. For a control, expression in B220+ Igκ+ Igλ− B cells from WT mice is also shown. Data are representative of four independent FACS analyses. Shaded areas represent isotype control staining.
In vivo evidence that Igλ genes rearrange in B220+ Igκ− Igλ− splenic cells of E3′F/F Ed−/− CD23-Cre mice.
We found that Igλ expression is increased in splenic cells, but not bone marrow cells, from E3′F/F Ed−/− CD23-Cre mice compared to the levels in control mice (Fig. 3D). Because Igκ− Igλ− B cells from E3′F/F Ed−/− CD23-Cre mice expressed RAG-1 and -2 (Fig. 4C), we hypothesized that these cells might be able to initiate new rearrangements in Igλ genes. If they successfully rearrange and express Igλ, they may survive because of the restored BCR on their cell surface, and they may represent the origin of increased Igλ expression in the spleen. However, we needed to rule out the possibility that the increased number of Igλ+ B cells might be from expansion of marginal zone B cells as has been reported previously in an animal model of autoimmunity (32). Importantly, as shown in Fig. 5A, we did not find differences in the populations of marginal zone (CD21high CD23low) and follicular B cells (CD21low CD23high) in the spleens of WT, Ed−/− CD23-Cre, or E3′F/F Ed−/− or E3′F/F Ed−/− CD23-Cre mice.
Fig 5.
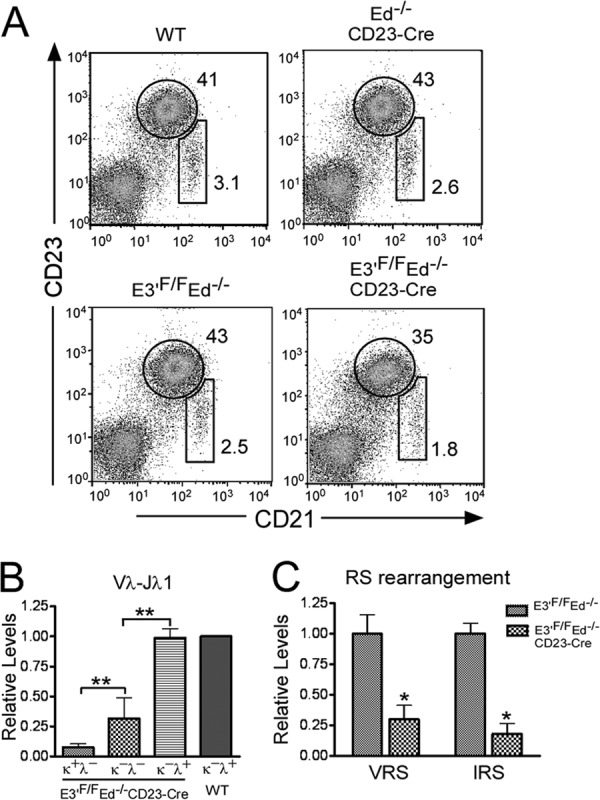
In vivo rearrangement of Igλ genes in B220+ Igκ− Igλ− splenic cells of E3′F/F Ed−/− CD23-Cre mice. (A) The development of marginal zone (CD21high CD23low) and follicular B cells (CD21low CD23high) in the spleens of WT, Ed−/− CD23-Cre, or E3′F/F Ed−/− or E3′F/F Ed−/− CD23-Cre mice was analyzed by FACS. (B) B220+ Igκ+ Igλ−, B220+ Igκ− Igλ−, and B220+ Igκ− Igλ+ splenic B cells from E3′F/F Ed−/− CD23-Cre mice were sorted by FACS, and real-time PCR was performed to assay for Vλ-Jλ1 gene rearrangements in purified DNA samples. B220+ Igκ−Igλ+ splenic B cells from WT mice were used as control. Data are presented as means plus SD (P < 0.01 ∗∗ compared to Igκ+ Igλ− or Igκ− Igλ+ cells [n = 3]). (C) B220+ Igκ− Igλ+ splenic cells were isolated from E3′F/F Ed−/− or E3′F/F Ed−/− CD23-Cre mice, and real-time PCR assays were used to measure the indicated two types of RS rearrangement in Igκ gene alleles. Data are presented as means plus SD (∗, P < 0.05 refers to differences between E3′F/F Ed−/− and E3′F/F Ed−/− CD23-Cre samples [n = 3]).
To gain support for the hypothesis that Igκ− Igλ− cells gave rise to Igκ− Igλ+ cells, we isolated Igκ+ Igλ−, Igκ− Igλ−, and Igκ− Igλ+ B cells from the spleens of E3′F/F Ed−/− CD23-Cre mice and measured the levels of Vλ-Jλ1 gene rearrangement products by real-time PCR. As expected, we found high levels of Vλ-Jλ1 gene rearrangement in Igκ− Igλ+ control cells from both WT or E3′F/F Ed−/− CD23-Cre mice (Fig. 5B). The level of Vλ-Jλ1 rearrangement in the Igκ+ Igλ− cell population was quiet low but was significantly increased in Igκ− Igλ− cells (Fig. 5B). Thus, some Igκ− Igλ− cells had already completed Igλ gene rearrangement but apparently had not yet successfully switched from Igκ to Igλ expression. In addition, because TdT is expressed in the Igκ− Igλ− B cells and is known to be responsible for inserting non-germ line nucleotides (N regions) between V(D)J junctions during the normal pro-B cell stage of development (33), we assayed for the presence of N regions in Vλ-Jλ1 junctions after cloning and sequencing PCR products. We found that 1/40 clones of Igλ+ splenic B cells from control WT mice exhibited N regions, whereas 5/40 clones of Igλ+ splenic B cells from E3′F/F Ed−/− CD23-Cre mice exhibited N regions. This increase in the occurrence of N regions in the mutant mice is consistent with the interconversion of Igκ− Igλ− cells to Igκ− Igλ+ cells in these animals during their expression of RAG-1 and -2 and TdT.
During early B cell development in bone marrow, if pre-B cells express autoreactive BCRs or fail to express functionally rearranged Igκ genes, these Igκ alleles are inactivated by RS rearrangement, and Igλ gene rearrangement is initiated (34, 35). Hence, most Igλ+ B cells in healthy mice carry RS rearrangements (reference 2 and references therein). Using RS rearrangement as an indicator of bone marrow-derived Igλ+ B cells, we employed real-time PCR assays to compare RS rearrangement levels in splenic Igκ− Igλ+ B cells between E3′F/F Ed−/− CD23-Cre and E3′F/F Ed−/− control mice (Fig. 5C). We found significantly lower levels of RS rearrangement in B220+ Igκ− Igλ+ cells in E3′F/F Ed−/− CD23-Cre mice compared with the counterparts from E3′F/F Ed−/− mice. These results provide evidence that the switch from Igκ to Igλ gene expression in the E3′F/F Ed−/− CD23-Cre mouse animal model occurs primarily in peripheral mature B cells and not before cells leave the bone marrow.
In vitro evidence that Igλ genes rearrange in B220+ Igκ− Igλ− splenic cells of E3′F/F Ed−/− CD23-Cre mice.
To address further our hypothesis that Igκ− Igλ− cells can give rise to Igκ− Igλ+ cells in E3′F/F Ed−/− CD23-Cre mice, we determined whether such an interconversion could be triggered ex vivo. For this purpose, we isolated the Igκ− Igλ− cells from the spleens of E3′F/F Ed−/− CD23-Cre mice and cultured them in the presence of BAFF. After 24 h of culture, live cells could be easily distinguished by their forward light scatter (FSC) and side light scatter (SSC) based on the 7-amino-actinomycin D (7-AAD) staining of dead cells (Fig. 6, top panel). We found that the live cells still expressed normal levels of B220 after 24 h of culture (Fig. 6, middle panels). We analyzed cell surface Igκ and Igλ levels in these B220+-gated cells and found that 12.1% ± 3.6% (n = 3) of the cultured live cells expressed Igλ (Fig. 6, bottom panel). Significantly, we found a high frequency of N regions, 14/58, in the Igκ− Igλ− cells cultured for 24 h. However, 87% of the Igλ rearrangement products were out of frame, thus providing an explanation of why most of these cells did not express Igλ. In contrast, we found that 70% of the Igλ rearrangement products in WT Igλ+ B cells were functionally rearranged. In conclusion, we have evidence from both in vivo and in vitro experiments that strongly support our hypothesis that Igκ− Igλ− cells give rise to Igκ− Igλ+ cells in E3′F/F Ed−/− CD23-Cre mice.
Fig 6.
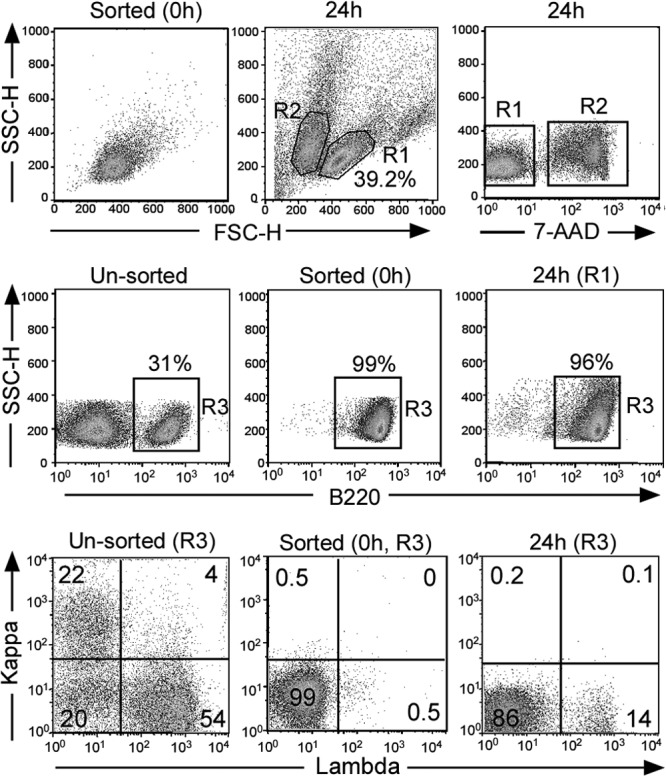
Ex vivo rearrangement of Igλ genes in B220+ Igκ− Igλ− splenic cells of E3′F/F Ed−/− CD23-Cre mice. (Top) B220+ Igκ− Igλ− splenic B cells from E3′F/F Ed−/− CD23-Cre mice were isolated with high purity through FACS sorting (0 h). After culturing for 24 h in the presence of BAFF (66 ng/ml), around 40% of the cells remained viable based on their FSC and SSC patterns as assayed by FACS or 7-AAD staining (R1 components). (Middle) Surface expression of B220 of unsorted cells and in R1 gate-sorted cells (0 h) or cells cultured for 24 h was analyzed. (Bottom) Igκ and Igλ expression in R3-gated cells was assayed by FACS, and data are representative of three independent experiments.
DISCUSSION
The results presented here demonstrate that essential downstream enhancers are indispensable for continued Igκ gene expression in mature B cells in vivo in nonreplicating chromatin and that the Igκ gene activity established at the immature B cell stage does not exhibit cellular memory after E3′ deletion. The Igκ gene is a highly complex locus spanning 3.2 Mb on mouse chromosome 6, and our previous studies have indicated that during its active transcription, looping occurs between E3′ and rearranged Vκ gene promoters (6, 7). Such looping may be required for continuous gene expression, and disruption of such looping may result in quite rapid gene silencing. Furthermore, we found that the continued presence of E3′ was required for maintaining epigenetic modifications in the Igκ gene.
BCR signaling is required for survival of mature B cells, because ablation of either Ig heavy chain or Igα/β heterodimer expression results in massive B cell death in the periphery (30, 36). However, engineered repression of Ig light chain expression in a mouse model showed that light chains are not required for expression of Ig heavy chains or cell survival (31). Here, we showed that loss of Igκ gene expression upon tamoxifen-induced enhancer deletion resulted in significant cell death of the mature B cell population. In marked contrast, we observed the persistence of B cells after E3′ deletion in the CD23-Cre model. We believe that the explanation for this apparent discrepancy is that B cells are continually being supplied from the bone marrow in CD23 mice and that interconversion of Igκ+ Igλ− cells to Igκ− Igλ− and then to Igκ− Igλ+ cells allows the trace of these “edited” Igλ+ cells to be continually generated, which would be expected to accumulate over time since mature B cells have long lifetimes (29).
Surprisingly, by staining with specific antibodies, we detected substantial levels of μ and δ chains on the surfaces of the Igκ− Igλ− B cells, indicating that Ig heavy chains can be expressed in the absence of Ig light chains as reported previously (31). While Shaffer and Schlissel (37) reported that truncated heavy chains, but not their full-length counterparts, can be expressed on the surfaces of pro-B cells in the absence of surrogate light chain expression, a later study showed that such surface expression did not require expression of surrogate light chains. Both studies used μ chain transgenic mice (38). Our results support the proposal that at least some endogenous heavy chains can be expressed in the absence of light chains in engineered mature B cells. Although the average level of μ chain expression was reduced in the Igκ− Igλ− B cells compared to the Igκ+ Igλ− or Igκ− Igλ+ B cells, the remaining μ chain might mimic BCR signaling and suppress cell death until cells can successfully rearrange and express their Igλ genes.
Repeated V(D)J rearrangement can occur in immature B cells in the bone marrow and in germinal center B cells in the periphery; these two events are accompanied by waves of RAG reexpression and have been termed receptor editing and receptor revision, respectively (39). Receptor editing occurs in immature B cells that have migrated to the periphery (40, 41) and in germinal center B cells developing responses to specific model antigens (19, 42, 43). The observation that Igκ− Igλ− B cells redifferentiate into Igκ− Igλ+ B cells in the spleens of E3′F/F Ed−/− CD23-Cre mice is surprising and suggests that receptor editing is not limited to immature B cells but can be induced in mature B cells as well. It has been thought that most autoreactive B cells are silenced by anergy and are unresponsive to antigen stimulation in the periphery (44). We propose that mature B cells retain the ability to edit their BCRs in peripheral tissues, thus providing an alternative mechanism for B cell tolerance. In the future, one way to address the relative importance of receptor revision versus editing would be to create conditional RAG knockout mice separately with mature B cells and germinal center B cells and test whether abrogation of peripheral receptor editing and receptor revision breaks B cell tolerance.
ACKNOWLEDGMENTS
We are indebted to Meinrad Busslinger of the Research Institute of Molecular Pathology, Vienna, Austria, for providing mice and to Martin Weigert of Princeton University for providing anti-VλX monoclonal antibody.
This investigation was supported by grants GM29935 and AI067906 from the National Institutes of Health and grant I-0823 from the Robert A. Welch Foundation to W.T.G.
Footnotes
Published ahead of print 18 March 2013
REFERENCES
- 1. Schatz DG, Ji Y. 2011. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 11:251–263 [DOI] [PubMed] [Google Scholar]
- 2. Vela JL, Ait-Azzouzene D, Duong BH, Ota T, Nemazee D. 2008. Rearrangement of mouse immunoglobulin kappa deleting element recombining sequence promotes immune tolerance and lambda B cell production. Immunity 28:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu ZM, George-Raizen JB, Li S, Meyers KC, Chang MY, Garrard WT. 2002. Chromatin structural analyses of the mouse Igkappa gene locus reveal new hypersensitive sites specifying a transcriptional silencer and enhancer. J. Biol. Chem. 277:32640–32649 [DOI] [PubMed] [Google Scholar]
- 4. Queen C, Baltimore D. 1983. Immunoglobulin gene transcription is activated by downstream sequence elements. Cell 33:741–748 [DOI] [PubMed] [Google Scholar]
- 5. Meyer KB, Neuberger MS. 1989. The immunoglobulin kappa locus contains a second, stronger B-cell-specific enhancer which is located downstream of the constant region. EMBO J. 8:1959–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Z, Garrard WT. 2005. Long-range interactions between three transcriptional enhancers, active Vkappa gene promoters, and a 3′ boundary sequence spanning 46 kilobases. Mol. Cell. Biol. 25:3220–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Z, Ma Z, Terada LS, Garrard WT. 2009. Divergent roles of RelA and c-Rel in establishing chromosomal loops upon activation of the Igkappa gene. J. Immunol. 183:3819–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gorman JR, van der Stoep N, Monroe R, Cogne M, Davidson L, Alt FW. 1996. The Ig(kappa) enhancer influences the ratio of Ig(kappa) versus Ig(lambda) B lymphocytes. Immunity 5:241–252 [DOI] [PubMed] [Google Scholar]
- 9. Xu Y, Davidson L, Alt FW, Baltimore D. 1996. Deletion of the Ig kappa light chain intronic enhancer/matrix attachment region impairs but does not abolish V kappa J kappa rearrangement. Immunity 4:377–385 [DOI] [PubMed] [Google Scholar]
- 10. Inlay M, Alt FW, Baltimore D, Xu Y. 2002. Essential roles of the kappa light chain intronic enhancer and 3′ enhancer in kappa rearrangement and demethylation. Nat. Immunol. 3:463–468 [DOI] [PubMed] [Google Scholar]
- 11. Xiang Y, Garrard WT. 2008. The downstream transcriptional enhancer, Ed, positively regulates mouse Ig kappa gene expression and somatic hypermutation. J. Immunol. 180:6725–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou X, Xiang Y, Garrard WT. 2010. The Igkappa gene enhancers, E3′ and Ed, are essential for triggering transcription. J. Immunol. 185:7544–7552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sen R, Grosschedl R. 2010. Memories of lost enhancers. Genes Dev. 24:973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reik A, Teling A, Zitnik G, Cimbora D, Epner E, Groudine M. 1998. The locus control region is necessary for gene expression in the human β-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol. Cell. Biol. 18:5992–6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grosschedl R, Marx M. 1988. Stable propagation of the active transcriptional state of an immunoglobulin μ gene requires continuous enhancer function. Cell 55:645–654 [DOI] [PubMed] [Google Scholar]
- 16. Lieberson R, Ong J, Shi X, Eckhardt LA. 1995. Immunoglobulin gene transcription ceases upon deletion of a distant enhancer. EMBO J. 14:6229–6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chong MM, Simpson N, Ciofani M, Chen G, Collins A, Littman DR. 2010. Epigenetic propagation of CD4 expression is established by the Cd4 proximal enhancer in helper T cells. Genes Dev. 24:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luning Prak ET, Monestier M, Eisenberg RA. 2011. B cell receptor editing in tolerance and autoimmunity. Ann. N. Y. Acad. Sci. 1217:96–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang YH, Diamond B. 2008. B cell receptor revision diminishes the autoreactive B cell response after antigen activation in mice. J. Clin. Invest. 118:2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zouali M. 2008. Receptor editing and receptor revision in rheumatic autoimmune diseases. Trends Immunol. 29:103–109 [DOI] [PubMed] [Google Scholar]
- 21. Yu W, Nagaoka H, Jankovic M, Misulovin Z, Suh H, Rolink A, Melchers F, Meffre E, Nussenzweig MC. 1999. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature 400:682–687 [DOI] [PubMed] [Google Scholar]
- 22. Badea TC, Hua ZL, Smallwood PM, Williams J, Rotolo T, Ye X, Nathans J. 2009. New mouse lines for the analysis of neuronal morphology using CreER(T)/loxP-directed sparse labeling. PLoS One 4:e7859 doi:10.1371/journal.pone.0007859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, Busslinger M. 2008. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28:751–762 [DOI] [PubMed] [Google Scholar]
- 24. Sanchez P, Nadel B, Cazenave PA. 1991. V lambda-J lambda rearrangements are restricted within a V-J-C recombination unit in the mouse. Eur. J. Immunol. 21:907–911 [DOI] [PubMed] [Google Scholar]
- 25. Kalinina O, Doyle-Cooper CM, Miksanek J, Meng W, Prak EL, Weigert MG. 2011. Alternative mechanisms of receptor editing in autoreactive B cells. Proc. Natl. Acad. Sci. U. S. A. 108:7125–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaw AC, Swat W, Davidson L, Alt FW. 1999. Induction of Ig light chain gene rearrangement in heavy chain-deficient B cells by activated Ras. Proc. Natl. Acad. Sci. U. S. A. 96:2239–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tallquist MD, Soriano P. 2000. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26:113–115 [DOI] [PubMed] [Google Scholar]
- 28. Farley FW, Soriano P, Steffen LS, Dymecki SM. 2000. Widespread recombinase expression using FLPeR (Flipper) mice. Genesis 28:106–110 [PubMed] [Google Scholar]
- 29. Forster I, Rajewsky K. 1990. The bulk of the peripheral B-cell pool in mice is stable and not rapidly renewed from the bone marrow. Proc. Natl. Acad. Sci. U. S. A. 87:4781–4784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. 2004. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117:787–800 [DOI] [PubMed] [Google Scholar]
- 31. Geraldes P, Rebrovich M, Herrmann K, Wong J, Jäck H-M, Wabl M, Cascalho M. 2007. Ig heavy chain promotes mature B cell survival in the absence of light chain. J. Immunol. 179:1659–1668 [DOI] [PubMed] [Google Scholar]
- 32. Grimaldi CM, Michael DJ, Diamond B. 2001. Expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J. Immunol. 167:1886–1890 [DOI] [PubMed] [Google Scholar]
- 33. Komori T, Sugiyama H. 1993. N sequences, P nucleotides and short sequence homologies at junctional sites in VH to VHDJH and VHDJH to JH joining. Mol. Immunol. 30:1393–1398 [DOI] [PubMed] [Google Scholar]
- 34. Durdik J, Moore MW, Selsing E. 1984. Novel kappa light-chain gene rearrangements in mouse lambda light chain-producing B lymphocytes. Nature 307:749–752 [DOI] [PubMed] [Google Scholar]
- 35. Daitch LE, Moore MW, Persiani DM, Durdik JM, Selsing E. 1992. Transcription and recombination of the murine RS element. J. Immunol. 149:832–840 [PubMed] [Google Scholar]
- 36. Lam KP, Kühn R, Rajewsky K. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90:1073–1083 [DOI] [PubMed] [Google Scholar]
- 37. Shaffer AL, Schlissel MS. 1997. A truncated heavy chain protein relieves the requirement for surrogate light chains in early B cell development. J. Immunol. 159:1265–1275 [PubMed] [Google Scholar]
- 38. Schuh W, Meister S, Roth E, Jäck H-M. 2003. Signaling and cell surface expression of a mu H chain in the absence of lambda 5: a paradigm revisited. J. Immunol. 171:3343–3347 [DOI] [PubMed] [Google Scholar]
- 39. Nemazee D. 2006. Receptor editing in lymphocyte development and central tolerance. Nat. Rev. Immunol. 6:728–740 [DOI] [PubMed] [Google Scholar]
- 40. Gartner F, Alt FW, Monroe RJ, Seidl KJ. 2000. Antigen-independent appearance of recombination activating gene (RAG)-positive bone marrow B cells in the spleens of immunized mice. J. Exp. Med. 192:1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagaoka H, Gonzalez-Aseguinolaza G, Tsuji M, Nussenzweig MC. 2000. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J. Exp. Med. 191:2113–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han S, Dillon SR, Zheng B, Shimoda M, Schlissel MS, Kelsoe G. 1997. V(D)J recombinase activity in a subset of germinal center B lymphocytes. Science 278:301–305 [DOI] [PubMed] [Google Scholar]
- 43. Igarashi H, Kuwata N, Kiyota K, Sumita K, Suda T, Ono S, Bauer SR, Sakaguchi N. 2001. Localization of recombination activating gene 1/green fluorescent protein (RAG1/GFP) expression in secondary lymphoid organs after immunization with T-dependent antigens in rag1/gfp knockin mice. Blood 97:2680–2687 [DOI] [PubMed] [Google Scholar]
- 44. Yarkoni Y, Getahun A, Cambier JC. 2010. Molecular underpinning of B-cell anergy. Immunol. Rev. 237:249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]